
Design and Synthesis of Fe3O4-Loaded Polymer Microspheres with Controlled Morphology




Abstract
1. Introduction
2. Experimental
2.1. Materials
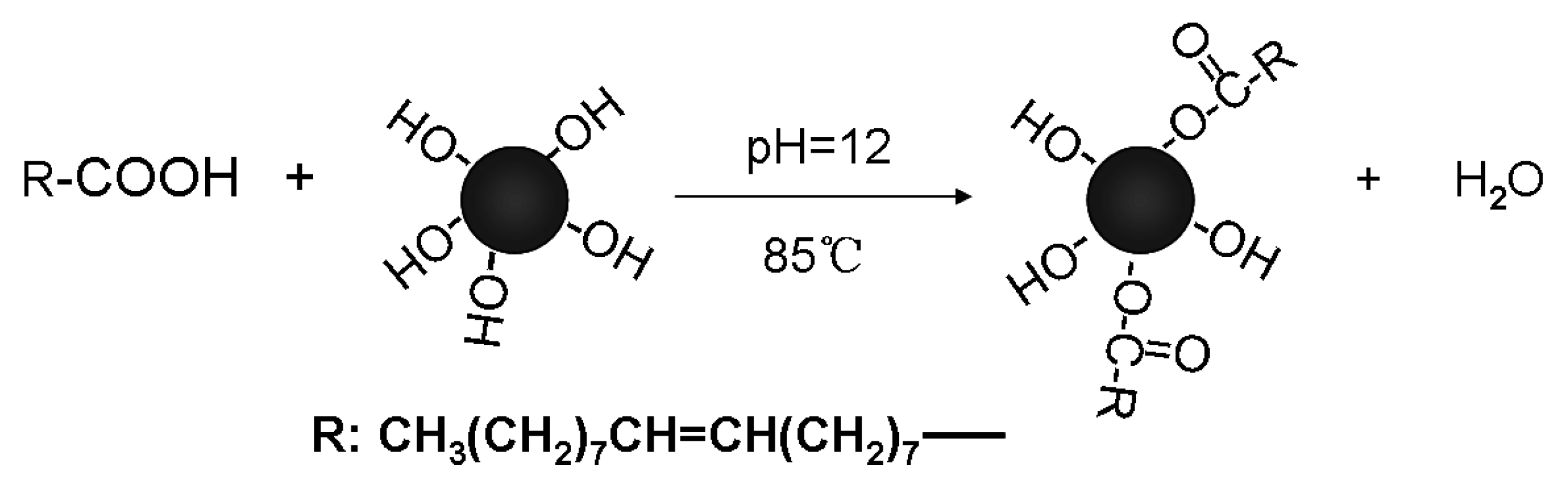
2.2. Magnetic Nanoparticle (MNP) Synthesis and Modification
2.3. Preparation of MPCPs
2.4. Characterizations
3. Results and Discussion
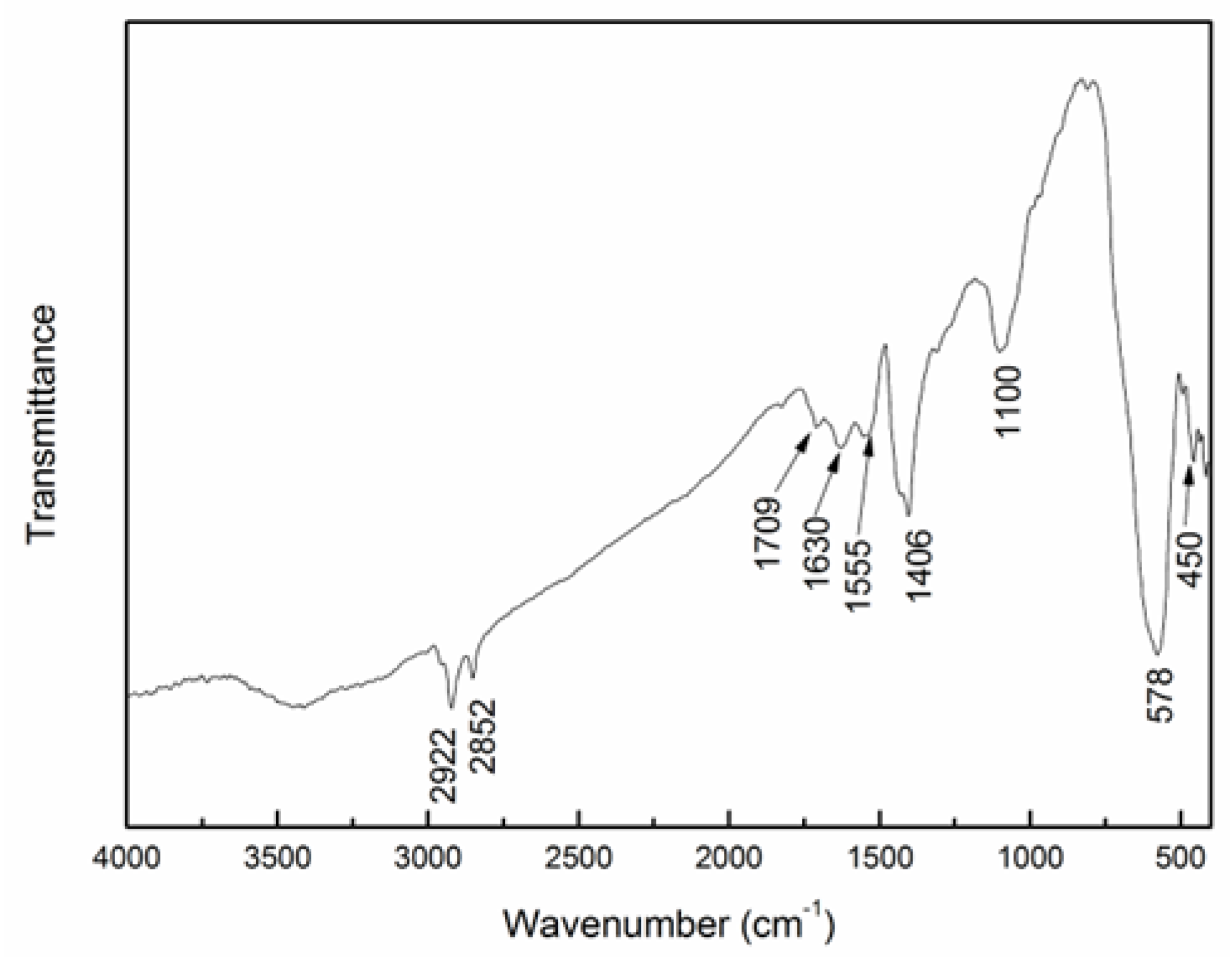
3.1. Characterization of Synthesized MNPs
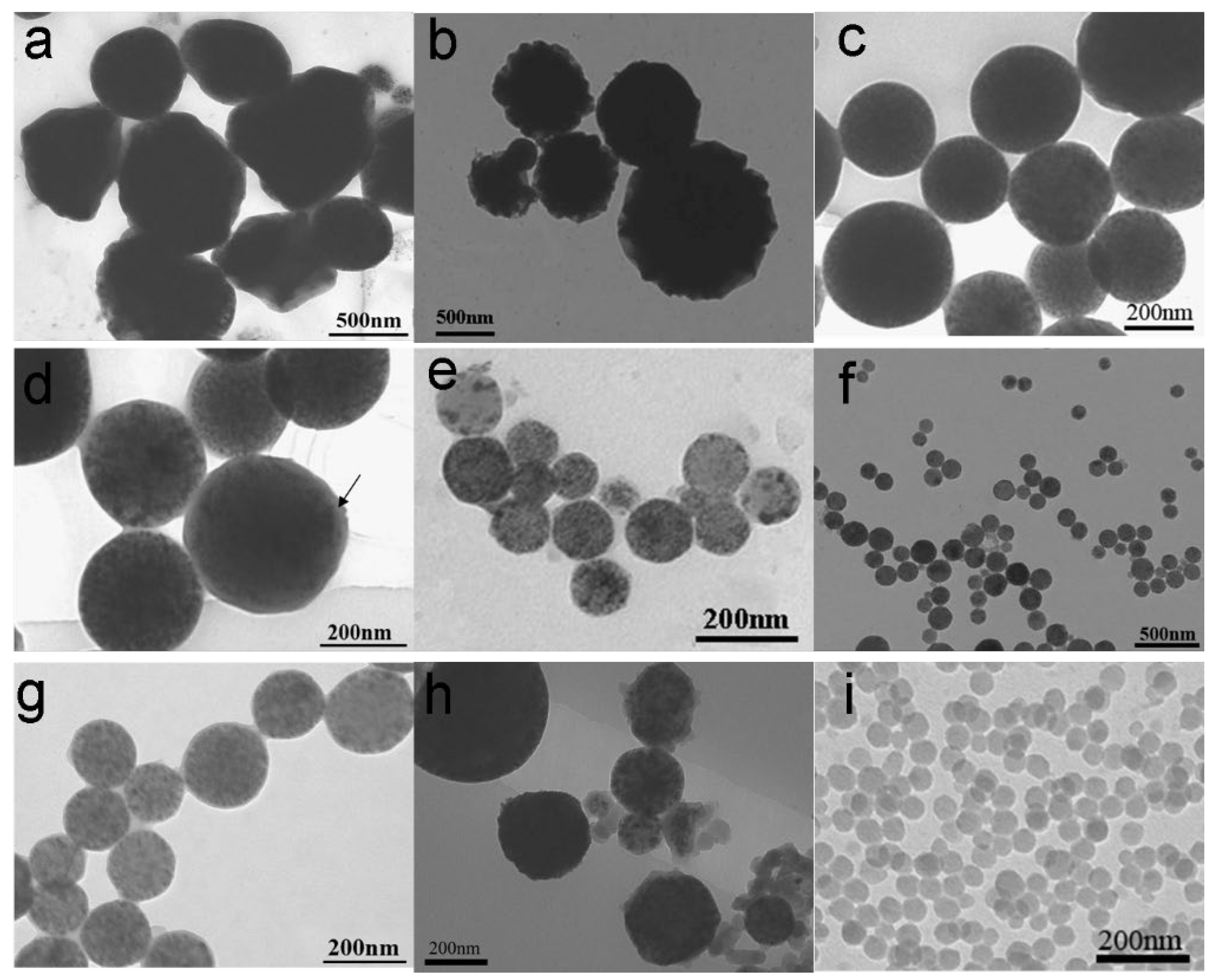
3.2. Morphology Characterization of MPCPs
3.3. Formation Mechanism of MPCPs
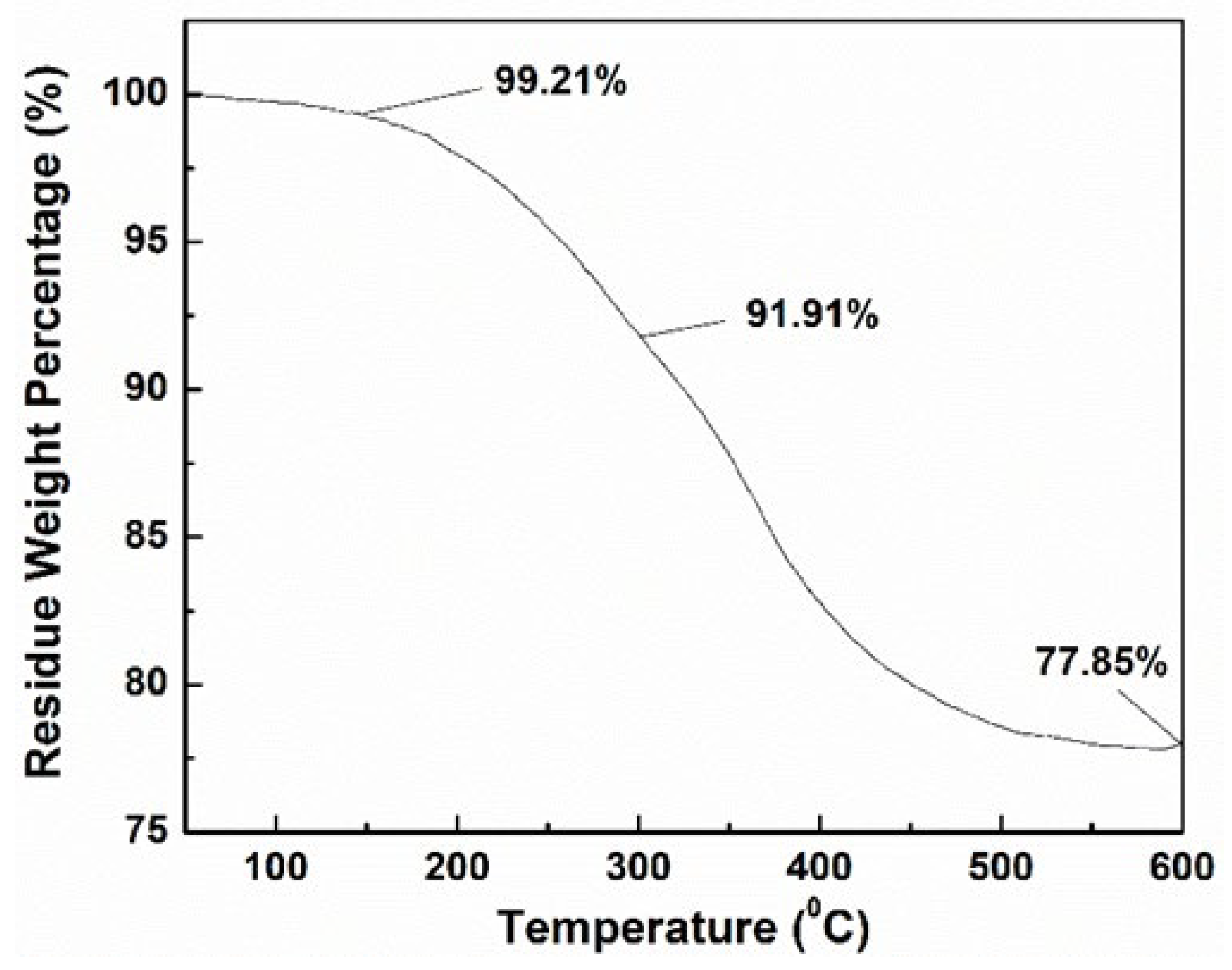
3.4. Properties Characterizations of MPCPs
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Dong, X.; Zheng, Y.; Huang, Y.; Chen, X.; Jing, X. Synthesis and characterization of multifunctional poly (glycidyl methacrylate) microspheres and their use in cell separation. Anal. Biochem. 2010, 405, 207–212. [Google Scholar] [PubMed]
- Hong, X.; Chu, X.; Zou, P.; Liu, Y.; Yang, G. Magnetic-field-assisted rapid ultrasensitive immunoassays using Fe3O4/ZnO/Au nanorices as Raman probes. Biosens. Bioelectron. 2010, 26, 918–922. [Google Scholar] [PubMed]
