2.2. The TSD Manifestation of Tg, the Tg,ρ Peak, and of TLL
Some peaks of the TSD outputs are caused by the motion of the dipoles at characteristic transitions in the material, such as its T
g. Some other peaks are due to the interactions between the fundamental morphological structure of the amorphous phase (and b-grains surrounded by F-conformers in the dual-phase model) and the electrical field, creating Wagner charges in F micro-voids.
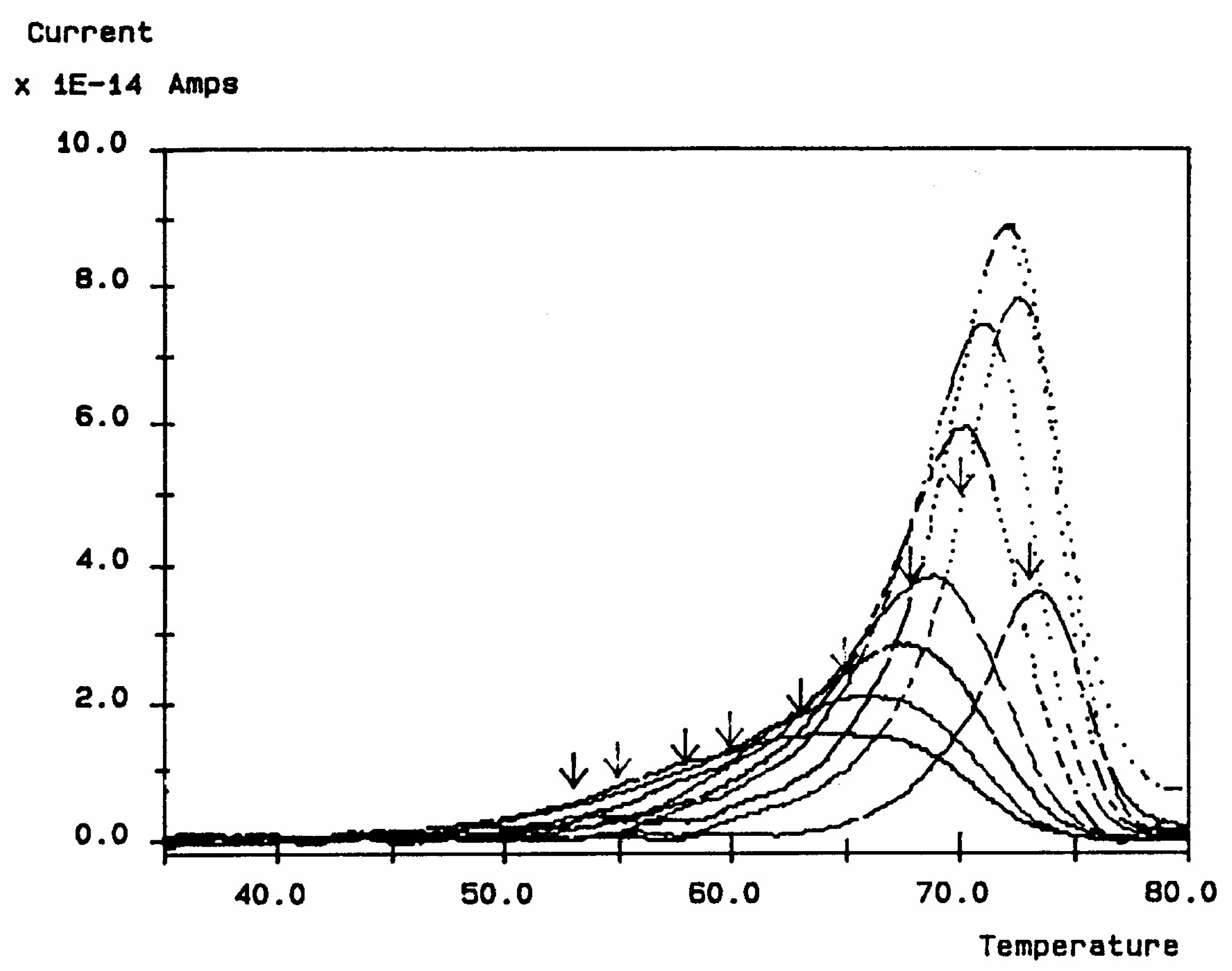
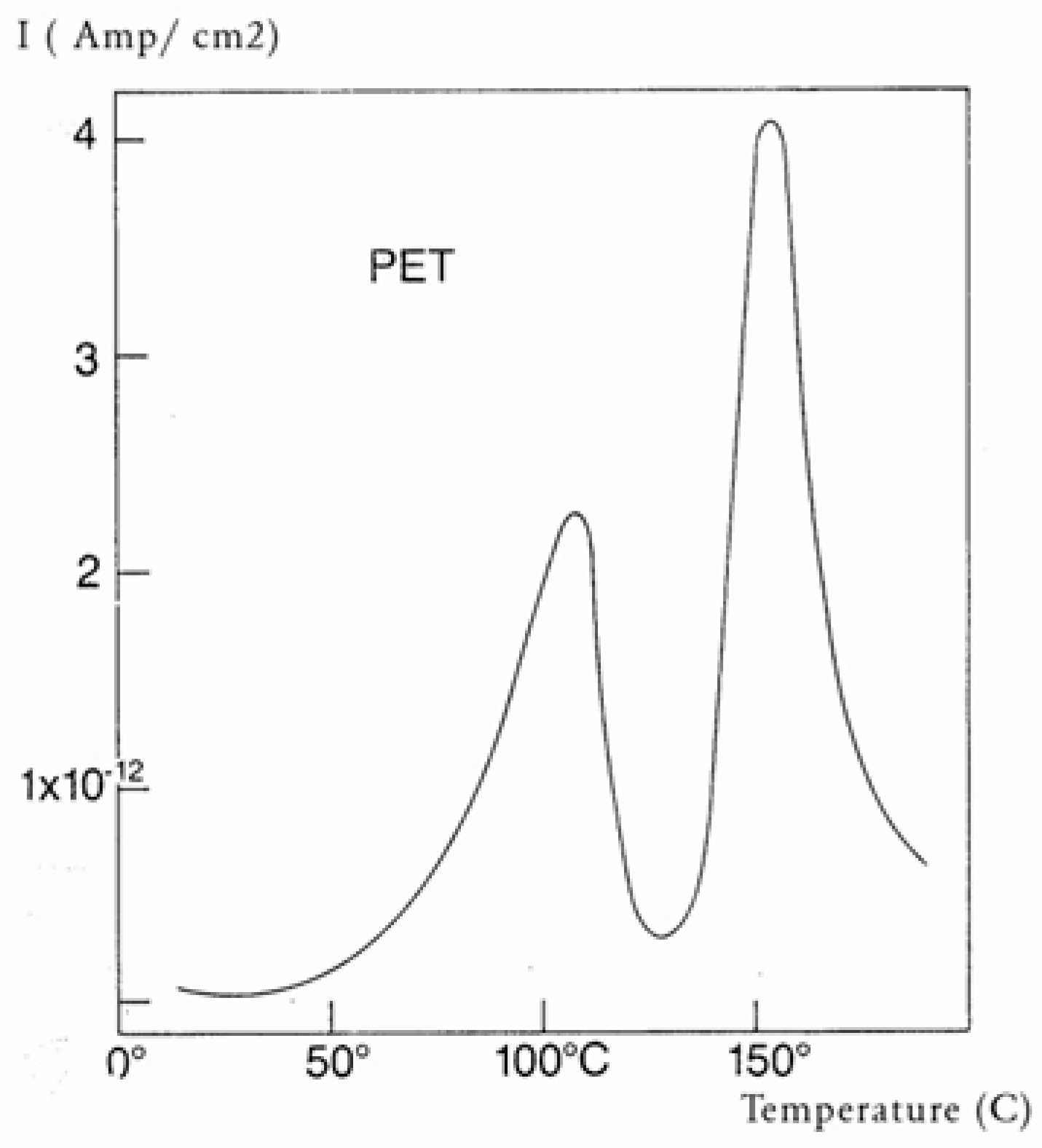
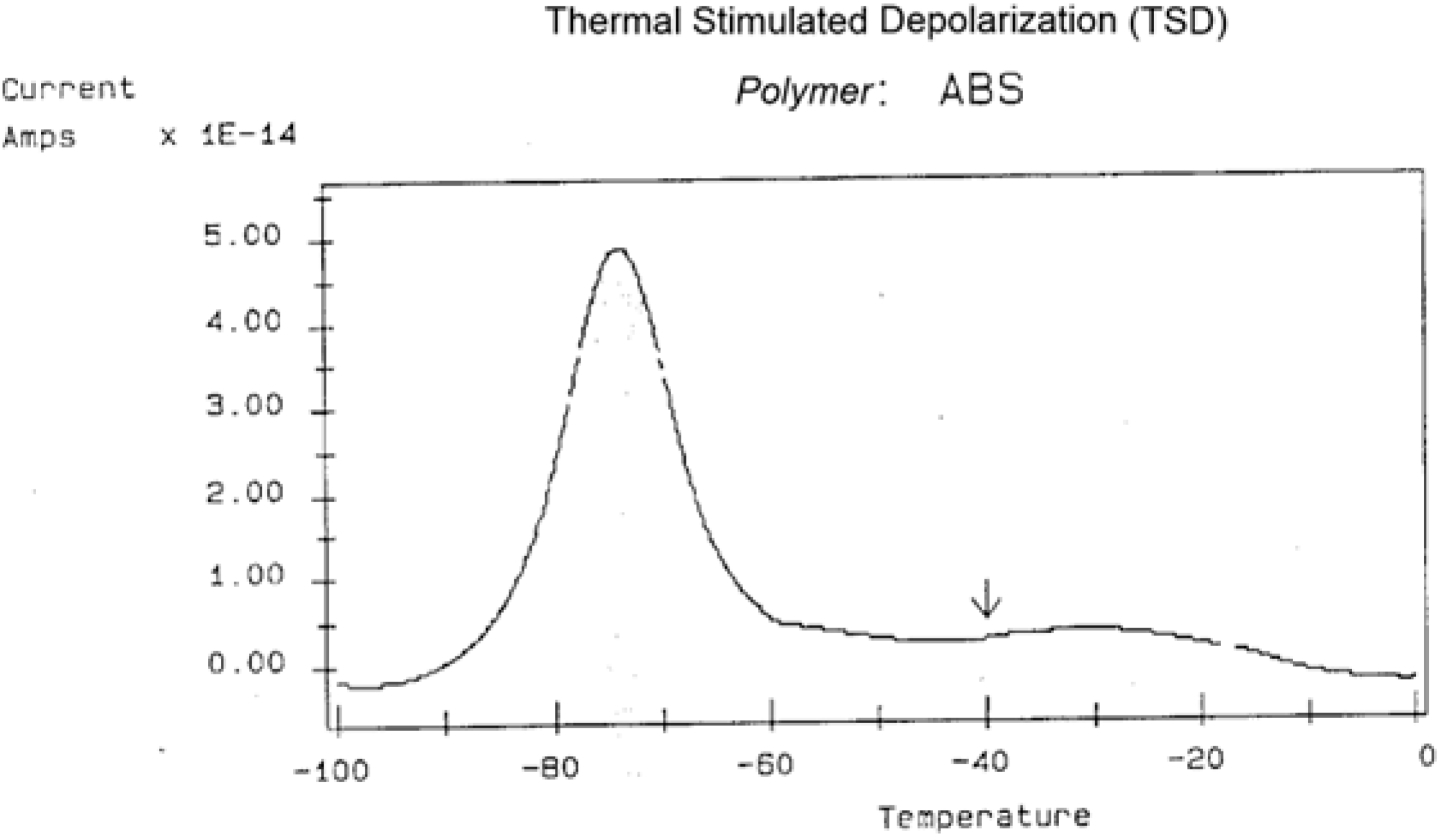
Figure 30 is a typical TSD curve for polyethylene terephthalate (PET). Two peaks are clearly visible, which we call T
g and T
g,ρ. The two peaks are separated by about 50 °C in this figure, and the intensity of the higher peak, T
g,ρ, is higher than the intensity of the first peak, T
g.
Figure 31 displays the TSD curve for polycarbonate (PC), showing that T
g,ρ is only 16 °C above the T
g peak.
