
Anion-Exchange Strategy for Ru/RuO2-Embedded N/S-Co-Doped Porous Carbon Composites for Electrochemical Nitrogen Fixation

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instruments
2.3. Preparation Procedure
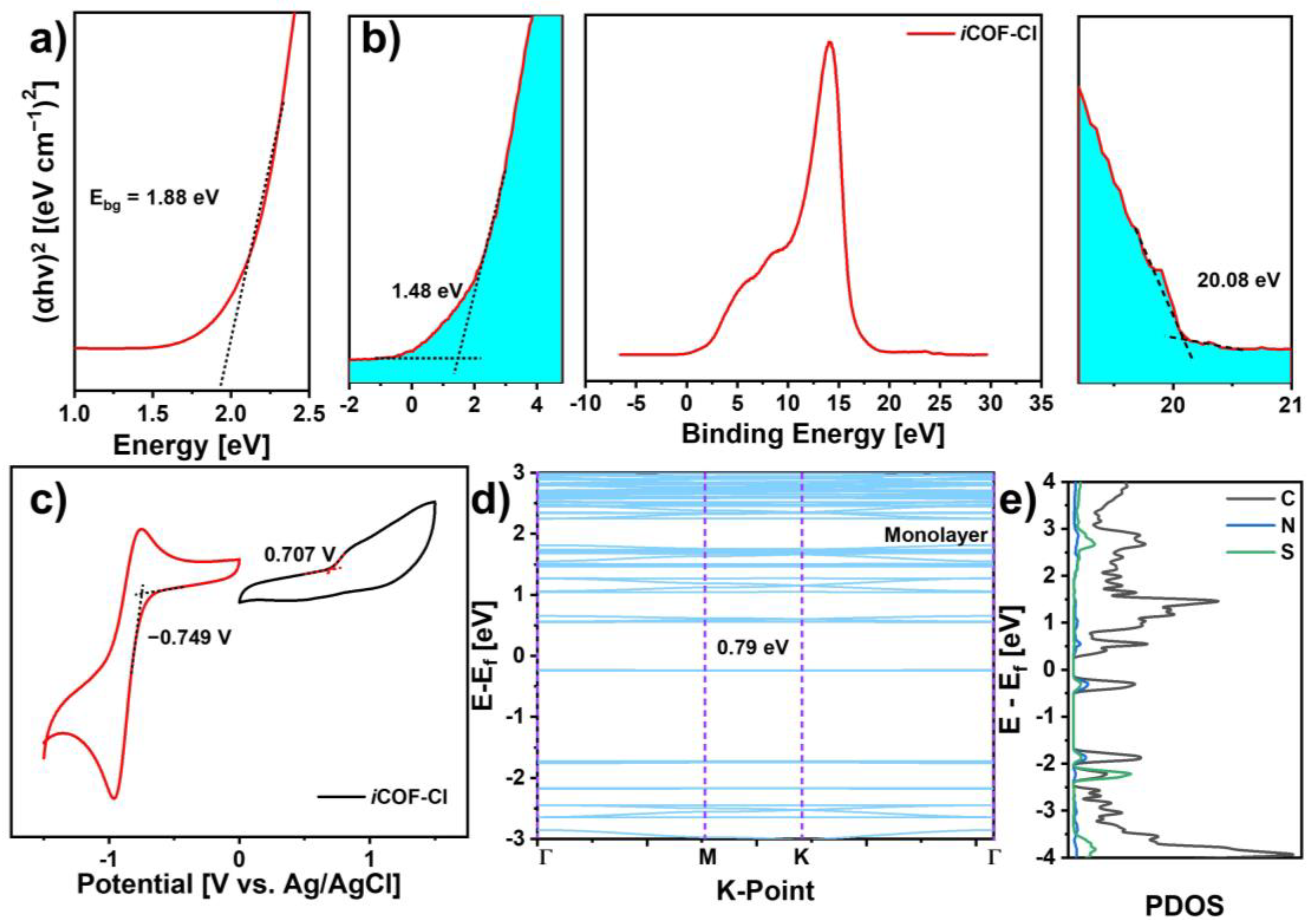
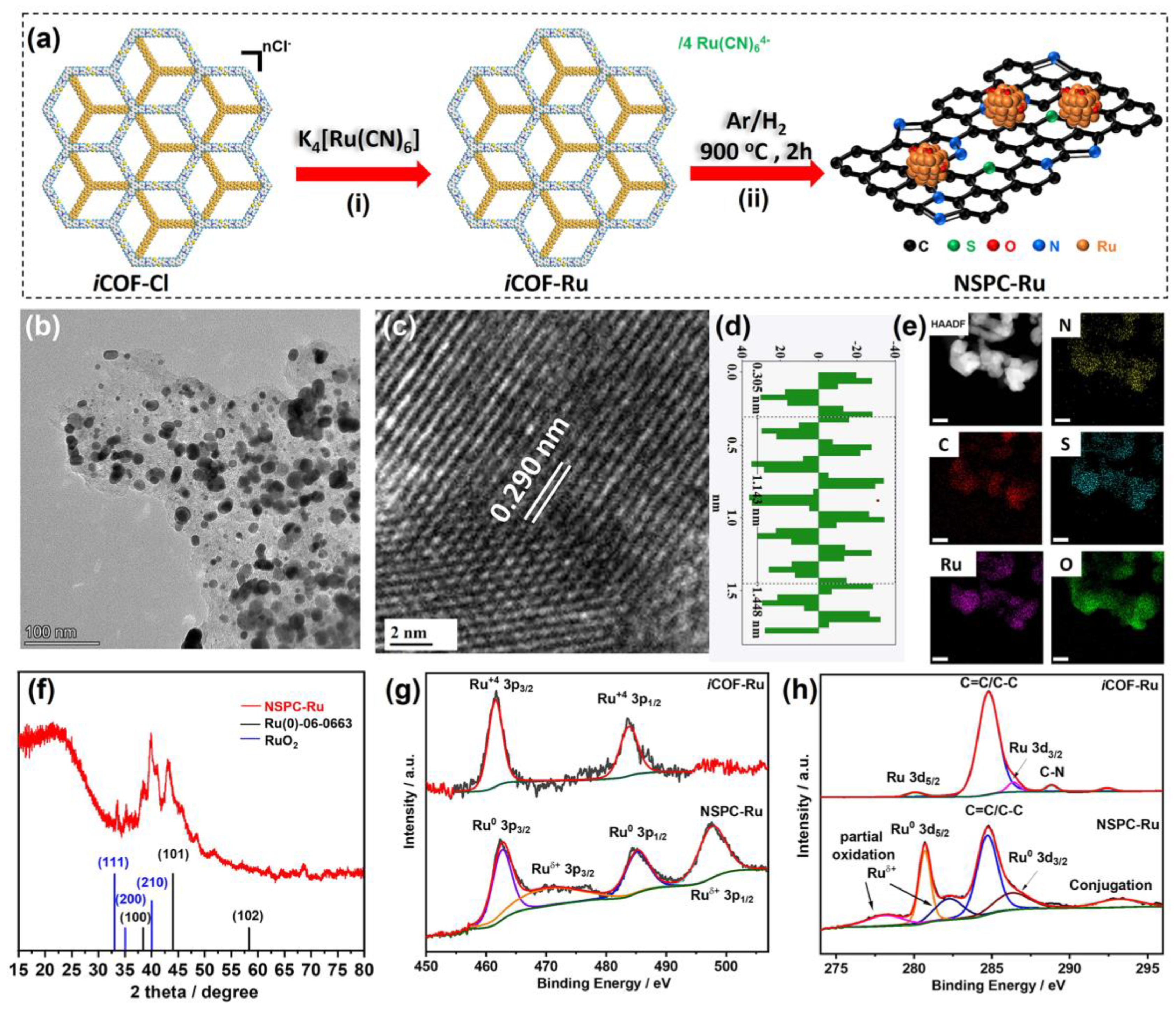
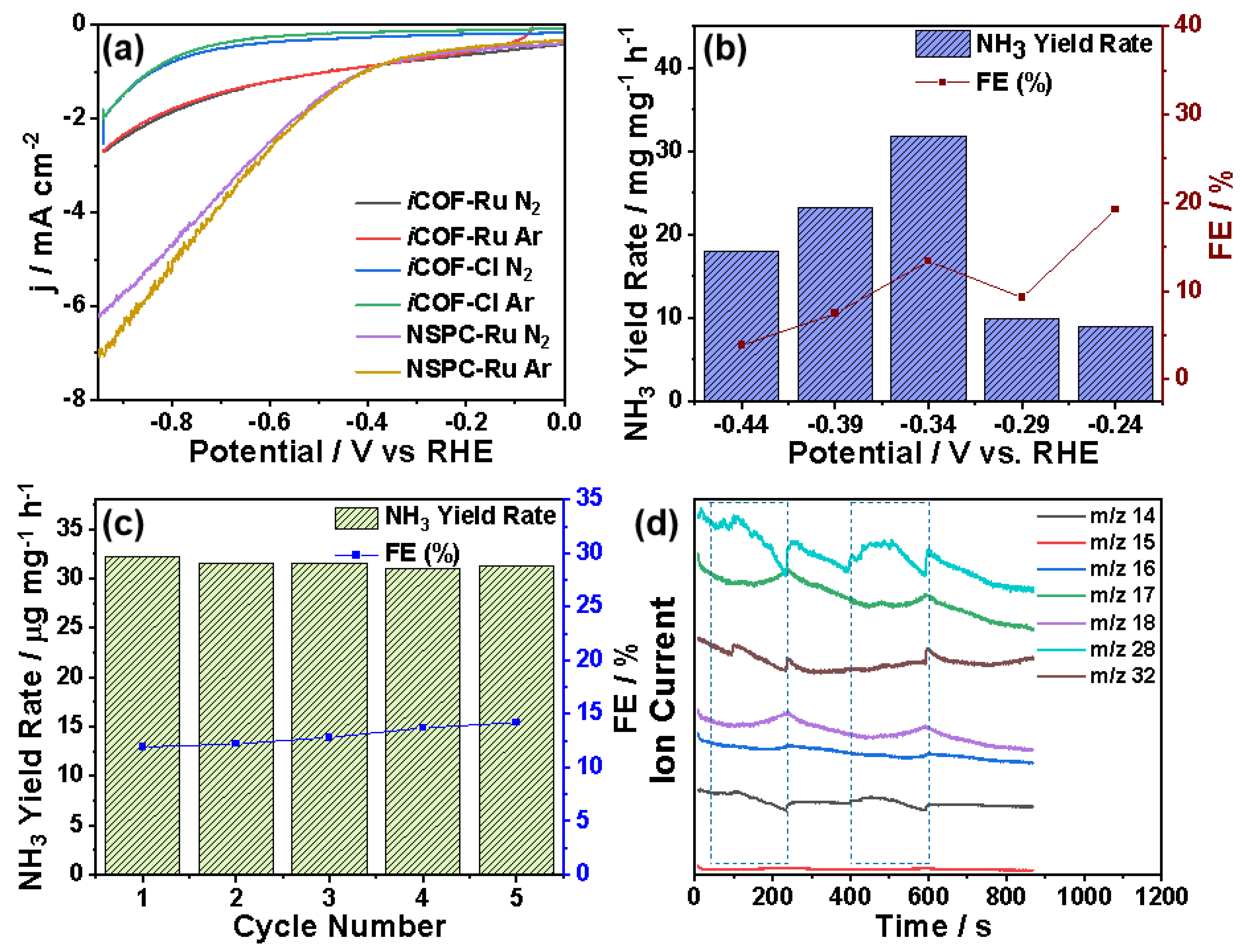
3. Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Kuriyama, S.; Arashiba, K.; Nakajima, K.; Matsuo, Y.; Tanaka, H.; Ishii, K.; Yoshizawa, K.; Nishibayashi, Y. Catalytic transformation of dinitrogen into ammonia and hydrazine by iron-dinitrogen complexes bearing pincer ligand. Nat. Commun. 2016, 7, 12181. [Google Scholar] [CrossRef] [PubMed]
- Kyriakou, V.; Garagounis, I.; Vourros, A.; Vasileiou, E.; Stoukides, M. An Electrochemical Haber-Bosch Process. Joule 2020, 4, 142–158. [Google Scholar] [CrossRef]
- Yu, X.; Han, P.; Wei, Z.; Huang, L.; Gu, Z.; Peng, S.; Ma, J.; Zheng, G. Boron-Doped Graphene for Electrocatalytic N2 Reduction. Joule 2018, 2, 1610–1622. [Google Scholar] [CrossRef]
- Ma, X.-L.; Liu, J.-C.; Xiao, H.; Li, J. Surface Single-Cluster Catalyst for N2-to-NH3 Thermal Conversion. J. Am. Chem. Soc. 2018, 140, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, M.; Qian, T.; Ji, H.; Liu, J.; Yan, C. Facilitating nitrogen accessibility to boron-rich covalent organic frameworks via electrochemical excitation for efficient nitrogen fixation. Nat. Commun. 2019, 10, 3898. [Google Scholar] [CrossRef]
- Jiang, M.; Han, L.; Peng, P.; Hu, Y.; Xiong, Y.; Mi, C.; Tie, Z.; Xiang, Z.; Jin, Z. Quasi-Phthalocyanine Conjugated Covalent Organic Frameworks with Nitrogen-Coordinated Transition Metal Centers for High-Efficiency Electrocatalytic Ammonia Synthesis. Nano Lett. 2022, 22, 372–379. [Google Scholar] [CrossRef]
- Ohashi, K.; Iwase, K.; Harada, T.; Nakanishi, S.; Kamiya, K. Rational Design of Electrocatalysts Comprising Single-Atom-Modified Covalent Organic Frameworks for the N2 Reduction Reaction: A First-Principles Study. J. Phys. Chem. C 2021, 125, 10983–10990. [Google Scholar] [CrossRef]