- Sur, K.; McFall, S.M.; Yeh, E.T.; Jangam, S.R.; Hayden, M.A.; Stroupe, S.D.; Kelso, D.M. Immiscible phase nucleic acid purification eliminates PCR inhibitors with a single pass of paramagnetic particles through a hydrophobic liquid. J. Mol. Diagn. 2010, 12, 620–628. [Google Scholar] [PubMed]
- Chen, F.; Shi, R.; Xue, Y.; Chen, L.; Wan, Q.H. Templated synthesis of monodisperse mesoporous maghemite/silica microspheres for magnetic separation of genomic DNA. J. Magn. 2010, 322, 2439–2445. [Google Scholar]
- Sureshkumar, M.; Lee, C.K. Polydopamine coated magnetic-chitin (MCT) particles as a new matrix for enzyme immobilization. Carbohydr. Polym. 2011, 84, 775–780. [Google Scholar]
- Brugger, E.C.; Prayer, D. Development of gastroschisis as seen by magnetic resonance imaging. Ultrasound Obstet. Gynecol. 2011, 37, 463–470. [Google Scholar]
- Maity, D.; Chandrasekharan, P.; Yang, C.T.; Chuang, K.H.; Shuter, B.; Xue, J.M.; Ding, J.; Feng, S.S. Facile synthesis of water-stable magnetite nanoparticles for clinical MRI and magnetic hyperthermia applications. Nanomedicine 2010, 5, 1571–1584. [Google Scholar]
- Gorti, V.M.; Shang, H.; Wereley, S.T.; Lee, G.U. Immunoassays in nanoliter volume reactors using fluorescent particle diffusometry. Langmuir 2008, 24, 2947–2952. [Google Scholar]