The value of T
g,ρ observed by TSD is designated differently by various authors: Lacabanne and Boyer designate this as the T
LL peak [
28] and associate its existence with the local order. Vanderschueren, Van Turnout, and other authors call it T
ρ [
2,
3,
29,
30] and speculate that it originates from the discharge of space charges delocalized in the structure. Extensive studies have shown that the relative intensity of the T
g and T
g,ρ peaks, as well as their respective position, depends on the thermal and mechanical history of the polymer, in particular physical aging. The magnitude of the T
g,ρ peak is also a function of the voltage field, increasing first and then leveling off as the voltage increases. The presence of the T
g,ρ peak is universal in the characterization of polymers by TSD. We found that peak just above T
g for all the polymers tested: from paints to thermoplastics, from thermosets to rubbers, and for non-polar plastics as well as polar ones. This peak is apparently absent in the characterization of the same polymers by dynamic mechanical analysis (DMA) or DEA. It is stipulated here that differential scanning calorimetry (DSC) experiments can show T
g,ρ under special cooling and processing conditions, which enhance its manifestation (see subsequent sections). The position of T
g,ρ with respect to T
g depends, to a large extent, on the choice of the polarization temperature T
p, which enhances the respective magnitude and the resolution of the peaks by the effect of polarization selectivity mentioned in the previous section. Thus, it is not uncommon to observe the T
g and T
g,ρ peaks merge into a broad intense peak for certain polarization temperatures, complicating their fine analysis. The transition T
LL is also found by TSD, but it is located at a higher temperature, and it should not be confused with T
g,ρ. In some instances, the mechanical history of the specimen and the temperature of polarization are such that T
g,ρ and T
LL merge or overlap, making the analysis even more complicated (
Figure 32). Fortunately, the thermal-windowing process normally allows for a good separation of all the peaks, which can be isolated and deconvoluted individually. The only drawback with thermal-windowing at temperatures of polarization above T
g is that it modifies and partially erases the kinetic effects, which are responsible for the relaxations under investigation. In other words, the manifestation of both T
LL and T
g,ρ is favored by thermal history treatments, which bring the specimen far out of equilibrium, but this is quickly erased by annealing above T
g, which occurs when the sample in the TSD cell is brought up to the polarization temperature. In
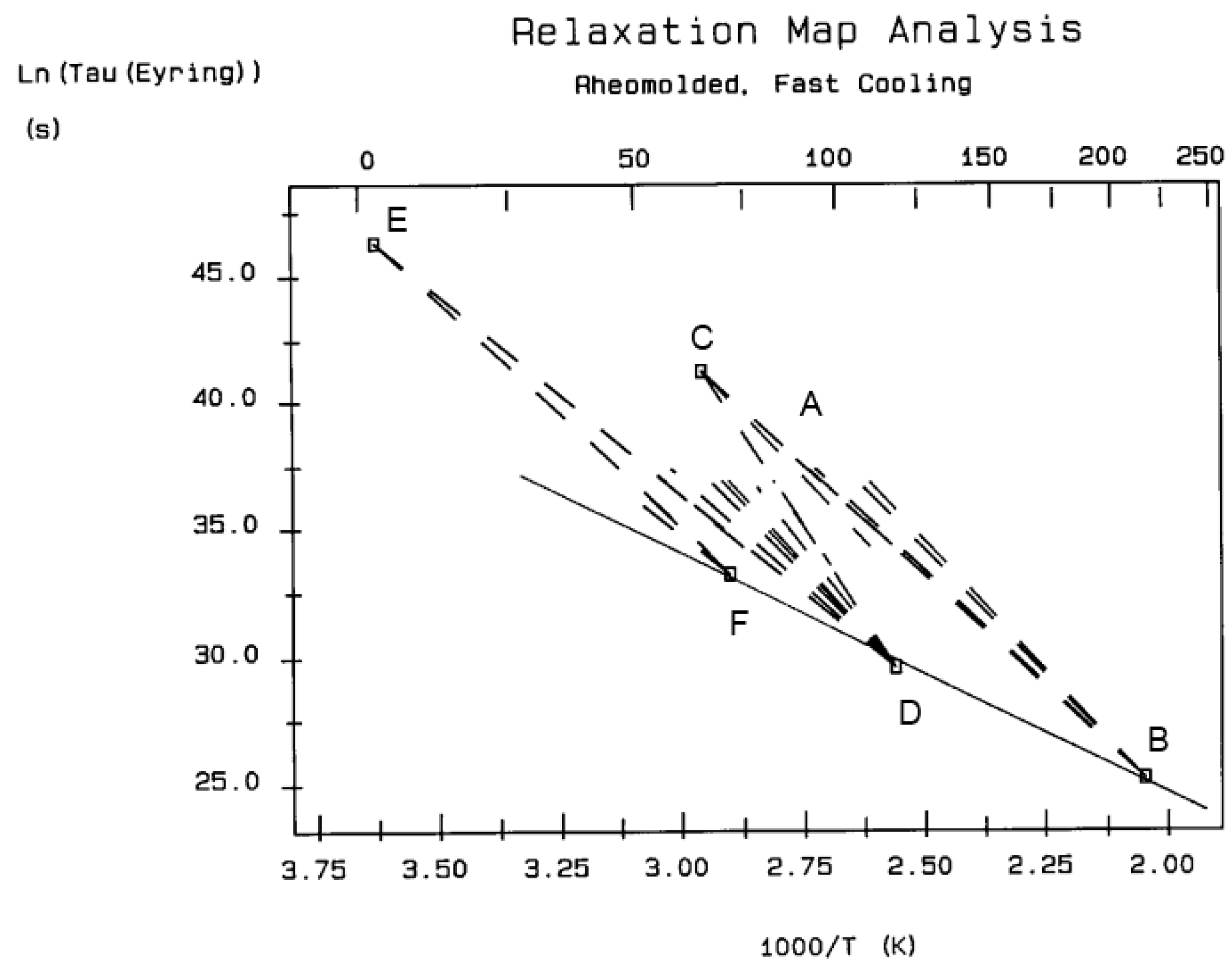
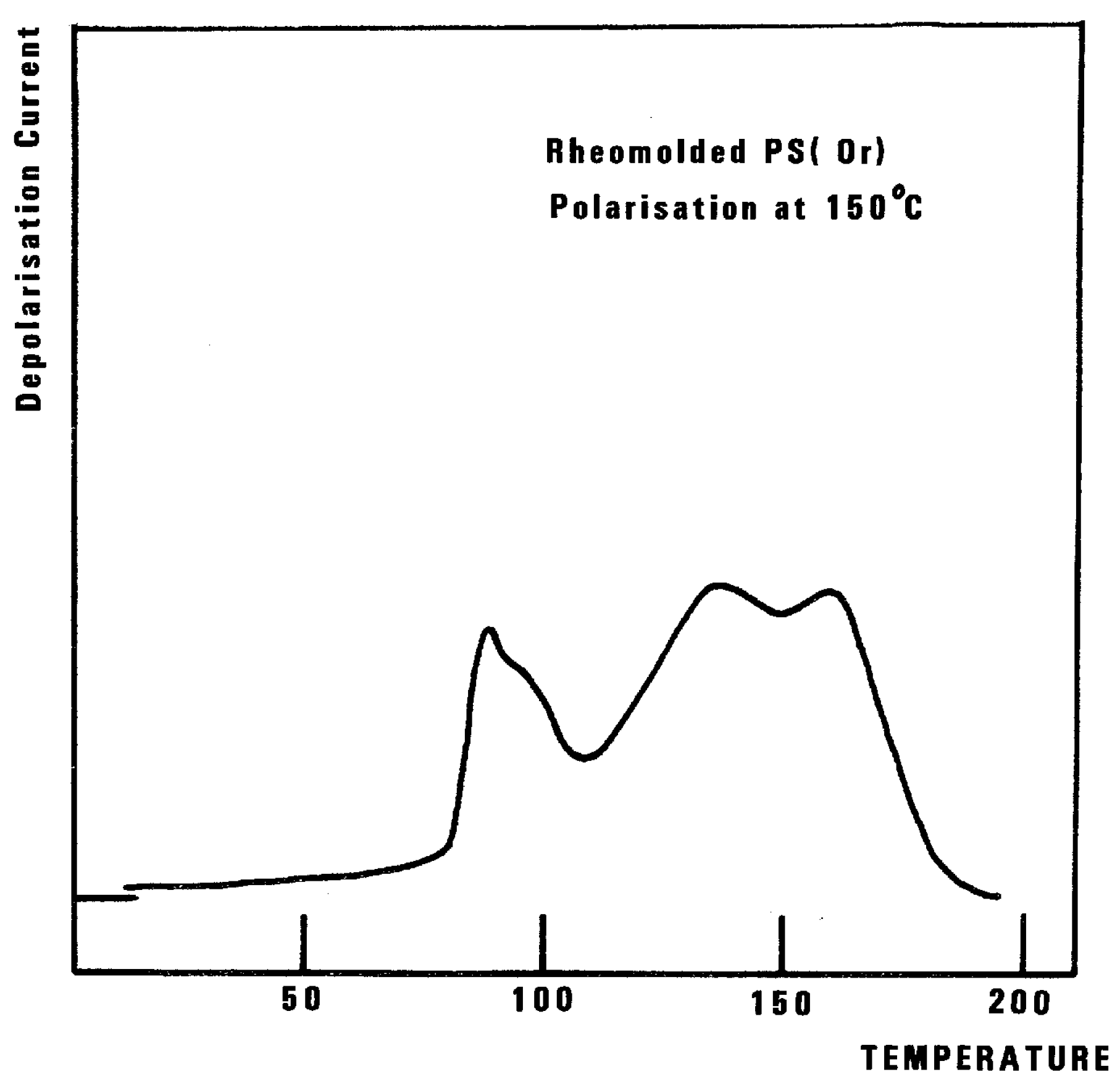
Figure 32, the 2 mm thick polystyrene specimen is compression-molded while rapidly cooled to room temperature. A platen pressure of 226 bars is applied at 122 °C prior to and during cooling. The polarization temperature is 115 °C. Three peaks are clearly visible: T
g at 97 °C, T
g,ρ at 112 °C, and T
LL at 150 °C.
The intensity of the T
g,ρ peak is enhanced by fast cooling or any other thermo-mechanical process, which creates a state of non-equilibrium in the polymer. The intensity of T
g,ρ decreases with annealing time, as demonstrated in
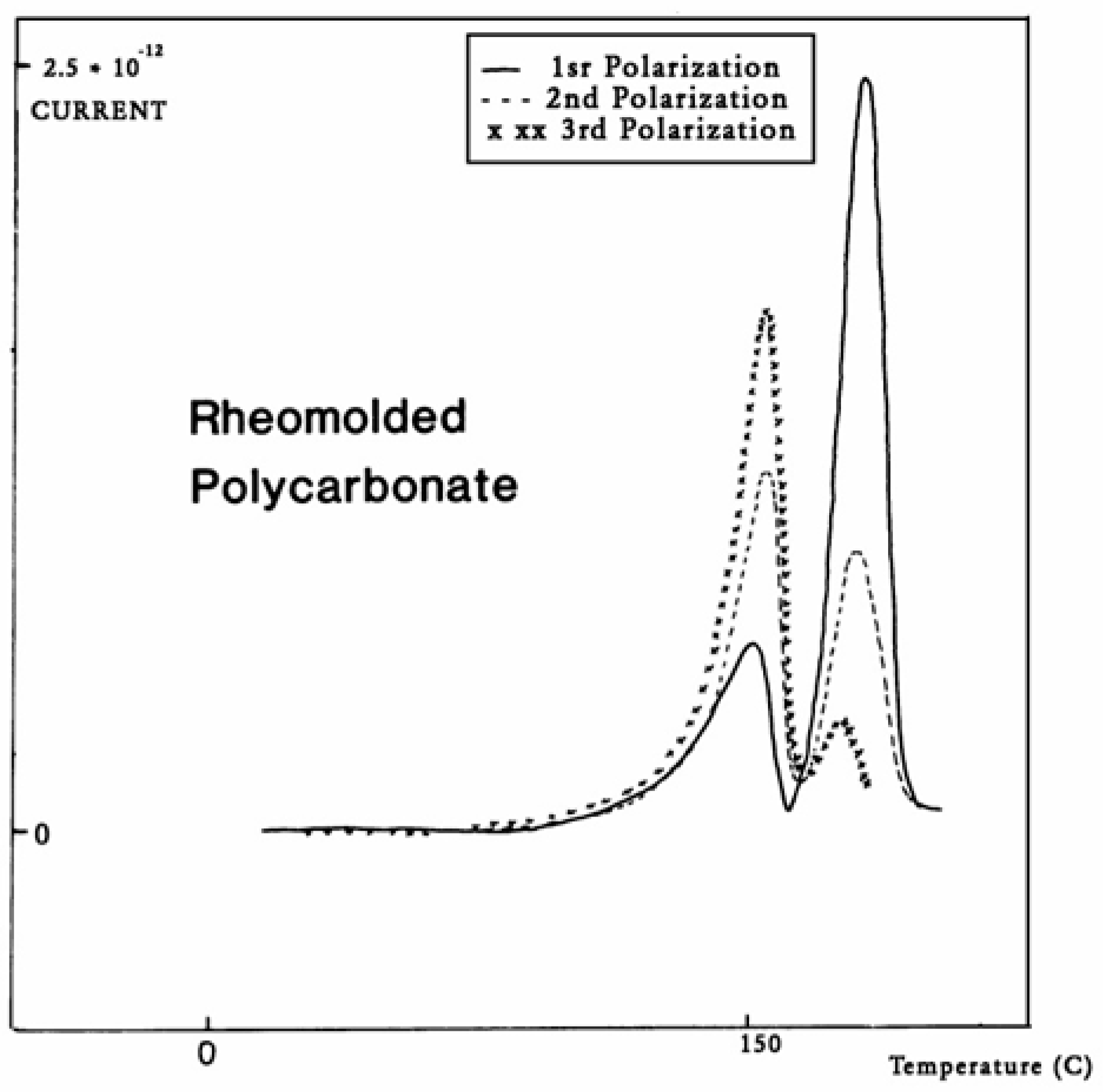
Figure 33. In this figure, PC is subjected to a compression molding treatment involving mechanical vibrations as it is being cooled. This type of treatment is called rheomolding and is described elsewhere [
8,
9]. In the TSD experiments shown in
Figure 33, a rheomolded PC is rerun three times in a row without changing the specimen. The same polarization conditions are used each time. Each run partially erases the initial thermal history of the specimen. We focus on the intensity and position of the T
g and T
g,ρ peaks. The first trace (first polarization) displays a very intense T
g,ρ peak relative to the T
g peak, but the intensity of this peak rapidly decreases for traces 2 and 3, as the initial state of PC returns to more traditional non-equilibrium conditions (cooling in the TSD cell, although rather fast, is no match to the severe cooling conditions imposed by rheomolding). It is interesting to observe that as the intensity of T
g,ρ decreases, the intensity of the T
g peak increases, in a kind of complementary manner. The position of the T
g and T
g,ρ peaks also changes in a reverse way: as T
g maximum increases slightly, T
g,ρ decreases, giving the impression of fusion of the two peaks.