- Song, P.; Wang, H.; Cao, X.; Liu, N.; Wang, Q.; Wang, R. Ambient Electrochemical N2 Reduction to NH3 on Nitrogen and Phosphorus Co-doped Porous Carbon with Trace Iron in Alkaline Electrolytes. Chem. Electro. Chem. 2020, 7, 212–216. [Google Scholar] [CrossRef]
- Chen, W.; Ge, C.; Li, J.T.; Beckham, J.L.; Yuan, Z.; Wyss, K.M.; Advincula, P.A.; Eddy, L.; Kittrell, C.; Chen, J.; et al. Heteroatom-Doped Flash Graphene. ACS Nano 2022, 16, 6646–6656. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Bai, H.; Cai, J.; Xiao, Y.; Talifu, D.; Abulizi, A. Efficient electrochemical reduction of nitrogen from biomass doped boron and nitrogen under ambient conditions. Surf. Interface Anal. 2023, 55, 176–183. [Google Scholar] [CrossRef]
- Zang, W.; Yang, T.; Zou, H.; Xi, S.; Zhang, H.; Liu, X.; Kou, Z.; Du, Y.; Feng, Y.P.; Shen, L.; et al. Copper Single Atoms Anchored in Porous Nitrogen-Doped Carbon as Efficient pH-Universal Catalysts for the Nitrogen Reduction Reaction. ACS Catal. 2019, 9, 10166–10173. [Google Scholar] [CrossRef]
- Huang, Y.-B.; Pachfule, P.; Sun, J.-K.; Xu, Q. From covalent–organic frameworks to hierarchically porous B-doped carbons: A molten-salt approach. J. Mater. Chem. A 2016, 4, 4273–4279. [Google Scholar] [CrossRef]
- Romero, J.; Rodriguez-San-Miguel, D.; Ribera, A.; Mas-Ballesté, R.; Otero, T.F.; Manet, I.; Licio, F.; Abellán, G.; Zamora, F.; Coronado, E. Metal-functionalized covalent organic frameworks as precursors of supercapacitive porous N-doped graphene. J. Mater. Chem. A 2017, 5, 4343–4351. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, L.; Chen, Z.; Liu, H.; Luque, R.; Li, Y. A covalent organic framework-based route to the in situ encapsulation of metal nanoparticles in N-rich hollow carbon spheres. Chem. Sci. 2016, 7, 6015–6020. [Google Scholar] [CrossRef]