- Guchhait, A.; Rath, A.K.; Pal, A.J. Hybrid core−shell nanoparticles: Photoinduced electron-transfer for charge separation and solar cell applications. Chem. Mater. 2009, 21, 5292–5299. [Google Scholar]
- Liao, Z.; Wang, H.; Lv, R.; Zhao, P.; Sun, X.; Wang, S.; Su, W.; Niu, R.; Chang, J. Polymeric liposomes-coated superparamagnetic iron oxide nanoparticles as contrast agent for targeted magnetic resonance imaging of cancer cells. Langmuir 2011, 27, 3100–3105. [Google Scholar]
- Yanase, N.; Noguchi, H.; Asakura, H.; Suzuta, T. Preparation of magnetic latex particles by emulsion polymerization of styrene in the presence of a ferrofluid. J. Appl. Polym. Sci. 1993, 50, 765–776. [Google Scholar] [CrossRef]
- Wormuth, K. Superparamagnetic latex via inverse emulsion polymerization. J. Colloid Interface Sci. 2001, 241, 366–377. [Google Scholar] [CrossRef]
- Deng, Y.H.; Yang, W.L.; Wang, C.C.; Fu, S.K. A novel approach for preparation of thermoresponsive polymer magnetic microspheres with core–shell structure. Adv. Mater. 2003, 15, 1729–1732. [Google Scholar] [CrossRef]
- Wang, P.C.; Chiu, W.Y.; Lee, C.F.; Young, T.H. Synthesis of iron oxide/poly (methyl methacrylate) composite latex particles: Nucleation mechanism and morphology. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 5695–5705. [Google Scholar] [CrossRef]