The understanding of the existence of T
g,ρ and T
LL is offered in the Discussion section (
Section 3.1). As far as we are concerned, the T
g,ρ peak is real, with a high value, often stronger than T
g, and located a few degrees after T
g. In this section, which deals with the applications of TSD, it is important to introduce the existence of peaks T
g, T
g,ρ, and T
LL and suggest that their presence is universal in the context of the TSD characterization technique. It is not essential, however, to speculate about their origin. Let us just mention that Part I, Chapter 3, suggests a common kinetic origin to T
g, T
g,ρ, and T
LL, and even to T
β. The apparent complexity of the relaxation behavior arises for two reasons: Even if T
g and T
g,ρ are kinetically related, their dielectric origin is different because the effect of the voltage field on the kinetic units responsible for their respective relaxation is different. In the case of T
g, the dipole moment’s relaxation is associated with a molecular dipole but probably not so for T
g,ρ. The electric moment for T
g,ρ seems to be associated with ionic dipoles or perhaps space charges. There is much evidence to confirm that the T
g,ρ peak is related to the “free volume” in the sample, which suggests that either local unstable ionic dipoles or space charges get trapped in the free volume of the polymer under the influence of the voltage field and relax at T
g,ρ. More details are provided in
Section 3.1 of the Discussion where the “free volume” is defined in the morphology in terms of statistical units of the polymer chains. The difference between the characteristics of T
g,ρ and T
LL is definitive: Hydrostatic pressure lowers the value of T
g,ρ and increases the value of T
LL. This is shown for T
g,ρ in
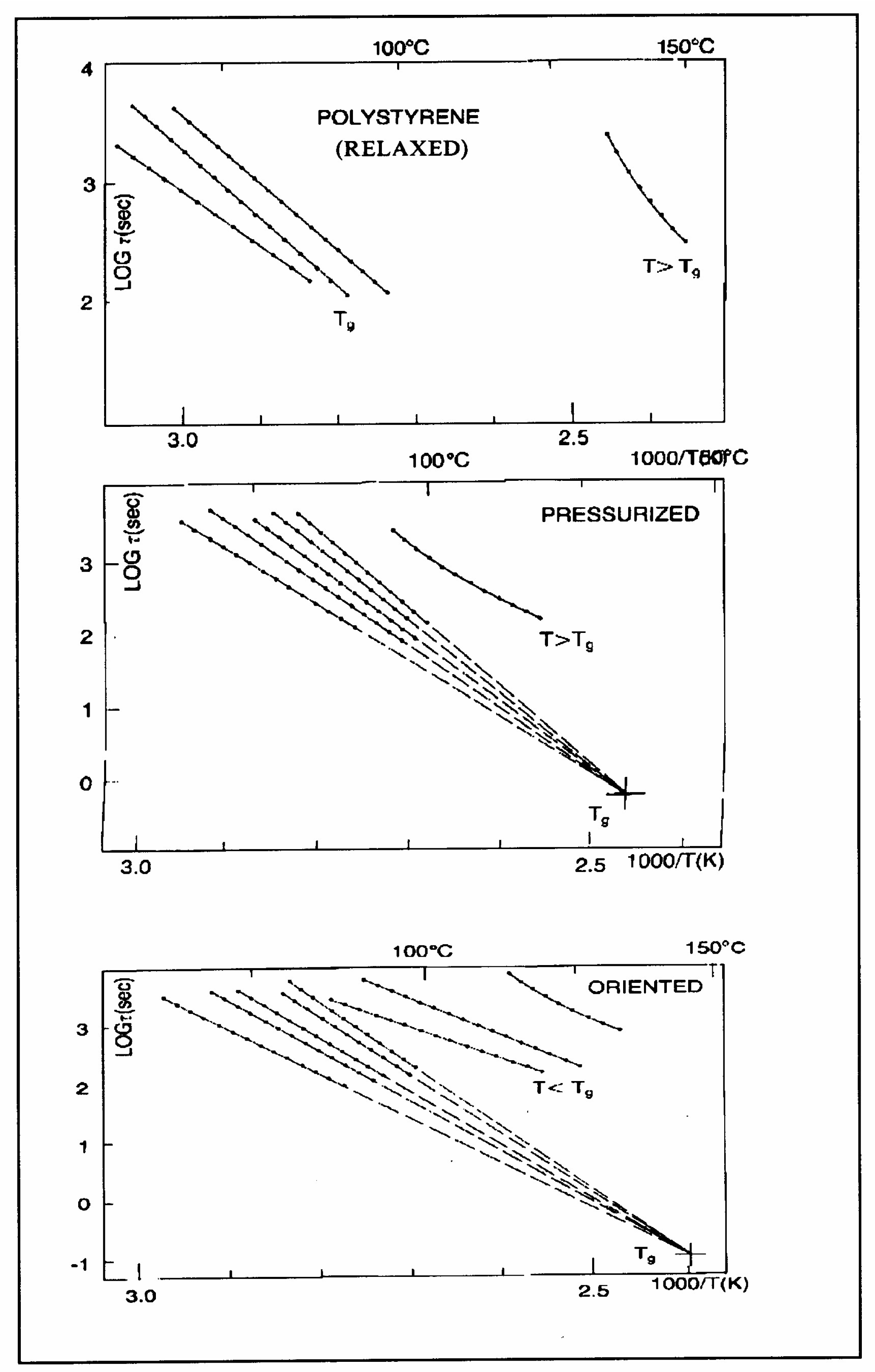
Figure 8 of the
Section 1. In this figure, polystyrene samples are prepared by compression molding under various conditions. In the top graph, a relaxed specimen is cooled under no mechanical stress at a very slow cooling rate. The Arrhenius transform of the T
g,ρ peak is shown at the extreme right of the graph; it is curved and can be fitted with a WLF type of equation, providing the free volume thermal expansion coefficient above T
g, and the temperature of infinite viscosity (the values found match viscosity data well). In the middle graph, corresponding to the “pressurized” specimen, the WLF curve is shifted toward a lower temperature. Another thermo-mechanical treatment is shown in the bottom graph.
Figure 34 displays the Arrhenius transforms of the T
g,ρ peak for several processing conditions: static pressure, rheomolding treatment, oriented sample, and relaxed polystyrene.
The relaxation mode obeys a free volume criterion for all conditions, but the content of the free volume and the mobility at a given temperature, given by the horizontal position of the WLF curve, are strong functions of the state of the polymer due to processing conditions. In some instances, the T
g,ρ peak is broad and contains a combined effect of molecular dipoles and free volume relaxations (
Figure 35). In such cases, the thermal-windowing around the T
g,ρ peak produces several relaxation modes (
Figure 8, bottom curve), and sometimes one or two additional compensations are observed in the (T > T
g) region up to T
LL.
We consider T
LL as the temperature, on heating, marking the end of a certain type of relaxation behavior due to cooperative kinetic interactions. Whether or not it actually corresponds to a TSD peak is irrelevant to our definition. For instance, we mentioned that T
LL shows up in
Figure 32 as a peak at 150 °C. It might be more appropriate to categorize this peak as one of the kinetic manifestations resulting from the cooperative kinetic process already giving rise to T
β, T
g, and T
g,ρ. In Part I, Chapter 3, as well as in references [
25,
31,
32], it is suggested that the mechanism of relaxation is due to the coherence between the collective behavior defining the statistical ensemble and the local existence of dual phases (the b and F phases), which describes the interactive coupling between the conformers belonging to the macromolecules modulating both the sub-T
g and the (T > T
g) kinetics. In that regard, T
LL is probably more at 175 °C than 150 °C in
Figure 32, and the peak at 150 °C should be considered as a manifestation of the modulation by the network (collective) of b/F interactions occurring in the phase richer in free volume and responsible for the T
g,ρ relaxation manifestation. This point is further illustrated in
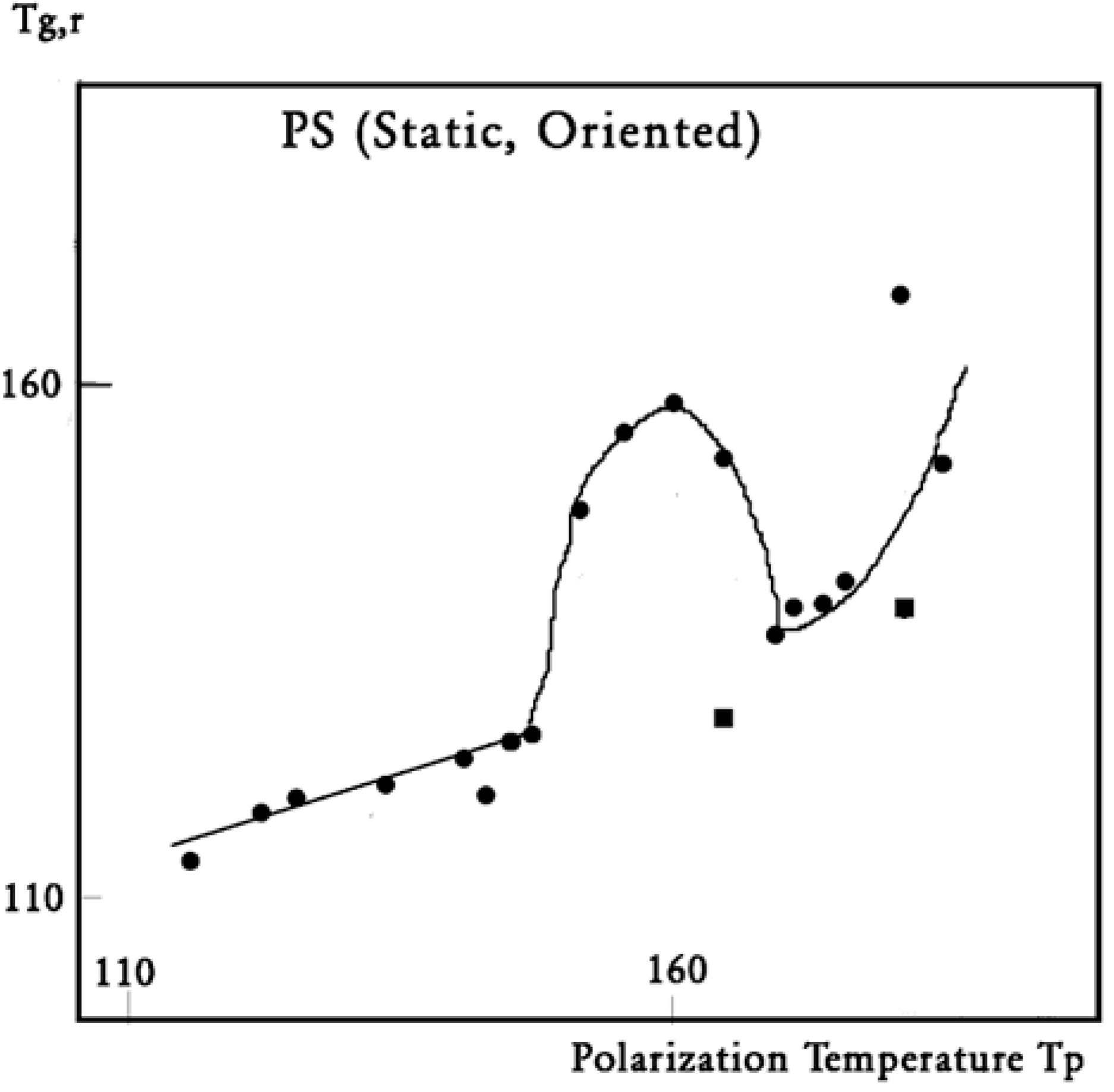
Figure 36.
In this figure, we plot the variation in T
g,ρ with T
p, T
g,ρ being considered the “first” peak observed above T
g. When a second peak appears, which we call T′
g,ρ, we indicate its temperature on this graph with square dots. One sees that T
g,ρ increases slowly and linearly with T
p (the slope is much less than 1), at least until T
p = 145 °C, a temperature at which T
g,ρ rises quickly to a maximum. The first split of T
g,ρ into two peaks occurs for T
p = 165 °C, and the second split occurs for 180 °C. In the case of a split, the temperatures of the two peaks are located on both sides of the “expected” T
g,ρ extrapolated from the lower values. The departure from the baseline at 145 °C reveals the kinetic presence of the network modulating collectively the interactions [
7]. Beyond T
p = 170 °C, the modulation by the elastic dissipative network of the inter–intramolecular interactions between the various dipoles is over. T
LL would be 170 °C, according to our definition.
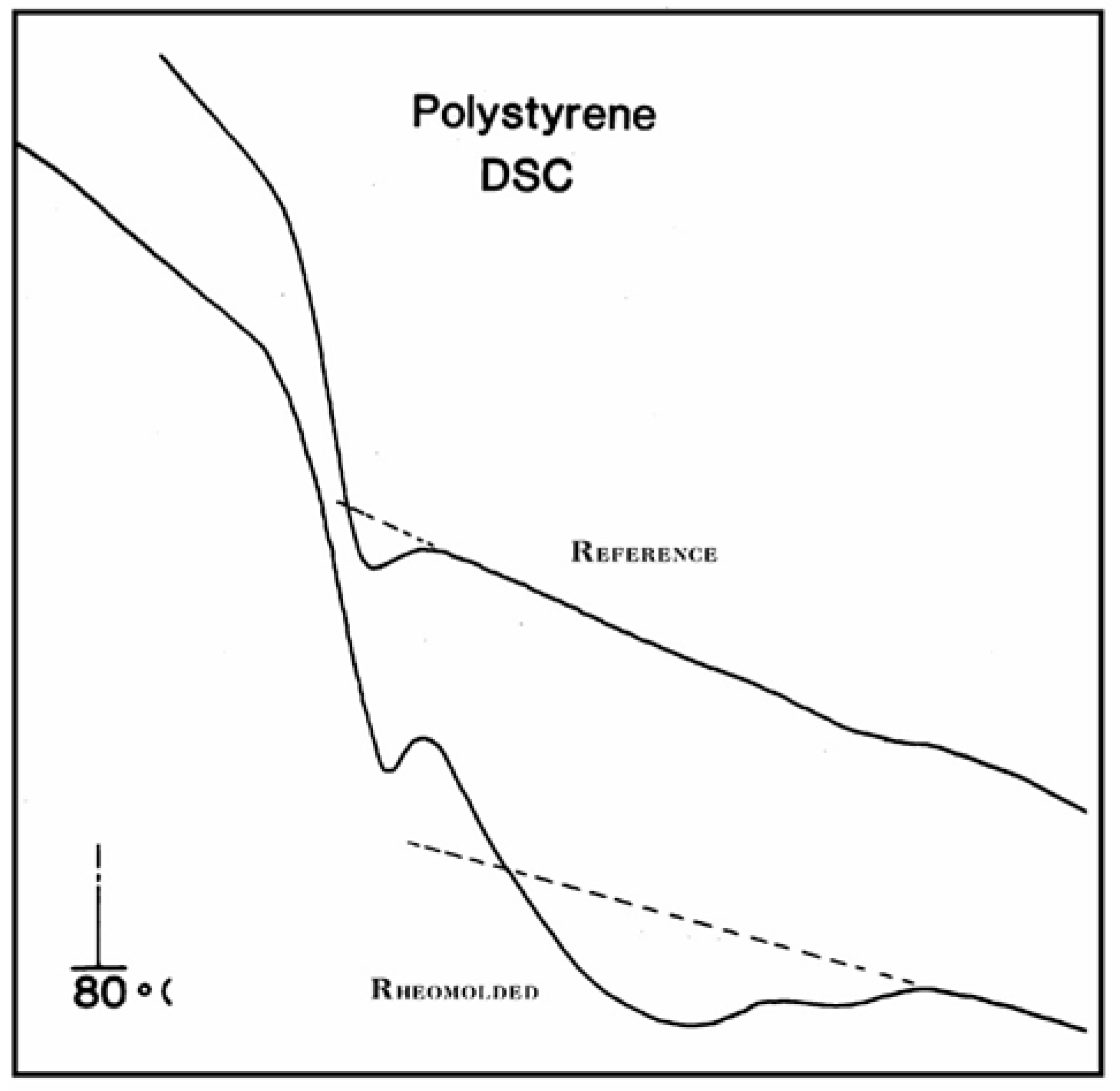
In
Figure 37, the DSC trace for a rheomolded polystyrene showing cooperative relaxation above T
g is compared with a reference trace (obtained by cooling the same sample in the DSC cell after the first run). The difference between the heat flow of the two curves is significant just above the T
g peak, and until the T
LL temperature is reached. The thermal history of the rheomolded sample, revealed during the first run, creates a thermal activity above T
g clearly similar to what is observed by TSD (
Figure 35). The thermal history is erased as the sample is heated in the DSC cell, and both the reference and rheomolded DSC traces are identical above T
LL. We consider
Figure 37 as an important illustration of the existence of T
g,ρ and T
LL using DSC. However, the sample needs to be severely out of equilibrium in order to reveal by DSC the T
g,ρ and T
LL transitions as clearly as in
Figure 37. TSD, which works at a very low-frequency equivalent (Part II, Chapter 1, [
5]), is capable of resolving the T
g/T
g,ρ kinetic presence with far higher resolution and sensitivity (note how thermal history of the rheomolded PC of
Figure 33 is still revealed after the successive runs). The thermal analyst should be gratified: the T
g,ρ peak gives us a new parameter to characterize polymers. It varies with the processing conditions and thus can be used to characterize the effect of processing variables (pressure, orientation, and thermal history); it is often stronger than Tg and exists for non-polar materials when the free volume can be trapped with charges. It is perhaps the only peak that one can observe to characterize thick non-polar polymers. In that case, the “T
g” value obtained is possibly a T
g,ρ value several degrees higher than what would have been expected from a DSC comparison (the TSD T
g peak matches the T
g obtained in a DSC experiment at a 20 °C/min heating rate well).
