
Electrocrystallization of Calcium Oxalate Mediated by Electrospun Polymer Fiber Using Poly(acrylic acid-co-4-styrene sulfonate)

 ,
,  and
and
Abstract
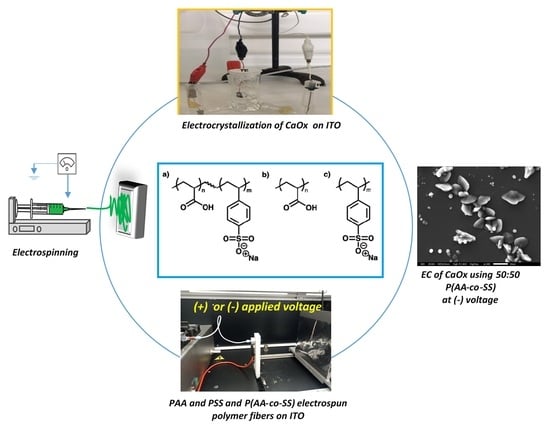
1. Introduction
2. Materials and Methods
2.1. Reactans
2.2. Synthesis of Homo- and Copolymers
2.3. Preparation of EPF by ES
2.4. In Vitro EC of CaOx
2.5. Characterization of EPF and CaOx Crystals
3. Results
3.1. FTIR of Homo- and Copolymer P(AA-co-SS) EPF Fibers
3.2. Chronopotentiometry Evaluation During CaOx EC Assays
3.3. Scanning Electron Microscopy (SEM) Analysis
3.4. X-Ray Diffraction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Daudon, M.; Letavernier, E.; Frochot, V.; Haymann, J.P.; Bazin, D.; Jungers, P. Respective influence of calcium and oxalate urine concentration on the formation of calcium oxalate monohydrate or dihydrate crystals. Comptes Rendus Chim. 2016, 19, 1504–1513. [Google Scholar] [CrossRef]
- Cai, X.H.; Lin, R.H.; Wu, J.; He, J.B.; Wu, Y.C.; Wang, X.Y. Adsorption of ethylenediaminetetraacetic dianhydride modified oxalate decarboxylase on calcium oxalate. Biotech. Histochem. 2018, 93, 220–229. [Google Scholar] [CrossRef]
- Chen, S.J.; Chiu, K.Y.; Chen, H.Y.; Lin, W.Y.; Chen, Y.H.; Chen, W.C. Animal models for studying stone disease. Diagnostics 2020, 10, 490. [Google Scholar] [CrossRef] [PubMed]
- Ake-Chiñas, M.; Mendoza-López, C.; Del-Angel-Caraza, J.; Quijano-Hernández, I.; Victoria-Mora, J. Factores demográficos y características clínicas en perros con urolitiasis de oxalato de calcio en México. Rev. MVZ Córdoba 2023, 28, 11–20. [Google Scholar] [CrossRef]
- Robinson, M.R.; Norris, R.D.; Sur, R.L.; Preminger, G.M. Urolithiasis: Not Just a 2-Legged Animal Disease. J. Urol. 2008, 179, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Del-Angel, J.; Mendoza, C.; Ake, M.; Aguiñaga, E.; Quijano, I. El gato con urolitiasis de oxalato de calcio o estruvita. Remevet 2020, 4, 5–10. [Google Scholar]
- Burggraaf, N.D.; Westgeest, D.B.; Corbee, R.J. Analysis of 7866 feline and canine uroliths submitted between 2014 and 2020 in the Netherlands. Res. Vet. Sci. 2021, 137, 86–93. [Google Scholar] [CrossRef]
- Asokan, D.; Kalaiselvi, P.; Muhammed Farooq, S.; Varalakshmi, P. Calcium oxalate monohydrate binding protein: A diagnostic biomarker for calcium oxalate kidney stone formers. Urol. Res. 2004, 32, 357–361. [Google Scholar] [CrossRef]
- Weber, E.; Verch, A.; Levy, D.; Fitch, A.N.; Pokroy, B. Amorphous biogenic calcium oxalate. ChemistrySelect 2016, 1, 132–135. [Google Scholar] [CrossRef]
- Daudon, M.; Jungers, P.; Bazin, D. Stone morphology: Implication for pathogenesis. AIP Conf. Proc. 2008, 1049, 199–215. [Google Scholar] [CrossRef]
- Ouyang, J.M.; Zheng, H.; Deng, S.P. Simultaneous formation of calcium oxalate (mono-, di-, and trihydrate) induced by potassium tartrate in gelatinous system. J. Cryst. Growth 2006, 293, 118–123. [Google Scholar] [CrossRef]