- Xu, Q.; Tang, Y.; Zhang, X.; Oshima, Y.; Chen, Q.; Jiang, D. Template Conversion of Covalent Organic Frameworks into 2D Conducting Nanocarbons for Catalyzing Oxygen Reduction Reaction. Adv. Mater. 2018, 30, 1706330. [Google Scholar] [CrossRef]
- Côté, A.P.; Benin, A.I.; Ockwig, N.W.; O’Keeffe, M.; Matzger, A.J.; Yaghi, O.M. Porous, Crystalline, Covalent Organic Frameworks. Science 2005, 310, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, Y.; Liang, H.; Wu, N.; Peng, X.; Wang, L.; Song, Y. A novel N,S-rich COF and its derived hollow N,S-doped carbon@Pd nanorods for electrochemical detection of Hg2+ and paracetamol. J. Hazard. Mater. 2021, 409, 124528. [Google Scholar] [CrossRef]
- Xu, Q.; Tang, Y.; Zhai, L.; Chen, Q.; Jiang, D. Pyrolysis of covalent organic frameworks: A general strategy for template converting conventional skeletons into conducting microporous carbons for high-performance energy storage. Chem. Commun. 2017, 53, 11690–11693. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Guo, D.; Hu, A.; Yang, X.; Shen, K.; Chen, L.; Li, Y. Resisting metal aggregation in pyrolysis of MOFs towards high-density metal nanocatalysts for efficient hydrazine assisted hydrogen production. Nano Res. 2023, 16, 6067–6075. [Google Scholar] [CrossRef]
- Feng, J.D.; Zhang, W.D.; Gu, Z.G. Covalent organic frameworks for electrocatalysis: Design, applications and perspective. Chem. Plus Chem. 2024, 89, e202400069. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Li, C.; Huang, J.; Yuan, Y.; Tian, Y.; Zhang, W. Ionic Covalent Organic Framework Solid-State Electrolytes. Adv. Mater. 2024, 36, 2407761. [Google Scholar] [CrossRef] [PubMed]
- Das, G.; Sharma, S.K.; Prakasam, T.; Gándara, F.; Mathew, R.; Alkhatib, N.; Saleh, N.i.; Pasricha, R.; Olsen, J.-C.; Baias, M.; et al. A polyrotaxanated covalent organic network based on viologen and cucurbit[7]uril. Commun. Chem. 2019, 2, 106. [Google Scholar] [CrossRef]