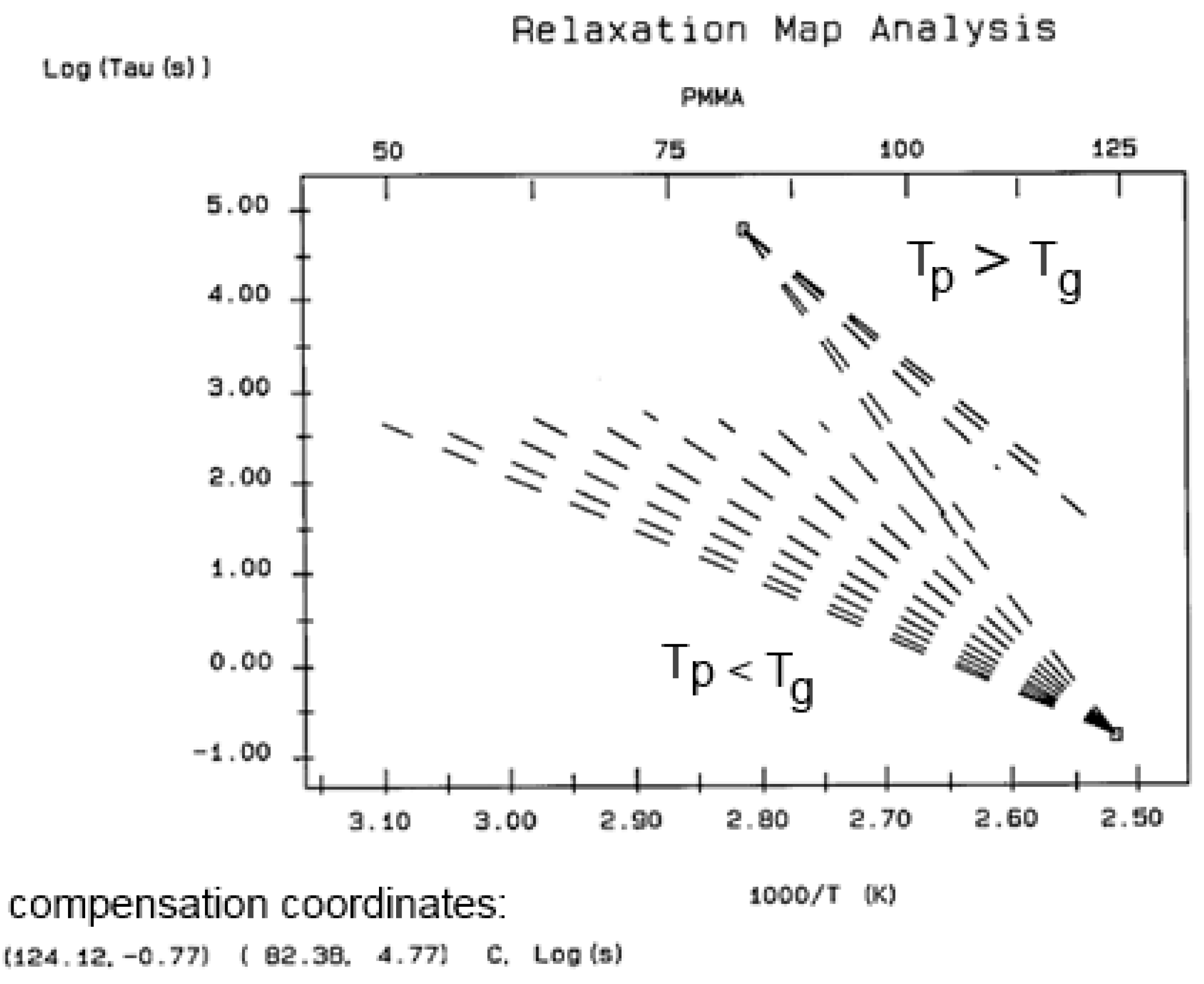
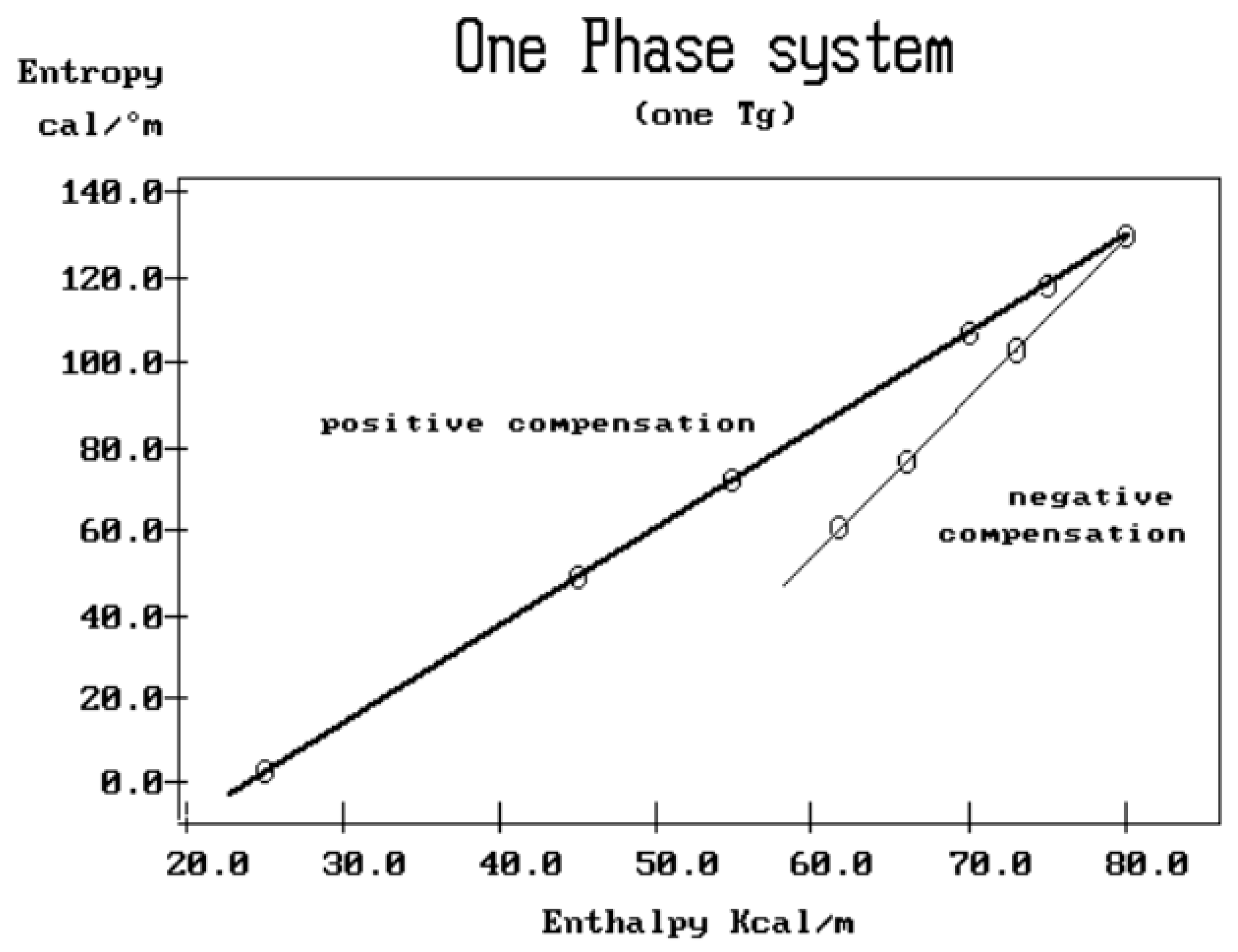
2.3. Super-Compensations Observed in the Amorphous State of Single-Phase Amorphous Samples
The objective is to explore the complexity of the phenomenon of compensation to better understand its origin. In the case of a multi-phase system, such as a segregated blend or block copolymers, the relaxation map displays multi-compensations, each associated with the T
g of the corresponding phase. In the case of crystallizable polymers or liquid crystal polymers, multi-compensations are often visible both below and above the glass transition temperature of the polymer. All these apparent complexities are expected because of the presence of multiple phases in the material.
Figure 38 and
Figure 39 illustrate, respectively, what is expected for the compensation search to look like for, a two-phase amorphous polymer (blend or copolymer) and a single-phase amorphous homopolymer such as polystyrene. In the case of a homopolymer (
Figure 39), only one T
g is observed. The compensation search leads to a single pair of compensation lines, one positive and one negative. The presence of two phases in the morphology is clearly revealed by two T
g s and thus two pairs of compensation lines (
Figure 38). A comparison of the respective position of the compensation lines for the homopolymer and the block copolymer leads to a better understanding of the segregation characteristics between the phases and, in particular, the degree of interpenetration and local compatibility (Part II, Chapter 4, [
5]). It is clear that, in this case, there is a direct relationship between the number of compensation lines and the number of phases in the morphology.
It will be shown in this section (e.g., Figure 55) that, for a true monophasic amorphous polymer (in this case, polystyrene), not only is it possible to observe several compensations but also to vary the number of compensations depending on the thermo-mechanical history: the processing variables, the cooling rate, the amount of pressure, vibration [
33], orientation, etc. This unexpected complex behavior has remained controversial: Lacabanne and Bernes [
34] have suggested that the several compensation points observed for polystyrene (and polycarbonate) were due to the presence of several amorphous phases, the indication of “local ordering”. Furthermore, these authors suggested that T
LL was the melting transition of these local micro-ordered structures [
35,
36,
37,
38]. In this
Section 2.3, we describe the multi-compensations in monophasic amorphous systems giving rise to super-compensations, and in D3, we propose an alternative explanation for the existence of super-compensations and of T
LL based on the dual-phase model of the dissipative interactions.
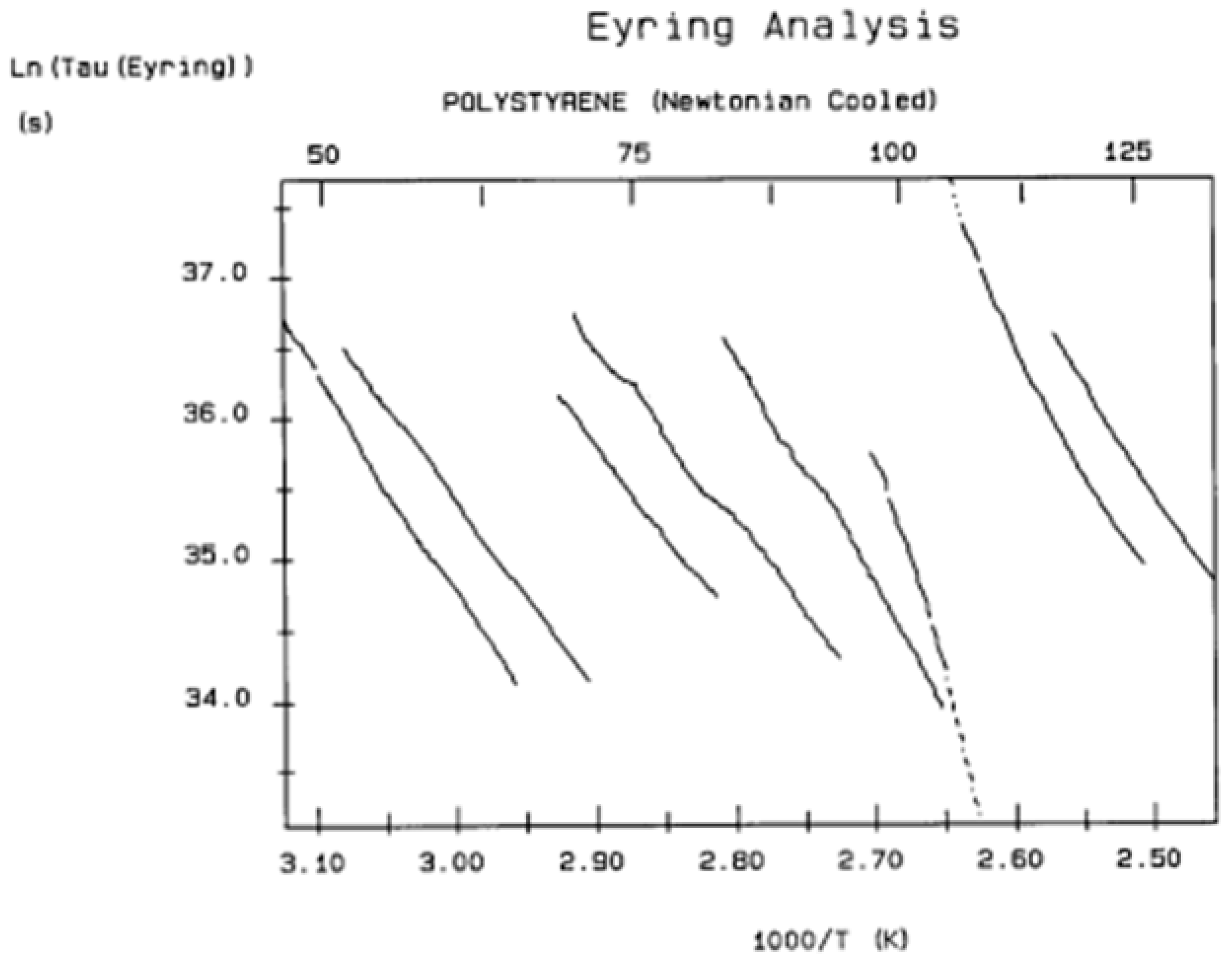
Figure 40 is a relaxation map in the “Eyring plane” (defined in A1) for a polystyrene disk (T
g~100 °C) very slowly cooled from 180 °C to 50 °C in a platen pressure mold with virtually no pressure applied on the specimen during the cooling stage. The mold is cooled very slowly by “Newtonian cooling”, i.e., by shutting off the power to the heater cartridges after thermal equilibrium is reached at 180 °C and letting it cool by itself, without the use of cooling fluids in the cooling channels of the mold halves. This procedure resulted in very slow cooling: it took approximately 7 h to cool the mold down to 50 °C.