- Neira-Carrillo, A.; Vásquez, P.; Sánchez, M.; Vargas, A.; Silva, J. Control of calcium oxalate morphology through electrocrystallization as an electrochemical approach for preventing pathological disease. Ionics 2015, 21, 3141–3149. [Google Scholar] [CrossRef]
- Pak, C.Y.C.; Adams-Huet, B.; Poindexter, J.R.; Pearle, M.S.; Peterson, R.D.; Moe, O.W. Rapid Communication: Relative effect of urinary calcium and oxalate on saturation of calcium oxalate. Kidney Int. 2004, 66, 2032–2037. [Google Scholar] [CrossRef]
- Bartges, J.W.; Callens, A.J. Urolithiasis. Vet. Clin. N. Am.—Small Anim. Pract. 2015, 45, 747–768. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Chan, M.; Brown, V.; Huo, Y.R.; Chan, L.; Ridley, L. Systematic review and meta-analysis of the diagnostic accuracy of low-dose computed tomography of the kidneys, ureters and bladder for urolithiasis. J. Med. Imaging Radiat. Oncol. 2017, 61, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Neira-Carrillo, A.; Zegers Arce, T.; Yazdani-Pedram, M. Controlled calcium oxalate crystals obtained by electrocrystallization on electrospun polycaprolactone fibers loaded with zarzaparrilla (Herreria stellata). ACS Omega 2024, 9, 17045–17053. [Google Scholar] [CrossRef]
- Sun, X.Y.; Zhang, C.Y.; Bhadja, P.; Ouyang, J.M. Preparation, properties, formation mechanisms, and cytotoxicity of calcium oxalate monohydrate with various morphologies. CrystEngComm 2018, 20, 75–87. [Google Scholar] [CrossRef]
- Ding, T.; Zhao, T.; Li, Y.; Liu, Z.; Ding, J.; Ji, B.; Guo, Z. Vitexin exerts protective effects against calcium oxalate crystal-induced kidney pyroptosis in vivo and in vitro. Phytomedicine 2021, 86, 153562. [Google Scholar] [CrossRef]
- Prochaska, M.L.; Moe, O.W.; Asplin, J.R.; Coe, F.L.; Worcester, E.M. Evidence for abnormal linkage between urine oxalate and citrate excretion in human kidney stone formers. Physiol. Rep. 2021, 9, e14943. [Google Scholar] [CrossRef]
- Lojanapiwat, B.; Tanthanuch, M.; Pripathanont, C.; Ratchanon, S.; Srinualnad, S.; Taweemonkongsap, T.; Lammongkolkul, S. Alkaline citrate reduces stone recurrence and regrowth after shockwave lithotripsy and percutaneous nephrolithotomy. Int. Braz. J. Urol. 2011, 37, 611–616. [Google Scholar] [CrossRef]
- Alford, A.; Furrow, E.; Borofsky, M.; Lulich, J. Animal models of naturally occurring stone disease. Nat. Rev. Urol. 2020, 17, 691–705. [Google Scholar] [CrossRef]
- Grohe, B.; Taller, A.; Vincent, P.L.; Tieu, L.D.; Rogers, K.A.; Heiss, A.; Hunter, G.K. Crystallization of calcium oxalates is controlled by molecular hydrophilicity and specific polyanion-crystal interactions. Langmuir 2009, 25, 11635–11646. [Google Scholar] [CrossRef]
- Bayir, Y.; Halici, Z.; Keles, M.S.; Colak, S.; Cakir, A.; Kaya, Y.; Akay, F. Helichrysum plicatum DC. subsp. plicatum extract as a preventive agent in experimentally induced urolithiasis model. J. Ethnopharmacol. 2011, 138, 408–414. [Google Scholar] [CrossRef]