- Mitra, S.; Kandambeth, S.; Biswal, B.P.; Khayum, M.A.; Choudhury, C.K.; Mehta, M.; Kaur, G.; Banerjee, S.; Prabhune, A.; Verma, S.; et al. Self-Exfoliated Guanidinium-Based Ionic Covalent Organic Nanosheets (iCONs). J. Am. Chem. Soc. 2016, 138, 2823–2828. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, H.; Guan, X.; Tang, J.; Yusran, Y.; Li, Z.; Xue, M.; Fang, Q.; Yan, Y.; Valtchev, V.; et al. Three-Dimensional Ionic Covalent Organic Frameworks for Rapid, Reversible, and Selective Ion Exchange. J. Am. Chem. Soc. 2017, 139, 17771–17774. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-B.; Lyu, H.; Tian, J.; Wang, H.; Zhang, D.-W.; Liu, Y.; Li, Z.-T. A polycationic covalent organic framework: A robust adsorbent for anionic dye pollutants. Polym. Chem. 2016, 7, 3392–3397. [Google Scholar] [CrossRef]
- Das, G.; Skorjanc, T.; Sharma, S.K.; Gándara, F.; Lusi, M.; Shankar Rao, D.S.; Vimala, S.; Krishna Prasad, S.; Raya, J.; Han, D.S.; et al. Viologen-Based Conjugated Covalent Organic Networks via Zincke Reaction. J. Am. Chem. Soc. 2017, 139, 9558–9565. [Google Scholar] [CrossRef]
- Samanta, P.; Chandra, P.; Dutta, S.; Desai, A.V.; Ghosh, S.K. Chemically stable ionic viologen-organic network: An efficient scavenger of toxic oxo-anions from water. Chem. Sci. 2018, 9, 7874–7881. [Google Scholar] [CrossRef]
- Fu, Y.; Li, Y.; Zhang, W.; Luo, C.; Jiang, L.; Ma, H. Ionic Covalent Organic Framework: What Does the Unique Ionic Site Bring to Us? Chem. Res. Chin. Univ. 2022, 38, 296–309. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, F.; Gao, J.; Fu, Y.; Zhao, W.; Tang, R.; Zhang, W.; Zhuang, X.; Feng, X. Synthesis and Properties of C2h-Symmetric BN-Heteroacenes Tailored through Aromatic Central Cores. J. Org. Chem. 2015, 80, 10127–10133. [Google Scholar] [CrossRef] [PubMed]