Table 1 gives the thermo-kinetic parameters as a function of T
p, namely ΔH
p(“enthalpy”), ΔS
p(“entropy”), and ΔG
p(“Gibbs”), as well as the value of the polarizing temperature, T
p, and T
m, the temperature at the maximum of the Debye depolarization peak.
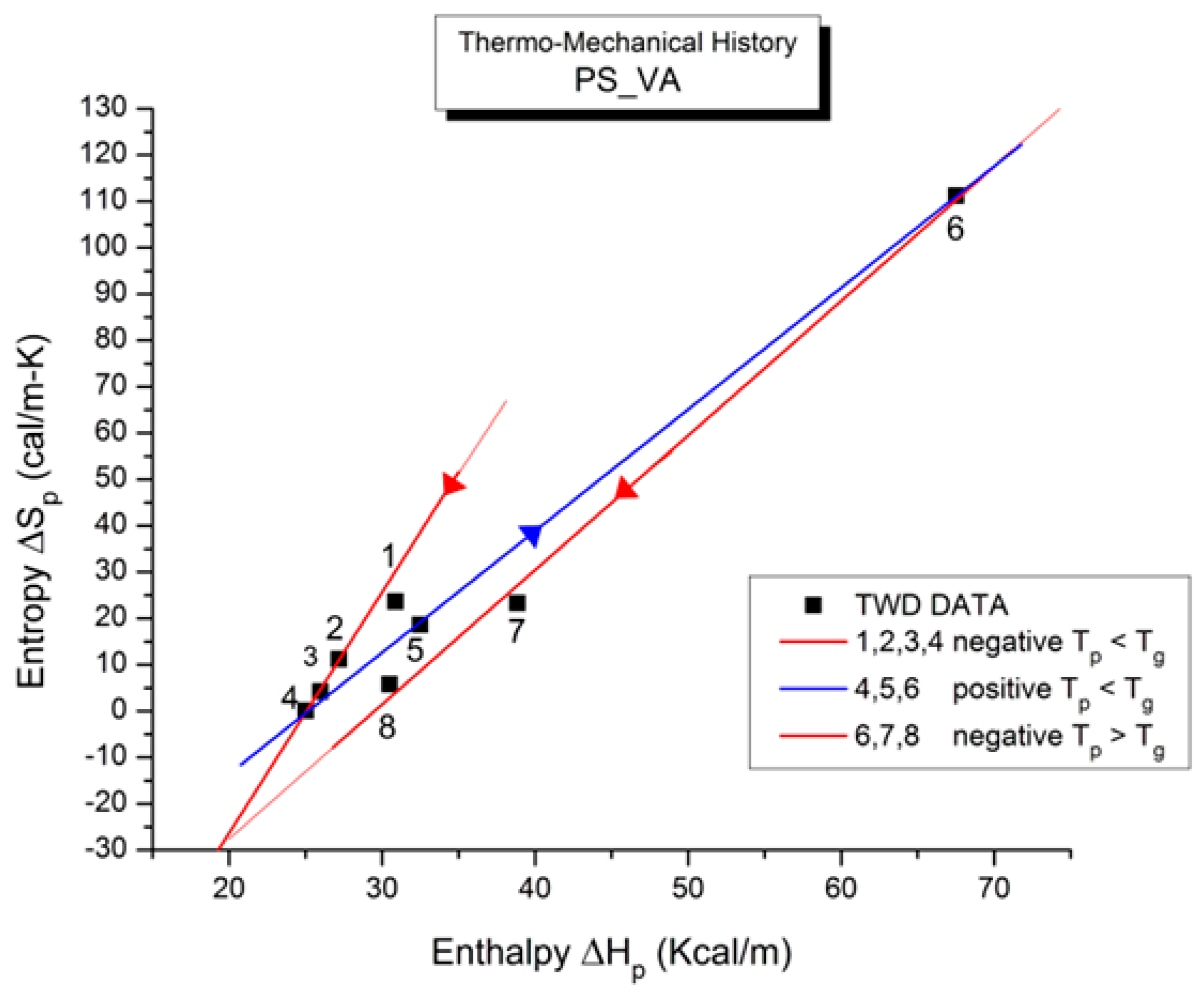
Figure 41 illustrates the compensation search in the EE plane (Entropy vs. Enthalpy). T
p varies between 60 °C and 125 °C (the window temperature is 10 °C). The numbers near the data in
Figure 41 apply to the row numbers in
Table 1, increasing with the polarization temperature T
p. We clearly observe two
negative compensation lines in
Figure 41. The first compensation line comprises points 1, 2, 3, and 4, all with T
p < T
g, and the second compensation line aligns points 6, 7, and 8, all with T
p > T
g; point 5 (at T
p = 95 °C) seems to be “lost” between the two negative compensation lines. We will go back to the analysis of this PS_VA treatment in Figure 56 and Figure 57 and Table 5 after the study of the relaxation maps for other thermo-mechanical histories, which will have given us some insight into how to handle the analysis of these unusual and complex relaxation maps. Our first impression of the spectral lines in
Figure 40 is that the first five lines, starting from the low T
p end, are more or less parallel. This is reflected in the compensation search by the closeness of their enthalpy values on the corresponding compensation line (from 31 to 25 Kcal/m). We also note that the entropy decreases from 24 cal/m-K to 0 (for point 4) and increases sharply to 111 for point 6.
Figure 40.
Relaxation map in the Eyring plane for a very slowly cooled compression-molded polystyrene sample with almost no pressure during cooling. The sample is called PS_VA in subsequent figures. The slope and intercept of the elementary deconvoluted relaxations provide the enthalpy and entropy values in
Table 1. The data are analyzed in
Figure 41, Figure 56, and Figure 57. Reproduced with permission from [
4], SLP Press, 1993.
Figure 40.
Relaxation map in the Eyring plane for a very slowly cooled compression-molded polystyrene sample with almost no pressure during cooling. The sample is called PS_VA in subsequent figures. The slope and intercept of the elementary deconvoluted relaxations provide the enthalpy and entropy values in
Table 1. The data are analyzed in
Figure 41, Figure 56, and Figure 57. Reproduced with permission from [
4], SLP Press, 1993.
Table 1.
Thermo-kinetics for sample PS_VA of
Figure 40.
Table 1.
Thermo-kinetics for sample PS_VA of
Figure 40.
| Row # | Tp (K) | Tm (K) | Enthalpy △Hp | Entropy △Sp | △Gp |
|---|
| | (K) | (K) | (Kcal/m) | cal/m-K | Kcal/m |
|---|
| 1 | 333.2 | 344 | 30.8906 | 23.6368 | 23.0148 |
| 2 | 338.2 | 349.8 | 27.1823 | 11.1923 | 23.3971 |
| 3 | 348.2 | 367.5 | 25.9793 | 4.211 | 24.5130 |
| 4 | 353.2 | 372.7 | 25.0293 | 0.0593 | 25.0084 |
| 5 | 368.2 | 380.5 | 32.4905 | 18.5915 | 25.6451 |
| 6 | 378.2 | 382 | 67.5262 | 111.1845 | 25.4762 |
| 7 | 388.2 | 411.9 | 38.8583 | 23.3365 | 29.7991 |
| 8 | 398.2 | 418.1 | 30.5001 | 5.8074 | 28.1876 |
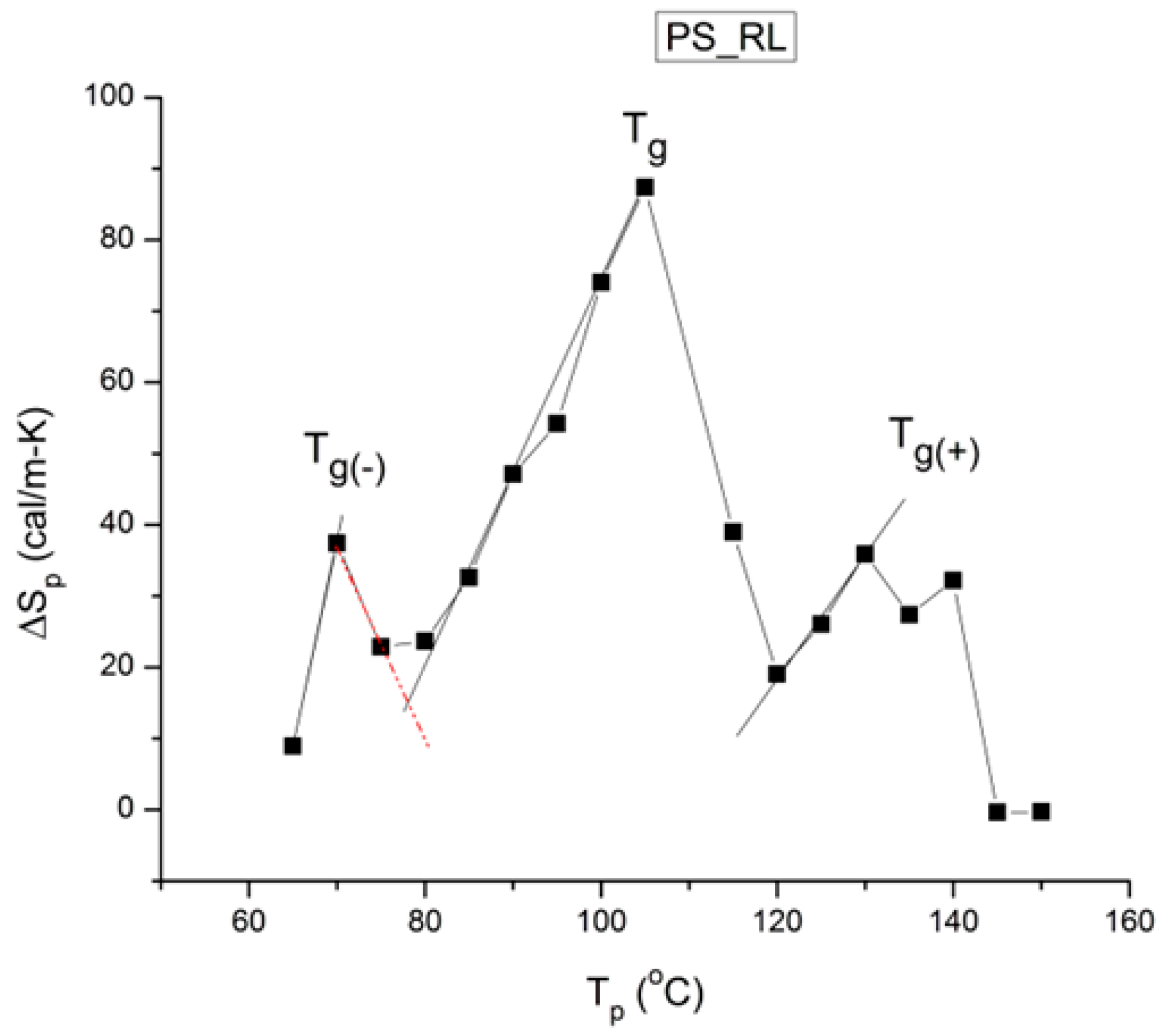
Figure 41.
Compensation search in the EE plane for the data of
Figure 40. The numbers near the data relate to the row position in
Table 1. Reproduced with permission from [
4], SLP Press, 1993.
Figure 41.
Compensation search in the EE plane for the data of
Figure 40. The numbers near the data relate to the row position in
Table 1. Reproduced with permission from [
4], SLP Press, 1993.
Figure 42,
Figure 43,
Figure 44,
Figure 45,
Figure 46 and
Figure 47 relate to the same polystyrene but mechanically pressurized during a fast cooling treatment. The mechanical treatment includes the application of vibrational positive pressure to induce specific thermal history patterns, a process known as “rheomolding” [
39]. The mold is cooled in approximately 30 s. The rate of cooling when the glass transition temperature is crossed is around 7 °C/s, considerably faster than for the PS_VA treatment in
Figure 40.