- Farmanesh, S.; Ramamoorthy, S.; Chung, J.; Asplin, J.R.; Karande, P.; Rimer, J.D. Specificity of growth inhibitors and their cooperative effects in calcium oxalate monohydrate crystallization. J. Am. Chem. Soc. 2014, 136, 367–376. [Google Scholar] [CrossRef] [PubMed]
- García-Perdomo, H.; Benavidez, P.; Posada, P. Pathophysiology associated with forming urinary stones. Urol. Colomb. 2016, 25, 118–125. [Google Scholar] [CrossRef]
- Yuan, P.; Sun, X.; Liu, X.; Hutterer, G.; Pummer, K.; Hager, B.; Chen, Z. Kaempferol alleviates calcium oxalate crystal-induced renal injury and crystal deposition via regulation of the AR/NOX2 signaling pathway. Phytomedicine 2021, 86, 153555. [Google Scholar] [CrossRef] [PubMed]
- Weiner, S.; Addadi, L. Crystallization pathways in biomineralization. Annu. Rev. Mater. Res. 2011, 41, 21–40. [Google Scholar] [CrossRef]
- Jung, T.; Kim, J.N.; Kim, W.S.; Chang, K.C. Study of polymeric additive effect on calcium oxalate dihydrate crystal growth using real-time atomic force microscopy. J. Cryst. Growth 2011, 327, 167–172. [Google Scholar] [CrossRef]
- Kleinman, J.G.; Alatalo, L.J.; Beshensky, A.M.; Wesson, J.A. Acidic polyanion poly(acrylic acid) prevents calcium oxalate crystal deposition. Kidney Int. 2008, 74, 919–924. [Google Scholar] [CrossRef][Green Version]
- Saxena, P.; Shukla, P. A comprehensive review on fundamental properties and applications of poly (vinylidene fluoride) (PVDF). Adv. Compos. Hybrid Mater. 2021, 4, 8–26. [Google Scholar] [CrossRef]
- Aminyan, R.; Bazgir, S. Fabrication and characterization of nanofibrous polyacrylic acid superabsorbent using gas-assisted electrospinning technique. React. Funct. Polym. 2019, 141, 133–144. [Google Scholar] [CrossRef]
- Díaz-Soler, F.; Rodriguez-Navarro, C.; Ruiz-Agudo, E.; Neira-Carrillo, A. Stabilization of calcium oxalate precursors during the pre-and post-nucleation stages with poly(acrylic acid). Nanomaterials 2021, 11, 235. [Google Scholar] [CrossRef] [PubMed]
- Jung, T.; Kim, W.S.; Kyun Choi, C. Crystal structure and morphology control of calcium oxalate using biopolymeric additives in crystallization. J. Cryst. Growth 2005, 279, 154–162. [Google Scholar] [CrossRef]
- Boerkamp, M.; Lamb, D.W.; Lye, P.G. Monitoring the kinetics of heterogeneous crystal growth using an Intrinsic Exposed Core Optical Fibre Sensor. Sens. Actuators B Chem. 2017, 240, 168–173. [Google Scholar] [CrossRef]
- Selim, K.A.; Farghaly, M.G.; Abdallah, S.S.; Abdel-khalek, M.A. Effect of polyacrylic acid molar mass as a surface modifier on rheological properties of calcium carbonate suspensions. Physicochem. Probl. Miner. Process. 2021, 57, 18–26. [Google Scholar] [CrossRef]
- Sridharan, B.; Ganesh, R.N.; Viswanathan, P. Polyacrylic acid attenuates ethylene glycol induced hyperoxaluric damage and prevents crystal aggregation in vitro and in vivo. Chem.-Biol. Interact. 2016, 252, 36–46. [Google Scholar] [CrossRef]