- Scott, H.S.; Shivanna, M.; Bajpai, A.; Madden, D.G.; Chen, K.-J.; Pham, T.; Forrest, K.A.; Hogan, A.; Space, B.; Perry Iv, J.J.; et al. Highly Selective Separation of C2H2 from CO2 by a New Dichromate-Based Hybrid Ultramicroporous Material. ACS Appl. Mater. Interfaces 2017, 9, 33395–33400. [Google Scholar] [CrossRef]
- Alesanco, Y.; Viñuales, A.; Palenzuela, J.; Odriozola, I.; Cabañero, G.; Rodriguez, J.; Tena-Zaera, R. Multicolor Electrochromics: Rainbow-Like Devices. ACS Appl. Mater. Interfaces 2016, 8, 14795–14801. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, M.; Dong, C.; Yu, H.; Lu, Y.; Huang, X.; Paasch, S.; Brunner, E.; Heine, T.; Song, F.; et al. A thienyl-benzodithiophene-based two-dimensional conjugated covalent organic framework for fast photothermal conversion. J. Polym. Sci. 2023, 61, 1843–1848. [Google Scholar] [CrossRef]
- Vattikuti, S.V.P.; Reddy, P.A.K.; Shim, J.; Byon, C. Synthesis of vanadium-pentoxide-supported graphitic carbon nitride heterostructure and studied their hydrogen evolution activity under solar light. J. Mater. Sci. Mater. Electron. 2018, 29, 18760–18770. [Google Scholar] [CrossRef]
- Jhariat, P.; Warrier, A.; Sasmal, A.; Das, S.; Sarfudeen, S.; Kumari, P.; Nayak, A.K.; Panda, T. Reticular synthesis of two-dimensional ionic covalent organic networks as metal-free bifunctional electrocatalysts for oxygen reduction and evolution reactions. Nanoscale 2024, 16, 5665–5673. [Google Scholar] [CrossRef]
- Zhang, S.; Mei, D.; Li, K.; Yan, B. β-Ketoenamine covalent organic frameworks with abundant carboxyl groups for the efficient removal of malachite green in actual water environments. Sep. Purif. Technol. 2024, 348, 127812. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, Y.; Zhao, Y.; Qiu, F.; Jiang, K.; Huang, S.; Ke, C.; Zhu, J.; Tranca, D.; Zhuang, X. B/N-Enriched Semi-Conductive Polymer Film for Micro-Supercapacitors with AC Line-Filtering Performance. Langmuir 2021, 37, 2523–2531. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Chen, X.; Yan, P.; Huang, S.; Lu, C.; Ji, H.; Zhu, J.; Yang, Z.; Cao, K.; Zhuang, X. Isomeric Dual-Pore Two-Dimensional Covalent Organic Frameworks. J. Am. Chem. Soc. 2023, 145, 26871–26882. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.-M.; Dong, L.-Z.; Qin, J.-S.; Guan, W.; Liu, J.; Li, S.-L.; Lu, M.; Lan, Y.-Q.; Su, Z.-M.; Zhou, H.-C. Effect of Imidazole Arrangements on Proton-Conductivity in Metal–Organic Frameworks. J. Am. Chem. Soc. 2017, 139, 6183–6189. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, H.; Liu, F.; Kang, J.; Qiu, F.; Ke, C.; Huang, Y.; Han, S.; Zhang, F.; Zhuang, X. Self-Assembly Approach Towards MoS2-Embedded Hierarchical Porous Carbons for Enhanced Electrocatalytic Hydrogen Evolution. Chem. Eur. J. 2021, 27, 2155–2164. [Google Scholar] [CrossRef]
- Tu, K.; Tranca, D.; Rodríguez-Hernández, F.; Jiang, K.; Huang, S.; Zheng, Q.; Chen, M.-X.; Lu, C.; Su, Y.; Chen, Z.; et al. A Novel Heterostructure Based on RuMo Nanoalloys and N-doped Carbon as an Efficient Electrocatalyst for the Hydrogen Evolution Reaction. Adv. Mater. 2020, 32, 2005433. [Google Scholar] [CrossRef]
- Gao, X.; Chen, J.; Sun, X.; Wu, B.; Li, B.; Ning, Z.; Li, J.; Wang, N. Ru/RuO2 Nanoparticle Composites with N-Doped Reduced Graphene Oxide as Electrocatalysts for Hydrogen and Oxygen Evolution. ACS Appl. Nano Mater. 2020, 3, 12269–12277. [Google Scholar] [CrossRef]
- Wang, X.; Li, D.; Dai, J.; Xue, Q.; Yang, C.; Xia, L.; Qi, X.; Bao, B.; Yang, S.; Xu, Y.; et al. Blocking Metal Nanocluster Growth through Ligand Coordination and Subsequent Polymerization: The Case of Ruthenium Nanoclusters as Robust Hydrogen Evolution Electrocatalysts. Small 2024, 20, 2309176. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Zhang, H.; Liu, K.; Zhang, Q.; Chen, K.-J. Biowaste-Derived Bimetallic Ru–MoOx Catalyst for the Direct Hydrogenation of Furfural to Tetrahydrofurfuryl Alcohol. ACS Sustain. Chem. Eng. 2019, 7, 12858–12866. [Google Scholar] [CrossRef]