- Csetneki, I.; Faix, M.K.; Szilagyi, A.; Kovacs, A.L.; Nemeth, Z.; Zrinyi, M. Preparation of magnetic polystyrene latex via the miniemulsion polymerization technique. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 4802–4808. [Google Scholar] [CrossRef]
- Zaitsev, V.S.; Filimonov, D.S.; Presnyakov, I.A.; Gambino, R.J.; Chu, B. Physical and chemical properties of magnetite and magnetite-polymer nanoparticles and their colloidal dispersions. J. Colloid Interface Sci. 1999, 212, 49–57. [Google Scholar] [CrossRef]
- Vestal, C.R.; Zhang, Z.J. Atom transfer radical polymerization synthesis and magnetic characterization of MnFe2O4/polystyrene core/shell nanoparticles. J. Am. Chem. Soc. 2002, 124, 14312–14313. [Google Scholar] [CrossRef]
- Deng, Y.; Wang, L.; Yang, W.; Fu, S.; Elaıssari, A. Preparation of magnetic polymeric particles via inverse microemulsion polymerization process. J. Magn. Magn. Mater. 2003, 257, 69–78. [Google Scholar] [CrossRef]
- Xu, Z.Z.; Wang, C.C.; Yang, W.L.; Deng, Y.H.; Fu, S.K. Encapsulation of nanosized magnetic iron oxide by polyacrylamide via inverse miniemulsion polymerization. J. Magn. Magn. Mater. 2004, 277, 136–143. [Google Scholar] [CrossRef]