- Alebrahim, M.A.; Ahmad, A.A.; Alakhras, L.A.; Al-Bataineh, Q.M. Self-healing nanocomposite film of polyvinyl alcohol—Polyacrylic acid incorporated with iodine. Appl. Polym. Sci. 2025, 142, e56454. [Google Scholar] [CrossRef]
- Boyjoo, Y.; Pareek, V.K.; Liu, J. Synthesis of micro and nano-sized calcium carbonate particles and their applications. J. Mater. Chem. A 2014, 2, 14270–14288. [Google Scholar] [CrossRef]
- Shen, J.W.; Li, C.; Van Der Vegt, N.F.A.; Peter, C. Understanding the control of mineralization by polyelectrolyte additives: Simulation of preferential binding to calcite surfaces. J. Phys. Chem. C 2013, 117, 6904–6913. [Google Scholar] [CrossRef]
- Vásquez-Quitral, P.; Toledo Arana, J.; Miras, M.C.; Acevedo, D.F.; Barbero, C.A.; Neira-Carrillo, A. Effect of diazotated sulphonated polystyrene films on the calcium oxalate crystallization. Crystals 2017, 7, 70. [Google Scholar] [CrossRef]
- Neira-Carrillo, A.; Krishna Pai, R.; Fernández, M.S.; Carreño, E.; Vasquez Quitral, P.; Arias, J.L. Synthesis and characterization of sulfonated polymethylsiloxane polymer as template for crystal growth of CaCO3. Colloid Polym. Sci. 2009, 287, 385–393. [Google Scholar] [CrossRef]
- Sun, X.Y.; Shankar, R.; Börner, H.G.; Ghosh, T.K.; Spontak, R.J. Field-driven biofunctionalization of polymer fiber surfaces during electrospinning. Adv. Mater. 2007, 19, 87–91. [Google Scholar] [CrossRef]
- Stachewicz, U.; Stone, C.A.; Willis, C.R.; Barber, A.H. Charge assisted tailoring of chemical functionality at electrospun nanofiber surfaces. J. Mater. Chem. 2012, 22, 22935–22941. [Google Scholar] [CrossRef]
- Angammana, C.; Jayaram, S. Fundamentals of electrospinning and processing technologies. Part. Sci. Technol. 2016, 34, 72–82. [Google Scholar] [CrossRef]
- Cho, S.H.; Kim, J.I.; Kim, C.S.; Park, C.H.; Kim, I.G. Harnessing the topography of 3D spongy-like electrospun bundled fibrous scaffold via a sharply inclined array collector. Polymers 2019, 11, 1444. [Google Scholar] [CrossRef]
- Bhardwaj, N.; Kundu, S.C. Electrospinning: A fascinating fiber fabrication technique. Biotechnol. Adv. 2010, 28, 325–347. [Google Scholar] [CrossRef]
- Ura, D.P.; Rosell-Llompart, J.; Zaszczyńska, A.; Vasilyev, G.; Gradys, A.; Szewczyk, P.K.; Knapczyk-Korczak, J.; Avrahami, R.; Šišková, A.O.; Arinstein, A.; et al. The role of electrical polarity in electrospinning and on the mechanical and structural properties of as-spun fibers. Materials 2020, 13, 4169. [Google Scholar] [CrossRef]
- Metwally, S.; Stachewicz, U. Surface potential and charges impact on cell responses on biomaterials interfaces for medical applications. Mater. Sci. Eng. C 2019, 104, 109883. [Google Scholar] [CrossRef]
- Robinson, A.J.; Pérez-Nava, A.; Ali, S.C.; González-Campos, J.B.; Holloway, J.L.; Cosgriff-Hernandez, E.M. Comparative analysis of fiber alignment methods in electrospinning. Matter 2021, 4, 821–844. [Google Scholar] [CrossRef]