- Han, Z.; Tranca, D.; Rodríguez-Hernández, F.; Jiang, K.; Zhang, J.; He, M.; Wang, F.; Han, S.; Wu, P.; Zhuang, X. Embedding Ru Clusters and Single Atoms into Perovskite Oxide Boosts Nitrogen Fixation and Affords Ultrahigh Ammonia Yield Rate. Small 2023, 19, 2208102. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Huang, S.; Zhang, J.; Wang, F.; Han, S.; Wu, P.; He, M.; Zhuang, X. Single Ru–N4 Site-Embedded Porous Carbons for Electrocatalytic Nitrogen Reduction. ACS Appl. Mater. Interfaces 2023, 15, 13025–13032. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Huang, J.; Cai, J.; Wei, Y.; Cao, A.; Liu, B.; Lu, S. In Situ/Operando Methods for Understanding Electrocatalytic Nitrate Reduction Reaction. Small Methods 2023, 7, 2300169. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samad, S.A.; Ye, X.; Han, Z.; Huang, S.; Lu, C.; Hou, J.; Yang, M.; Zhang, Z.; Qiu, F.; Zhuang, X. Anion-Exchange Strategy for Ru/RuO2-Embedded N/S-Co-Doped Porous Carbon Composites for Electrochemical Nitrogen Fixation. Polymers 2025, 17, 543. https://doi.org/10.3390/polym17040543
Samad SA, Ye X, Han Z, Huang S, Lu C, Hou J, Yang M, Zhang Z, Qiu F, Zhuang X. Anion-Exchange Strategy for Ru/RuO2-Embedded N/S-Co-Doped Porous Carbon Composites for Electrochemical Nitrogen Fixation. Polymers. 2025; 17(4):543. https://doi.org/10.3390/polym17040543
Chicago/Turabian StyleSamad, Shahzeb Ali, Xuanzi Ye, Zhiya Han, Senhe Huang, Chenbao Lu, Junbo Hou, Min Yang, Zhenyu Zhang, Feng Qiu, and Xiaodong Zhuang. 2025. "Anion-Exchange Strategy for Ru/RuO2-Embedded N/S-Co-Doped Porous Carbon Composites for Electrochemical Nitrogen Fixation" Polymers 17, no. 4: 543. https://doi.org/10.3390/polym17040543
APA StyleSamad, S. A., Ye, X., Han, Z., Huang, S., Lu, C., Hou, J., Yang, M., Zhang, Z., Qiu, F., & Zhuang, X. (2025). Anion-Exchange Strategy for Ru/RuO2-Embedded N/S-Co-Doped Porous Carbon Composites for Electrochemical Nitrogen Fixation. Polymers, 17(4), 543. https://doi.org/10.3390/polym17040543