- Landfester, K.; Ramirez, L.P. Encapsulated magnetite particles for biomedical application. J. Phys. Condens. Matter 2003, 15, 1345. [Google Scholar] [CrossRef]
- Yang, S.; Liu, H.; Zhang, Z. Fabrication of novel multihollow superparamagnetic magnetite/polystyrene nanocomposite microspheres via water-in-oil-in-water double emulsions. Langmuir 2008, 24, 10395–10401. [Google Scholar] [PubMed]
- de Oliveira Reis, M.; de Sousa, R.G.; Batista, A.D.S.M. Synthesis and characterization of poly (styrene-co-divinylbenzene) and nanomagnetite structures. MethodsX 2022, 9, 101764. [Google Scholar]
- Lu, A.H.; Salabas, E.E.; Schüth, F. Magnetic nanoparticles: Synthesis, protection, functionalization, and application. Angew. Chem. Int. Ed. 2007, 46, 1222–1244. [Google Scholar]
- Sun, C.; Lee, J.S.; Zhang, M. Magnetic nanoparticles in MR imaging and drug delivery. Adv. Drug Deliv. Rev. 2008, 60, 1252–1265. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MPCPs | H2O (mL) | SDS (g) | DVB (g) | CA (g) | MNPs (g) (Modified) | MNPs (g) (Unmodified) |
|---|---|---|---|---|---|---|
| S-1 | 30 | 0.02 | 3 | 0.20 | 0.10 | / |
| S-2 | 30 | 0.04 | 3 | 0.20 | 0.10 | / |
| S-3 | 30 | 0.06 | 3 | 0.20 | 0.10 | / |
| S-4 | 30 | 0.06 | 3 | 0.30 | 0.10 | / |
| S-5 | 30 | 0.06 | 3 | 0.40 | 0.10 | / |