- Yu, J.; Tang, H.; Cheng, B. Influence of PSSS additive and temperature on morphology and phase structures of calcium oxalate. J. Colloid Interface Sci. 2005, 288, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Doherty, W.O.S.; Fellows, C.M.; Gorjian, S.; Senogles, E.; Cheung, W.H. Inhibition of calcium oxalate monohydrate by poly(acrylic acid)s with different end groups. J. Appl. Polym. Sci. 2004, 91, 2035–2041. [Google Scholar] [CrossRef]
- Dietrich, J.; Costa-Bauza, A.; Grases, F. Thermodynamic and kinetic aspects of calcium oxalate crystallization and renal lithiasis. Biomolecules 2025, 15, 1141. [Google Scholar] [CrossRef]
- Kim, D.; Chauhan, V.P.; Alamani, B.G.; Fisher, S.D.; Yang, Z.; Jones, M.R.; Terlier, T.; Vekilov, P.G.; Rimer, J.D. Bio-inspired multifunctional disruptors of calcium oxalate crystallization. Nat. Commun. 2025, 16, 5229. [Google Scholar] [CrossRef]
- Carees, F.O.; Sivadasan, K.; Somasundaran, P.; Turro, N.J. Interpolymer complexation of poly(acrylic acid) and poly(acrylamide): Structural and dynamic studies by solution-and solid-state NMR. Macromolecules 1994, 27, 272–278. [Google Scholar]
- Coughlin, J.E.; Reisch, A.; Markarian, M.Z.; Schlenoff, J.B. Sulfonation of polystyrene: Toward the “Ideal” polyelectrolyte. J. Polym. Sci. A Polym. Chem. 2013, 51, 2416–2424. [Google Scholar] [CrossRef]
- Kirwan, L.J.; Fawell, P.D.; Van Bronswijk, W. In situ FTIR-ATR examination of poly(acrylic acid) adsorbed onto hematite at low pH. Langmuir 2003, 19, 5802–5807. [Google Scholar] [CrossRef]
- Kausar, A. Fabrication and characteristics of poly(benzimidazole/fluoro/ether/siloxane/amide)/sulfonated polystyrene/silica nanoparticle-based proton exchange membranes doped with phosphoric acid. Int. J. Polym. Mater. Polym. Biomater. 2015, 64, 184–191. [Google Scholar] [CrossRef]
- Balding, P.; Borreli, R.; Volkovinsky, R.; Russo, P.S. Physical properties of sodium poly(styrene sulfonate): Comparison to incompletely sulfonated polystyrene. Macromolecules 2022, 55, 1747–1762. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, Y.; Zhang, Y.; Wu, C. pH-sensitive poly(acrylic acid) (PAA) hydrogels trapped with poly(sodium p-styrenesulfonate) (PSS). J. Macromol. Sci. 2006, 45 Pt B, 563–571. [Google Scholar] [CrossRef]
- M’Bareck, C.O.; Nguyen, Q.T.; Metayer, M.; Saiter, J.M.; Garda, M.R. Poly (acrylic acid) and poly (sodium styrenesulfonate) compatibility by Fourier transform infrared and differential scanning calorimetry. Polymer 2004, 45, 4181–4187. [Google Scholar] [CrossRef]
- Smith, B.C. Infrared spectroscopy of polymers X: Polyacrylates. Spectroscopy 2023, 38, 10–14. [Google Scholar] [CrossRef]
- Zorn, G.; Baio, J.E.; Weidner, T.; Migonney, V.; Castner, D.G. Characterization of poly(sodium styrene sulfonate) thin films grafted from functionalized titanium surfaces. Langmuir 2011, 27, 13104–133112. [Google Scholar] [CrossRef] [PubMed]