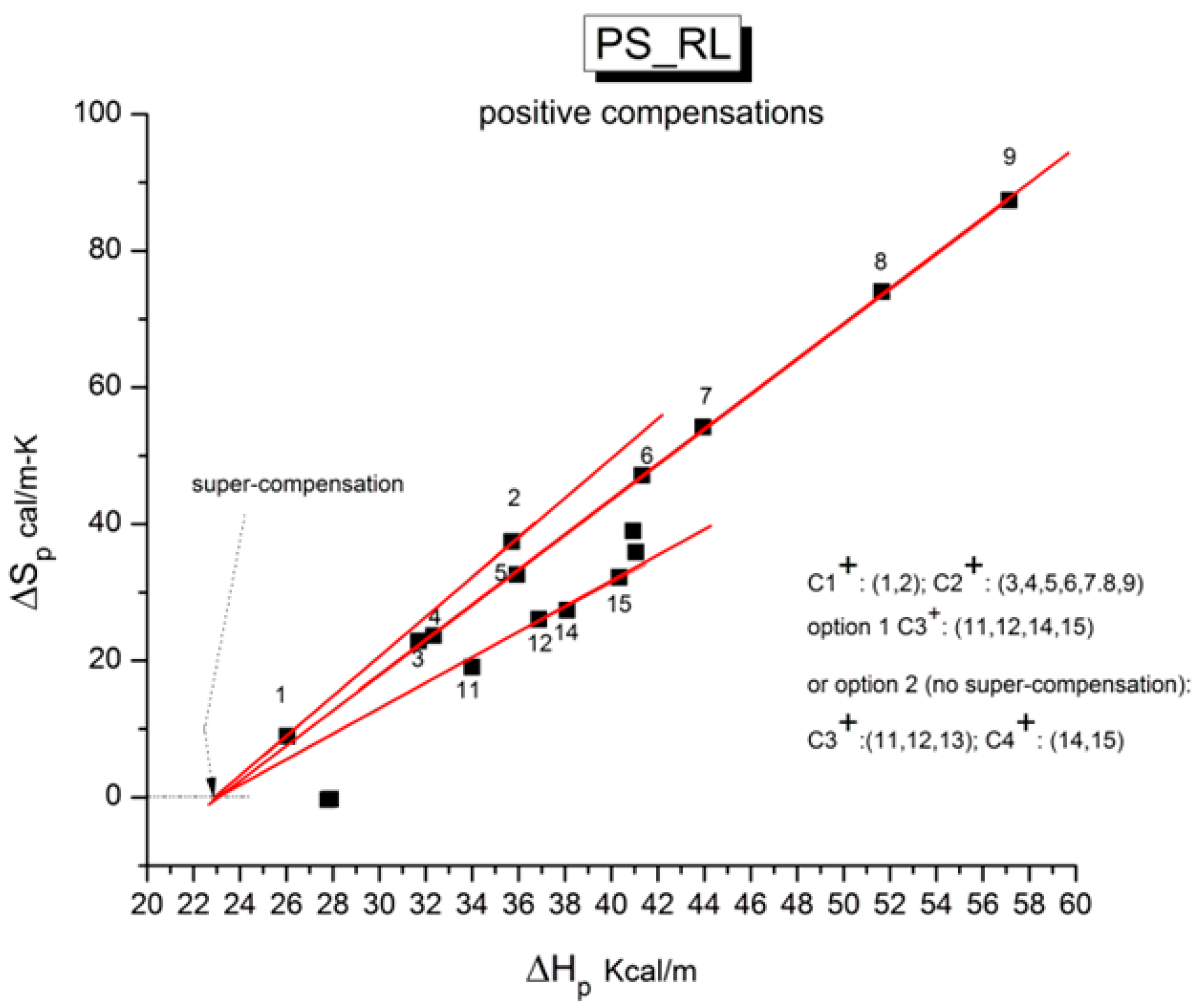
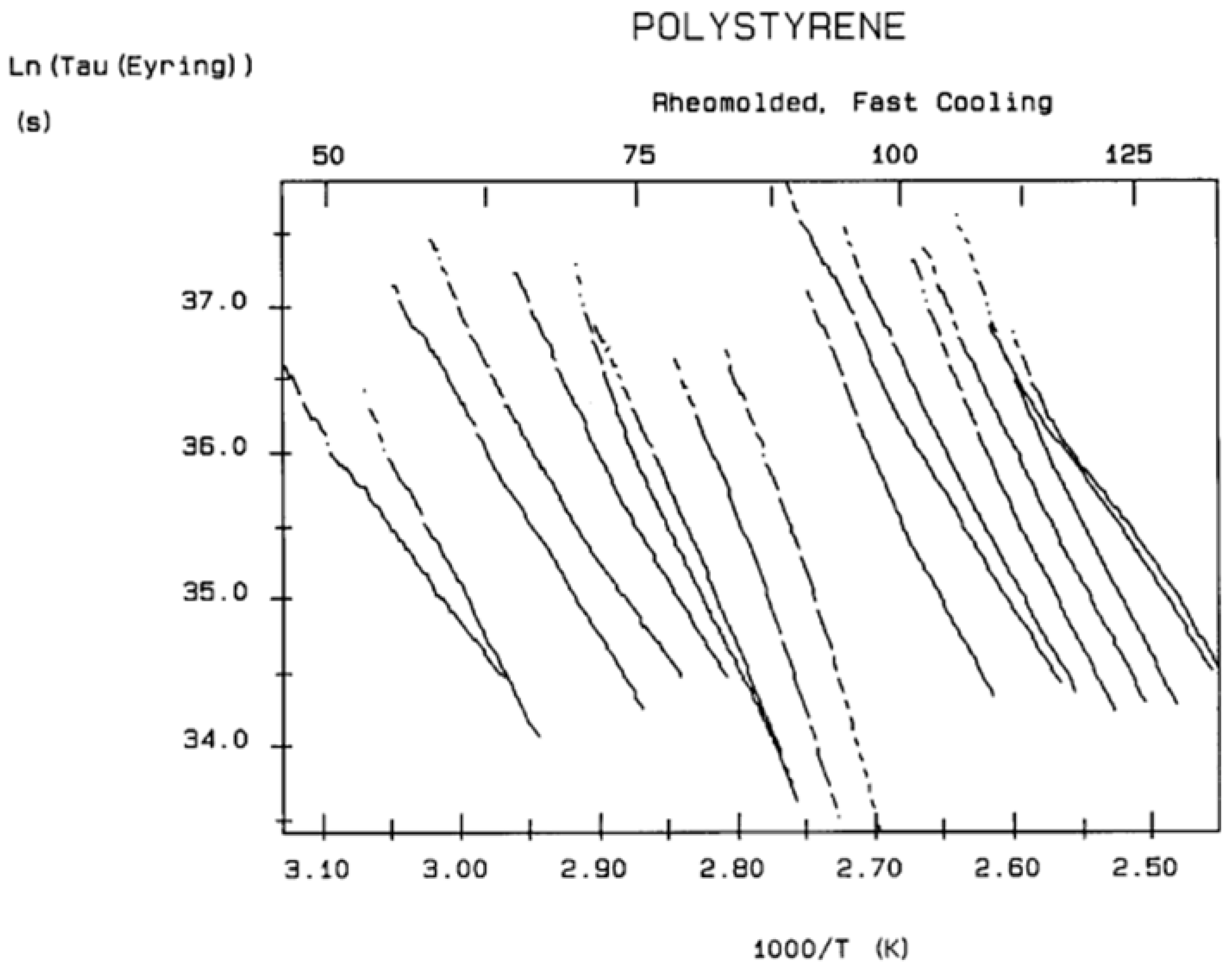
Figure 42 is the relaxation map in the Eyring plane.
Figure 43 is the compensation search in the EE plane based on the entropy and enthalpy data of
Table 2. The thermo-mechanical treatment is called “PS_RL”.
Figure 43 appears complex, unlike what is observed for stable amorphous states for which two compensation lines can be drawn passing through the data: one positive compensation line for T
p < T
g relaxations and one negative compensation line for T
p > T
g. This cannot be the correct solution in
Figure 43 because the two lines only solution would not join consecutive T
p data points, which is a sine qua non condition to define compensation. In fact, as
Figure 44,
Figure 45,
Figure 46 and
Figure 47 will demonstrate, the situation is only apparently more complex because we find several correlations and a network structure between the six compensations (three positive and three negative) that are necessary to comprehend the relaxation maps in
Figure 42 and
Figure 43.
Figure 44,
Figure 45,
Figure 46 and
Figure 47 illustrate our step-by-step method to clarify complex compensation searches, which we will only explain for PS_RL.
In the Eyring plane, the Debye relaxation times are normalized by the vibrational energy of the atoms (kT/h), where k is the Boltzmann constant, T is the absolute temperature, and h is the Planck constant. The slope and intercept of log τ (Eyring) vs. 1/T directly provide the activation enthalpy, ΔHp = slope*k, and the activation entropy, ΔSp= intercept*k, of the elementary Debye depolarization process: (k is replaced by R, the gas constant, equal to 1.987 cal/m-K, to express the activation energies per mole instead of per molecule).
Figure 43.
Compensation search in the EE plane for the PS_RL sample of
Figure 42. The analysis appears complex. The numbers near the data (black squares) are the row numbers in
Table 2 corresponding to the T
p value (shown in the inset) during TWD. For a simple amorphous state, such an EE plot has two straight lines going through the data, a positive and a negative compensation line intersecting at T
g, but we cannot represent such a simple solution since compensation lines should join consecutive increasing or decreasing T
p points for positive or negative compensations, respectively. The finding of the true positive and negative compensation lines for this complex situation is shown in
Figure 45 and
Figure 46.
Figure 43.
Compensation search in the EE plane for the PS_RL sample of
Figure 42. The analysis appears complex. The numbers near the data (black squares) are the row numbers in
Table 2 corresponding to the T
p value (shown in the inset) during TWD. For a simple amorphous state, such an EE plot has two straight lines going through the data, a positive and a negative compensation line intersecting at T
g, but we cannot represent such a simple solution since compensation lines should join consecutive increasing or decreasing T
p points for positive or negative compensations, respectively. The finding of the true positive and negative compensation lines for this complex situation is shown in
Figure 45 and
Figure 46.
A compensation search looks for an affinity between successive interactive coupling relaxation modes activated at different polarization temperatures T
p. In
Figure 43, the Eyring relaxation map of
Figure 42 is converted to the EE plane variables (entropy vs. enthalpy), and the affinity between the relaxation modes is determined by the alignment on a straight line (the “compensation”) of the thermo-kinetic coordinates (ΔH
p and ΔS
p) for neighboring T
p values. In
Table 2, we see that T
p varies every 5 degrees from 65 °C to 150 °C (17 values). Here, rows 1 and 2 for T
p = 65 and 70 °C correspond to the two first relaxation lines on the left of
Figure 42. These two relaxation modes appear to cross in that figure at an Eyring relaxation time, τ
c, and a temperature T = T
c. In the EE plane (
Figure 43), these two relaxation modes are points #1 and 2. The line (1,2) is the compensation line that allows for the calculation of the coordinates of the compensation points of these two elementary relaxations in
Figure 42. This is illustrated in
Figure 44, which shows a plot of ΔH
p vs. ΔS
p in which the x and y axes are swapped to allow for an easy determination of the compensation point in the ΔG plane: T
c, ΔG
c. In
Figure 44, the value of ΔG
c is simply the intercept of line (1,2), and the T
c value (in Kelvin degrees) is equal to 1000 times the slope of line (1,2). In
Figure 43, as T
p increases beyond point # 2, one sees that #3 is not located on line (1,2) but rather below it, although its T
p has increased; a clear reversal of the direction indicates the presence of a new compensation with a change in its “sign”, here between points #2 and #3. The first compensation (1,2) is called “1
+”since it is positive, while (2,3) is called “1
−” since it is the first negative compensation (we also use the appellations C
1+ and C
1− to designate these compensations). As T
p continues to increase, points #3 to #9 are aligned on a single straight line forming the second positive compensation, “2
+” (or C
2+). The second negative compensation lines down points #9, #10, and #11 and is designated “2
−” (or C
2−), and so on and so forth for the remaining points sub-grouping in compensations “3
+” and “3
−” or even in “4
+” and “4
−” in some instances. The rule followed to define the various compensation lines is that only successive points can participate in a compensation line. Sometimes, we hesitate between two options because of the errors that occur during the determination of the coordinates of the points: this happens at larger T
p values where the relaxation of non-equilibrium samples occurs faster. The compensation search is illustrated in
Figure 45 for positive compensations and
Figure 46 for negative compensations.
Table 2.
Thermo-kinetics for sample PS_RL.
Table 2.
Thermo-kinetics for sample PS_RL.
| Row # | Tp | Tm | Enthalpy ΔHp | Entropy ΔSp | ΔGp |
|---|
| | °C | °C | (Kcal/m) | cal/m-K | Kcal/m |
|---|
| 1 | 65 | 69.7 | 26.0444 | 8.917 | 23.03045 |
| 2 | 70 | 70.2 | 35.7053 | 37.4304 | 22.86667 |
| 3 | 75 | 79.8 | 31.6927 | 22.8775 | 23.73133 |
| 4 | 80 | 84.6 | 32.343 | 23.6549 | 23.99282 |
| 5 | 85 | 88.8 | 35.9299 | 32.599 | 24.25946 |
| 6 | 90 | 90.2 | 41.3195 | 47.1144 | 24.21697 |
| 7 | 95 | 90.6 | 43.9499 | 54.2181 | 23.99764 |
| 8 | 100 | 94.6 | 51.6457 | 74.0286 | 24.03303 |
| 9 | 105 | 98.8 | 57.1351 | 87.381 | 24.10508 |
| 10 | 115 | 113.5 | 40.9335 | 38.9967 | 25.80278 |
| 11 | 120 | 120.4 | 34.0003 | 19.0242 | 26.52379 |
| 12 | 125 | 121.9 | 36.8776 | 26.0567 | 26.50703 |
| 13 | 130 | 125.4 | 41.0596 | 35.889 | 26.59633 |
| 14 | 135 | 129 | 38.0933 | 27.3621 | 26.92956 |
| 15 | 140 | 132.5 | 40.3364 | 32.172 | 27.04936 |
| 16 | 145 | 137.3 | 27.7921 | −0.3743 | 27.94856 |
| 17 | 150 | 138.5 | 27.8984 | −0.3003 | 28.02543 |
Figure 44.
Compensation search
in the EE plane (Enthalpy vs. Entropy) plane for PS_RL to find the compensation(s) in
Figure 42. Several positive and negative compensations emerge from what initially looked complex (in
Figure 43). The down arrows indicate positive compensations, whereas the up arrows indicate negative compensations.
Figure 44.
Compensation search
in the EE plane (Enthalpy vs. Entropy) plane for PS_RL to find the compensation(s) in
Figure 42. Several positive and negative compensations emerge from what initially looked complex (in
Figure 43). The down arrows indicate positive compensations, whereas the up arrows indicate negative compensations.