| S-6 | 30 | 0.06 | 3 | 0.20 | / | 0.10 |
| S-7 | 30 | 0.06 | 3 | 0.2 | / | / |
| MPCPs | Average Size (nm) | Standard Deviation (nm) |
|---|---|---|
| S-1 | 500 | 150 |
| S-2 | 400 | 100 |
| S-3 | 200 | 25 |
| S-4 | 100 | 15 |
| S-5 | 80 | 10 |
| S-6 | 200 | 150 |
| S-7 | 50 | 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scheff, T.; Acha, F.; Armas, N.D.; Turkoglu, S.; Zhang, J. Design and Synthesis of Fe3O4-Loaded Polymer Microspheres with Controlled Morphology. Polymers 2025, 17, 865. https://doi.org/10.3390/polym17070865
Scheff T, Acha F, Armas ND, Turkoglu S, Zhang J. Design and Synthesis of Fe3O4-Loaded Polymer Microspheres with Controlled Morphology. Polymers. 2025; 17(7):865. https://doi.org/10.3390/polym17070865
Chicago/Turabian StyleScheff, Talya, Florence Acha, Nathalia Diaz Armas, Sevil Turkoglu, and Jinde Zhang. 2025. "Design and Synthesis of Fe3O4-Loaded Polymer Microspheres with Controlled Morphology" Polymers 17, no. 7: 865. https://doi.org/10.3390/polym17070865
APA StyleScheff, T., Acha, F., Armas, N. D., Turkoglu, S., & Zhang, J. (2025). Design and Synthesis of Fe3O4-Loaded Polymer Microspheres with Controlled Morphology. Polymers, 17(7), 865. https://doi.org/10.3390/polym17070865