- Bhutto, A.A.; Vesely, D.; Gabrys, B.J. Miscibility and interactions in polystyrene and sodium sulfonated polystyrene with poly(vinyl methyl ether) PVME blends. Part II. FTIR. Polymer 2003, 44, 6627–6631. [Google Scholar] [CrossRef]
- Amiri Khoshkar Vandani, S.; Farhadian, L.; Pennycuick, A.; Ji, H.-F. Polymerization of sodium 4-styrenesulfonate inside filter paper via dielectric barrier discharge plasma. Plasma 2024, 7, 867–876. [Google Scholar] [CrossRef]
- Haryono, A.; Ghozali, M.; Harmami, S.B.; Meliana, Y. Effect of concentration on the ionic interaction between polystyrene sulfonate and cationic surfactant in aqueous solution. Mater. Sci. Forum 2018, 916, 24–29. [Google Scholar] [CrossRef]
- Lu, Q.; Cai, Z.; Wang, S.; Lin, F.; Lu, B.; Chen, Y.; Huang, B. Controlled construction of nanostructured organic-inorganic hybrid material induced by nanocellulose. ACS Sustain. Chem. Eng. 2017, 5, 8456–8463. [Google Scholar] [CrossRef]
- Metwally, S.; Ferraris, S.; Spriano, S.; Krysiak, Z.J.; Kaniuk, Ł.; Marzec, M.M.; Stachewicz, U. Surface potential and roughness controlled cell adhesion and collagen formation in electrospun PCL fibers for bone regeneration. Mater. Des. 2020, 194, 108915. [Google Scholar] [CrossRef]
- Kletzmayr, A.; Mulay, S.R.; Motrapu, M.; Luo, Z.; Anders, H.J.; Ivarsson, M.E.; Leroux, J.C. Inhibitors of calcium oxalate crystallization for the treatment of oxalate nephropathies. Adv. Sci. 2020, 7, 1903337. [Google Scholar] [CrossRef]
- Naka, K.; Chujo, Y. Control of crystal nucleation and growth of calcium carbonate by synthetic substrates. Chem. Mater. 2001, 13, 3245–3259. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ES Parameters | PAA 8% | P(PAA-co-PSS) 70:30 | P(PAA-co-PSS) 50:50 | P(PAA-co-PSS) 30:70 | PSS 20% |
|---|---|---|---|---|---|
| Voltage (+) (kV) | 15 | 15 | 13 | 23.5 | 15 |
| Voltage (−) (kV) | −17 | −15 | −13 | −23.5 | −18 |
| Distance (cm) | 15 | 20 | 16 | 20 | 15 |
| Feed rate (µL/h) | 800 | 1200 | 300 | 300 | 1000 |
| Condition | C Wt% | O Wt% | S Wt% | Na Wt% | Si Wt% | In Wt% | Al Wt% | Sn Wt% |
|---|---|---|---|---|---|---|---|---|
| V (+) | V (+) | V (+) | V (+) | V (+) | V (+) | V (+) | V (+) | |
| V (−) | V (−) | V (−) | V (−) | V (−) | V (−) | V (−) | V (−) | |
| PAA | 48.0 | 28.1 | - | - | 7.8 | 9.8 | 2.4 | 3.9 |
| 41.8 | 27.8 | - | - | 10.7 | 11.5 | 3.5 | 4.6 | |
| (PAA-co-PSS) 70:30 | 37.5 | 28.4 | 1.3 | 1.4 | 10.5 | 12.6 | 3.4 | 5.0 |
| 37.1 | 29.0 | 1.2 | 1.4 | 10.4 | 12.7 | 3.3 | 5.0 | |
| (PAA-co-PSS) 50:50 | 51.0 | 21.5 | 0.8 | 1.2 | 8.5 | 10.5 | 2.7 | 2.0 |
| 42.4 | 25.0 | 0.8 | 1.1 | 10.1 | 12.3 | 3.3 | 4.9 | |
| (PAA-co-PSS) 30:70 | 40.9 | 25.2 | 0.2 | 0.3 | 13.0 | 12.2 | 4.1 | 1.2 |
| 35.7 | 28.9 | 0.6 | 0.8 | 12.1 | 13.3 | 3.9 | 2.5 | |
| PSS | 57.2 | 17.3 | 1.1 | 1.2 | 7.7 | 9.2 | 2.5 | 3.7 |
| 55.2 | 17.5 | 0.4 | 0.5 | 8.8 | 10.5 | 2.7 | 4.3 |
| Condition | PAA 8% | P(PAA-co-PSS) 70:30 | P(PAA-co-PSS) 50:50 | P(PAA-co-PSS) 30:70 | PSS 20% |