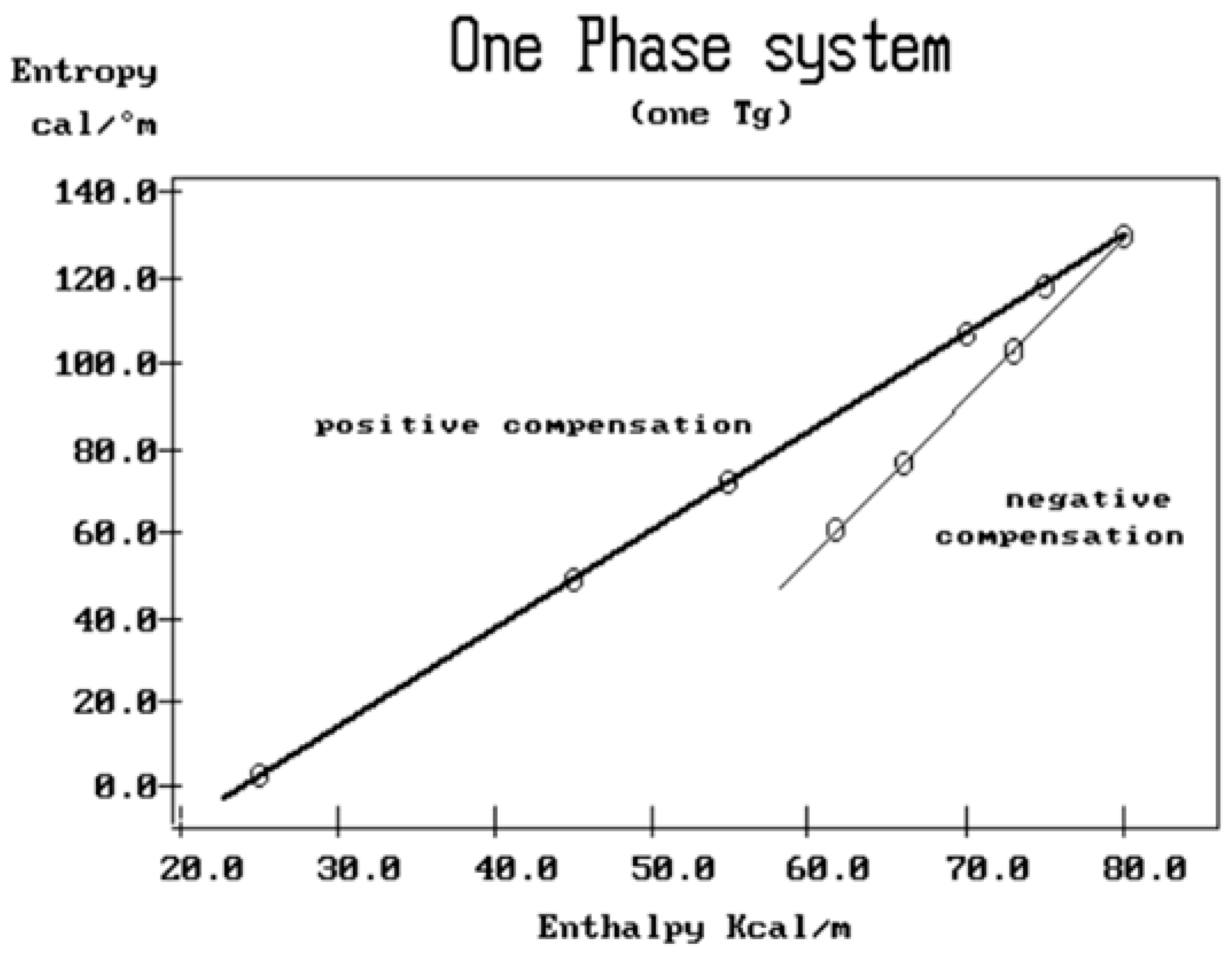
Figure 45 reveals that the
positive compensation lines (in red) appear to converge on the ΔS
p = 0 axis. The corresponding value of ΔH
p at the intercept point is 23 kcal/mole. This point is fully independent of the value of T
p: it is a
super-compensation, i.e., the compensation of all the positive compensation lines for sample PS_RL. When we say for “all” positive compensations, we should add that this is 100% sure for compensations C
1+ and C
2+, but that a different split of the points is possible for C
3+, as shown in the inset of
Figure 45: either C
3+ = (11,12,14,15) or C
3+ = (11,12,13) and C
4+ = (14,15).
Figure 45 shows the first choice, which has only three compensation lines.
Figure 45.
Compensation search in the EE plane for PS_RL showing the
positive compensation results only (lines joining successive T
p points with increasing values of ΔS
p and ΔH
p). The 3 positive compensation lines converge to a “super-compensation” point (whose y-coordinate is ΔS
sc(+) = 0) for option 1. For option 2, with 4 compensations, only 3 of them super-compensate. The analysis for the negative compensations is illustrated in
Figure 46.
Figure 45.
Compensation search in the EE plane for PS_RL showing the
positive compensation results only (lines joining successive T
p points with increasing values of ΔS
p and ΔH
p). The 3 positive compensation lines converge to a “super-compensation” point (whose y-coordinate is ΔS
sc(+) = 0) for option 1. For option 2, with 4 compensations, only 3 of them super-compensate. The analysis for the negative compensations is illustrated in
Figure 46.
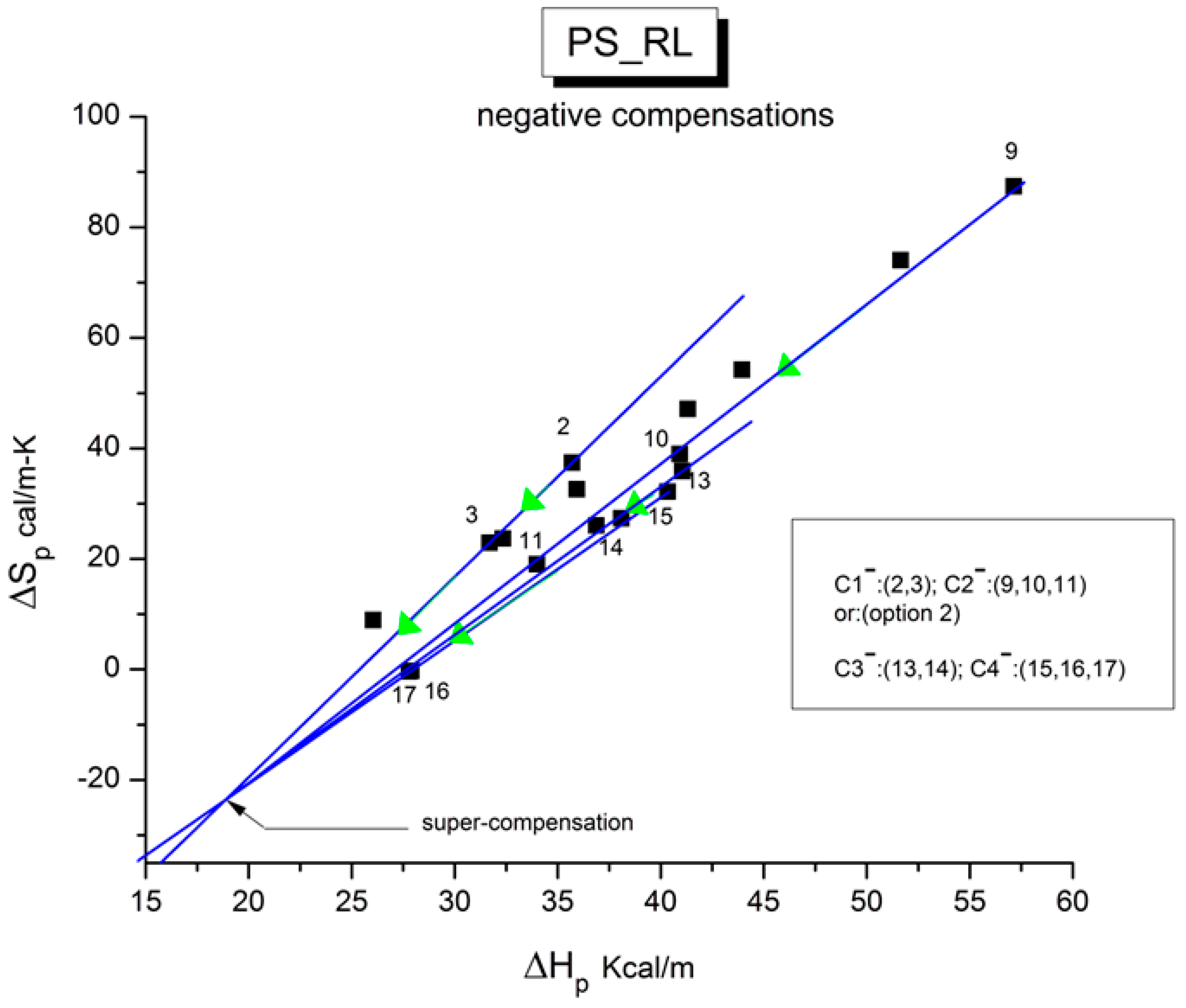
Figure 46 demonstrates the points interacting to generate negative compensation lines. This analysis is a little bit more difficult because there are fewer points to define the lines accurately. Nevertheless, it is possible to draw with confidence three negative compensation lines (option 1) or four compensation lines (option 2), and in the latter case, they themselves converge with confidence to a super-compensation (ΔH
c = 18.8 Kcal/m, ΔS
c = −23 cal/m-K).
Figure 46.
The compensation search in the EE plane for PS_RL showing only the
negative compensation results of the analysis conducted in
Figure 43. In option 2, the 4 negative compensation lines converge to a single point, a super-compensation point whose ΔH
pc(−) = 18.8 Kcal/m and Δ
Spc(−) = −23 cal/m-K. For option 1, with C
3− = (14,15,16,17)), there are only 3 negative compensations and the super-compensation is defined with less confidence (r
2 = 0.988). For option 2, with C
3− and C
4− defined in the inset, the convergence to a super-compensation is established with confidence (r
2 = 0.9987).
Figure 46.
The compensation search in the EE plane for PS_RL showing only the
negative compensation results of the analysis conducted in
Figure 43. In option 2, the 4 negative compensation lines converge to a single point, a super-compensation point whose ΔH
pc(−) = 18.8 Kcal/m and Δ
Spc(−) = −23 cal/m-K. For option 1, with C
3− = (14,15,16,17)), there are only 3 negative compensations and the super-compensation is defined with less confidence (r
2 = 0.988). For option 2, with C
3− and C
4− defined in the inset, the convergence to a super-compensation is established with confidence (r
2 = 0.9987).
In
Table 3, we summarize the results of the regressions of the compensation lines (ΔS
p vs. ΔH
p) shown in
Figure 45 and
Figure 46, with ΔS
p in cal/m-K and ΔH
p in kcal/m.
| ΔSp (cal/m-K) vs. ΔHp (Kcal/m) | |
|---|
| PS_RL | |
|---|
| Positive Compensations | Intercept | Slope | Negative Compensations | Intercept | Slope |
|---|
| (1,2) | 1(+) | −67.951 | 2.95142 | (2,3) | 1(−) | −92.0656 | 3.6268 |
| (3–9) | 2(+) | −59.1929 | 2.57205 | (9,10,11) | 2(−) | −81.8557 | 2.96034 |
| (11,12,13) | 3(+) | −62.0482 | 2.38628 | (14,15,16,17) | 3(−) | −73.74 | 2.6381 |
Figure 47.
Schematic reconstruction for PS_RL of the compensations of the spectral lines in
Figure 42 following the compensation search results of
Figure 45 and
Figure 46. This graph shows the full Debye spectral lines for the 3 positive compensations (with F, D, and B indicating the aligned compensation points); the spectral lines for the negative compensations are only visible for the lower T
p compensation points (E and C) and not for compensation point A.
Figure 47.
Schematic reconstruction for PS_RL of the compensations of the spectral lines in
Figure 42 following the compensation search results of
Figure 45 and
Figure 46. This graph shows the full Debye spectral lines for the 3 positive compensations (with F, D, and B indicating the aligned compensation points); the spectral lines for the negative compensations are only visible for the lower T
p compensation points (E and C) and not for compensation point A.
Figure 47 represents a summary of our complex compensation search, displaying the interactive coupling map of the relaxations revealed at different T
ps. The positive compensation points are shown at F, D, and B in order of increasing T
p. The line passing through F, D, and B is the super-compensation line whose intercept and slope provide the coordinates of the super-compensation point of the positive compensations. Note that we did not draw a line through the negative E, C, and A compensation points because the spectral lines for A were not drawn. Yet, this negative super-compensation line can be imagined passing through E and C and would appear to be parallel to the positive super-compensation line FDB. In other words, all the compensation points and the super-compensation lines appear to create a macro-network where the local spectral lines collectively belong, showing their collective dependence. We will explain this concept in the next figures below (
Figure 48,
Figure 49,
Figure 50,
Figure 51,
Figure 52,
Figure 53,
Figure 54 and
Figure 55). For this, we will focus on the ΔG
p(T) plane representation of the TWD relaxation map ([
16], p. 79 of [
5]), where ΔG
p(T) is the free activation energy of depolarization at the T of the elementary Debye relaxation after polarization at T
p according to the TWD protocol, namely ΔG
p(T) = ΔH
p − T ΔS
p, in the ΔG plane.
Table 3 provides the intercept and slope of the linear relationship between ΔS
p and ΔH
p for the compensation lines. The free energy at each of the compensation points, ΔG
c, and the temperature of compensation, T
c, can be calculated from the value of the slope (S
p) and the intercept (I
p) of the compensation lines when they are rewritten as ΔH
p = I
p + S
p ΔS
p; with the conversion of the energy units for enthalpy and entropy to become coherent, we obtain the following: ΔG
c = I
p and T
c(K) = 1000 S
p. Their values are tabulated in
Table 4 for each of the positive and negative compensations.
Table 4.
Values of ΔGc and Tc for the 3 positive (+) and the 3 negative (−) compensation points. These compensation point coordinates are the references that characterize the sub-groups of interactions, each comprising the relaxation modes belonging to the sub-group.
Table 4.