|---|---|---|---|---|---|
| Voltage (+) | 0.162 | 0.185 | 0.095 | 0.112 | 0.146 |
| Voltage (−) | 0.158 | 0.214 | 0.061 | 0.164 | −0.030 |
| Polymer films on ITO | 0.085 | 0.124 | 0.065 | 0.126 | 0.021 |
| Polymer in ECS | −0.028 | 0.172 | 0.195 | 0.157 | 0.118 |
| Condition | C Wt% | O Wt% | S Wt% | Na Wt% | Ca Wt% | Si Wt% | In Wt% | Al Wt% | Sn Wt% |
|---|---|---|---|---|---|---|---|---|---|
| V (+) | V (+) | V (+) | V (+) | V (+) | V (+) | V (+) | V (+) | V (+) | |
| V (−) | V (−) | V (−) | V (−) | V (−) | V (−) | V (−) | V (−) | V (−) | |
| PAA | 51.7 | 21.7 | - | - | 2.9 | 9.0 | 9.9 | 2.9 | 2.0 |
| 45.1 | 36.3 | - | - | 2.5 | 2.9 | 5.6 | 0.8 | 1.3 | |
| (PAA-co-PSS) 70:30 | 47.9 | 27.5 | 0.5 | 2.8 | 1.7 | 6.8 | 8.9 | 2.0 | 1.9 |
| 40.3 | 33.2 | 0.1 | 3.3 | 3.2 | 7.0 | 8.7 | 2.2 | 1.8 | |
| (PAA-co-PSS) 50:50 | 40.5 | 29.8 | 0.3 | 1.2 | 2.9 | 9.0 | 11.0 | 2.8 | 0.3 |
| 36.8 | 36.3 | 0.1 | 2.1 | 5.2 | 6.7 | 8.7 | 2.1 | 2.0 | |
| (PAA-co-PSS) 30:70 | 36.5 | 33.5 | 0.2 | 1.0 | 2.9 | 9.7 | 11.0 | 3.1 | 2.1 |
| 34.2 | 37.0 | 0.0 | 2.8 | 3.6 | 7.9 | 10.0 | 2.5 | 1.9 | |
| PSS | 40.1 | 27.9 | 0.0 | 0.2 | 3.3 | 11.0 | 11.6 | 3.4 | 2.5 |
| 36.8 | 28.5 | 0.1 | 0.3 | 3.8 | 12.0 | 12.4 | 3.7 | 2.4 |
| ES Parameters | PAA | P(PAA-co-PSS) 70:30 | P(PAA-co-PSS) 50:50 | P(PAA-co-PSS) 30:70 | PSS |
|---|---|---|---|---|---|
| Voltage (+) (kV) | COM (98%) | COM (100%) | COM (100%) | COM (100%) | COM (97%) |
| COD (2%) | COD (0%) | COD (0%) | COD (0%) | COD (3%) | |
| Voltage (−) (kV) | COM (85%) | COM (54%) | COM (63%) | COM (92%) | COM (90%) |
| COD (15%) | COD (46%) | COD (37%) | COD (8%) | COD (10%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neira-Carrillo, A.; Nieto, E.; Butto-Miranda, N.; Cataldo, D.; Urbano, B.F.; Yazdani-Pedram, M. Electrocrystallization of Calcium Oxalate Mediated by Electrospun Polymer Fiber Using Poly(acrylic acid-co-4-styrene sulfonate). Polymers 2025, 17, 2888. https://doi.org/10.3390/polym17212888
Neira-Carrillo A, Nieto E, Butto-Miranda N, Cataldo D, Urbano BF, Yazdani-Pedram M. Electrocrystallization of Calcium Oxalate Mediated by Electrospun Polymer Fiber Using Poly(acrylic acid-co-4-styrene sulfonate). Polymers. 2025; 17(21):2888. https://doi.org/10.3390/polym17212888
Chicago/Turabian StyleNeira-Carrillo, Andrónico, Eddie Nieto, Nicole Butto-Miranda, Dania Cataldo, Bruno F. Urbano, and Mehrdad Yazdani-Pedram. 2025. "Electrocrystallization of Calcium Oxalate Mediated by Electrospun Polymer Fiber Using Poly(acrylic acid-co-4-styrene sulfonate)" Polymers 17, no. 21: 2888. https://doi.org/10.3390/polym17212888
APA StyleNeira-Carrillo, A., Nieto, E., Butto-Miranda, N., Cataldo, D., Urbano, B. F., & Yazdani-Pedram, M. (2025). Electrocrystallization of Calcium Oxalate Mediated by Electrospun Polymer Fiber Using Poly(acrylic acid-co-4-styrene sulfonate). Polymers, 17(21), 2888. https://doi.org/10.3390/polym17212888