Values of ΔGc and Tc for the 3 positive (+) and the 3 negative (−) compensation points. These compensation point coordinates are the references that characterize the sub-groups of interactions, each comprising the relaxation modes belonging to the sub-group.
| | △Gc (Kcal/m) | Tc (°C) |
|---|
| Comp(1)+ | 23.02 | 65.7 |
| Comp(1)− | 25.38 | 2.57 |
| Comp(2)+ | 23.01 | 115.6 |
| Comp(2)− | 27.65 | 64.6 |
| Comp(3)+ | 26 | 145.9 |
| Comp(3)− | 27.95 | 105.9 |
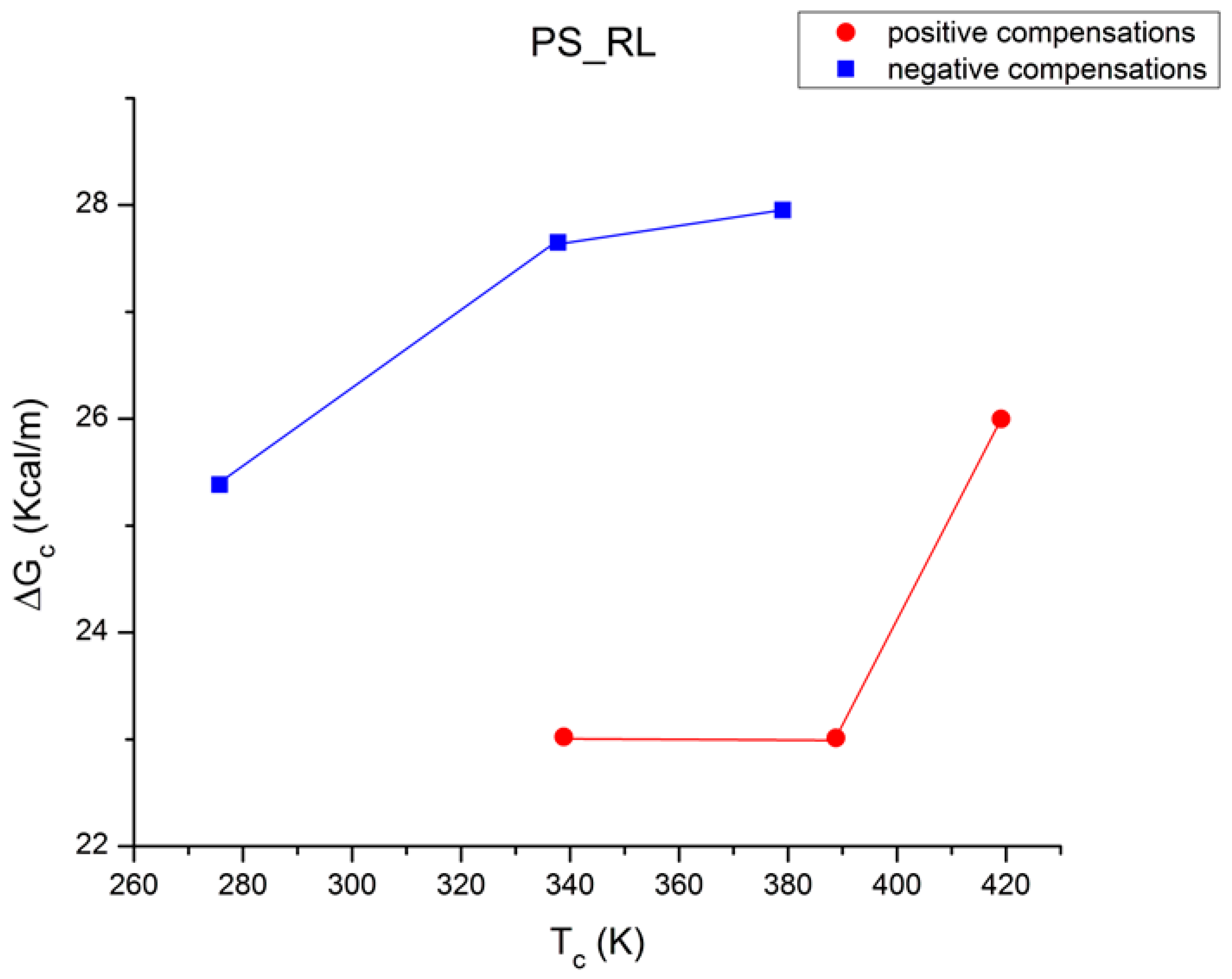
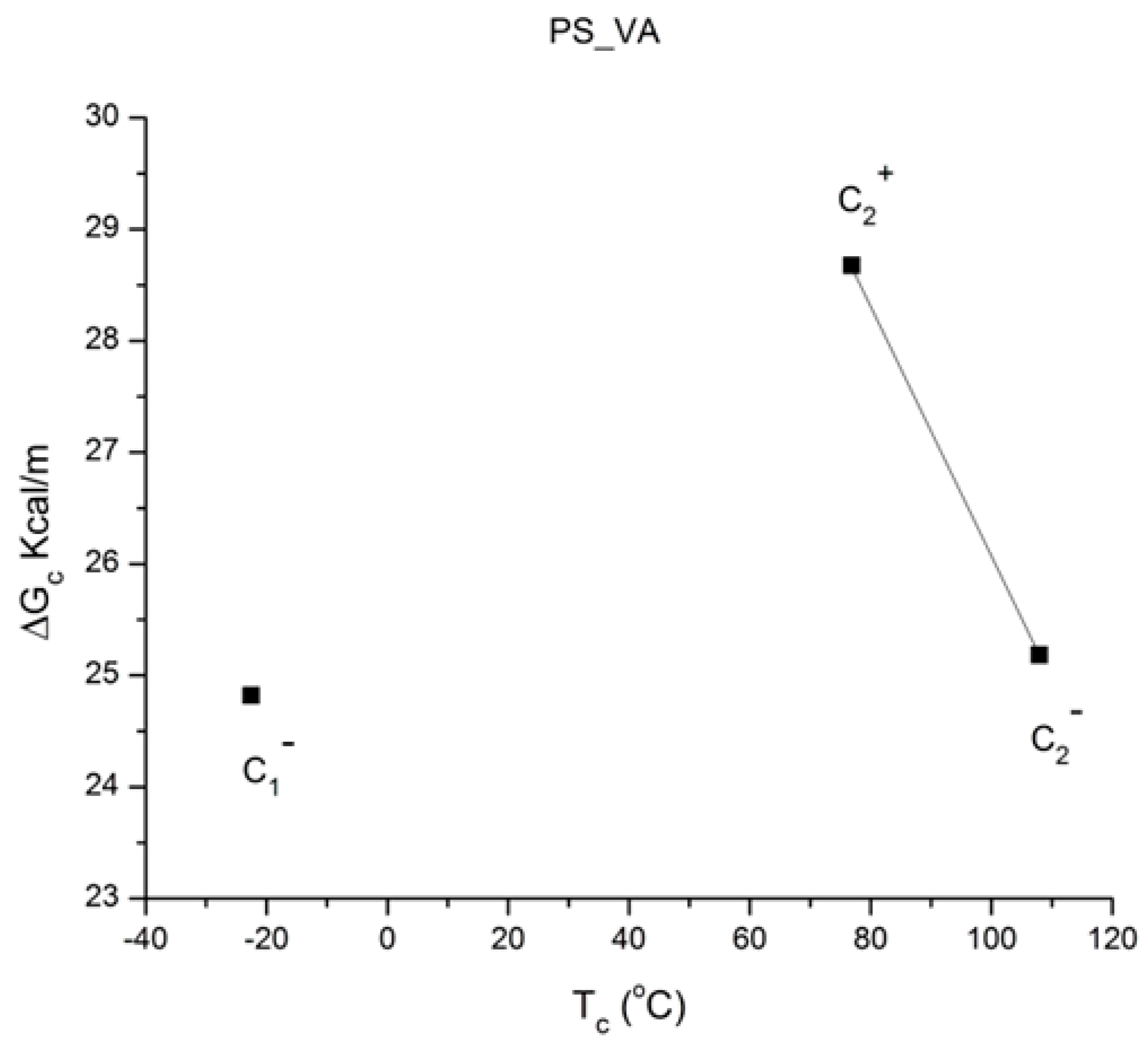
Figure 48 is a plot of ΔG
c vs. T
c for the six compensations in
Table 4, using a color change to differentiate the positive (red, bottom) from the negative (blue, top) compensation concave curves. One sees that T
c increases as the T
p range of the points belonging to the sub-group increases. The trace going through the points is monotonous for both “polarities”, but their curvature is the opposite, concave downwards for the top curve and concave upwards for the bottom one. The two traces of
Figure 49 link points with the same polarity, positive or negative, just like
Figure 45 and
Figure 47, separate the polarities, trying to determine correlations within a given polarity: we did find the super-compensation characteristic of the compensation lines within each polar group. In
Figure 49,
Figure 50,
Figure 51,
Figure 52 and
Figure 53, we investigate the possible correlations between the compensation points across polarities. For instance, in
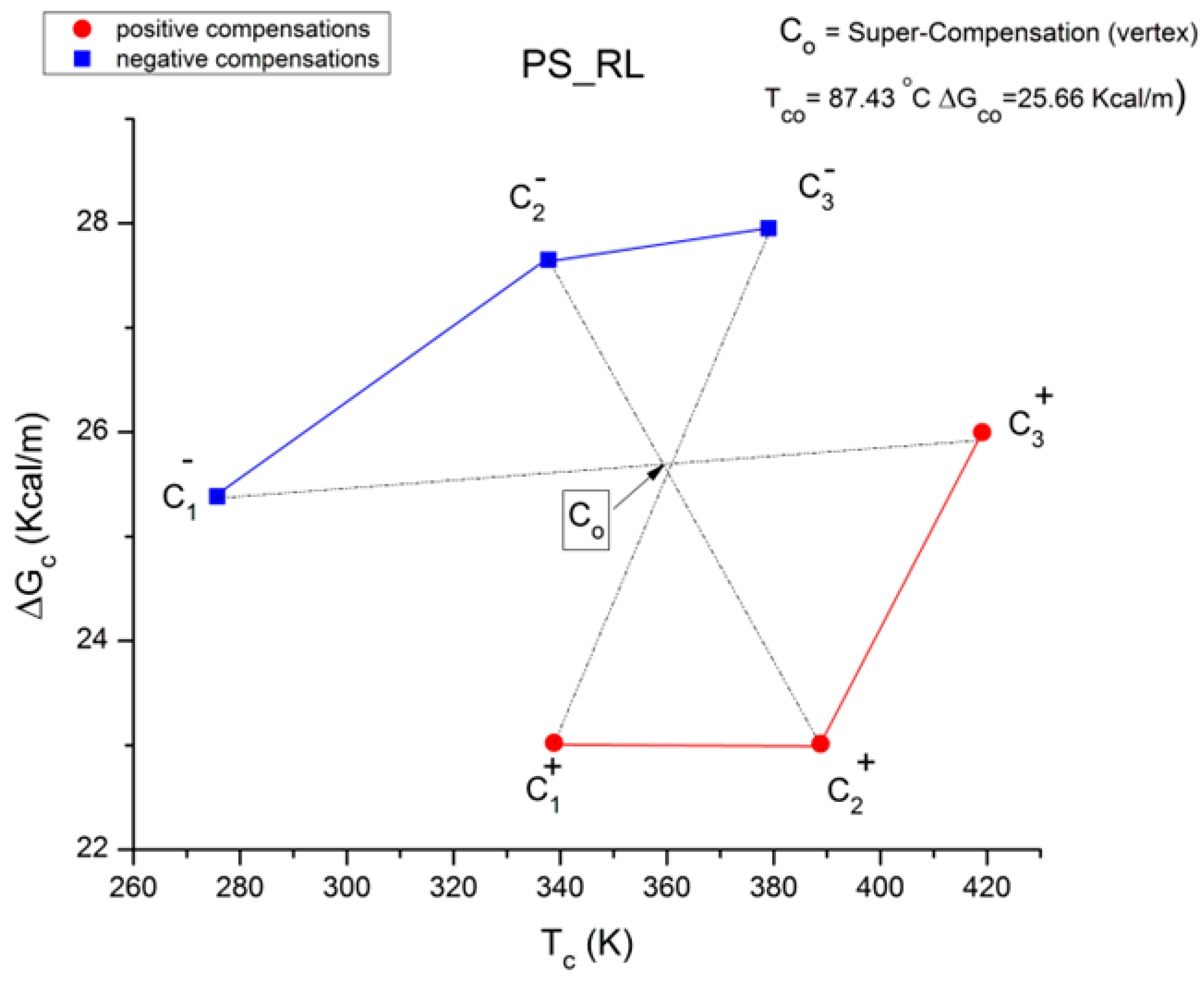
Figure 49, we show that the lines joining the “cross-compensation” points, such as C
1+ and C
3− or C
3+ and C
1−, cut each other at a point belonging to the line joining C
2+ and C
2−. In other words, the point designated C
o in
Figure 49 is a super-compensation point for the cross-polarity network of compensation points, i.e., the vertex of a pencil of lines formed by the 3 cross-polarities lines C
1+C
3−, C
2+C
2−, and C
1−C
3+. This appears to be quite a remarkable property of the depolarization process of a rheomolded sample occurring over time as the temperature follows the TWD sophisticated protocol (
Figure 3) and at the same time releases the internal stress induced by the thermal–mechanical treatment. The coordinates of C
o, the super-compensation point, are ΔG
co = 25.66 Kcal/m and T
co = 87.43 °C, which is 12.6 °C below the T
g of PS.
Figure 48.
Plot of ΔG
c vs. T
c for the 6 compensations in
Table 4. Red, bottom curve: positive compensation points; blue, top curve: negative compensation points.
Figure 48.
Plot of ΔG
c vs. T
c for the 6 compensations in
Table 4. Red, bottom curve: positive compensation points; blue, top curve: negative compensation points.
Figure 49.
Possible correlations between the compensation points across polarities.
Figure 49.
Possible correlations between the compensation points across polarities.
Figure 50 is an extension of our exploration in
Figure 49 of the geometrical features characterizing the network of the compensation points. We expanded the number of links to include C
3+C
3− and C
1+C
1−, in addition to C
2−C
2+, which was already included from the previous figure. We also extended these three links so that they could cut each other. C
1−C
1+ cuts C
2+C
2− at point O
12, and C
3−C
3+ cuts C
2+C
2− at point O
32. The new remarkable feature is that O
12 and O
32 are found vertically below and above C
3+ and C
1−, respectively. This may indicate that the position of the points in the geometrical network responds to symmetrical rules of dependence. We are aware of the possible errors in the determination of the coordinates of the compensation points, which is carried out at the beginning by regressions of the Debye elementary relaxation lines in the Eyring plane (
Figure 42), far from being perfect straight lines even visually, followed by new regressions in the EE planes after the selection of the compensation sub-groups (
Figure 45 and
Figure 46). This makes the quasi-perfectness standing of the geometrical features in
Figure 50 almost impossible to accept. At least some imperfection should exist. This imperfection may be actually present and could have resulted, in fact, in slightly distorting the network symmetry so that C
1+ should align horizontally with C
2+, C
2− should also align horizontally with C
3−, and C
3− should align vertically with C
2+. In
Figure 50, these alignments are almost there; for instance, C
2− already aligns vertically with C
1−, in addition to O
32 with C
1− and O
12 with C
3+, as already mentioned above. This is as if we had discovered a crystal-like perfection of the alignment of the compensation points in the network of compensations. This finding is new and was not presented in [
5], although the experimental evidence was already included (II.4.2 of [
5]). Obviously, these new findings remain at the stage of speculation, and the standard scientific procedure should be applied to affirm or disapprove such new discoveries. Other geometrical particularities of the “structure of the compensation network” are noticeable in
Figure 51.
- -
The line (black) joining the middle of the segment C1−C1+, M1 to the middle of C3−C3+, M3 passes through Co, the vertex of the pencil of lines C1+C3−, C1−C3+, and C2+C2−.
- -
Line C1−C2− is parallel to lines C1+C3+ and M1CoM3.
- -
distance O32C2− = distance CoO12.
Figure 50.
Other possible correlations between the compensation points across polarities. See the text.
Figure 50.
Other possible correlations between the compensation points across polarities. See the text.
Figure 51.
Z-structure-type correlations between the compensation points across polarities.
Figure 51.
Z-structure-type correlations between the compensation points across polarities.
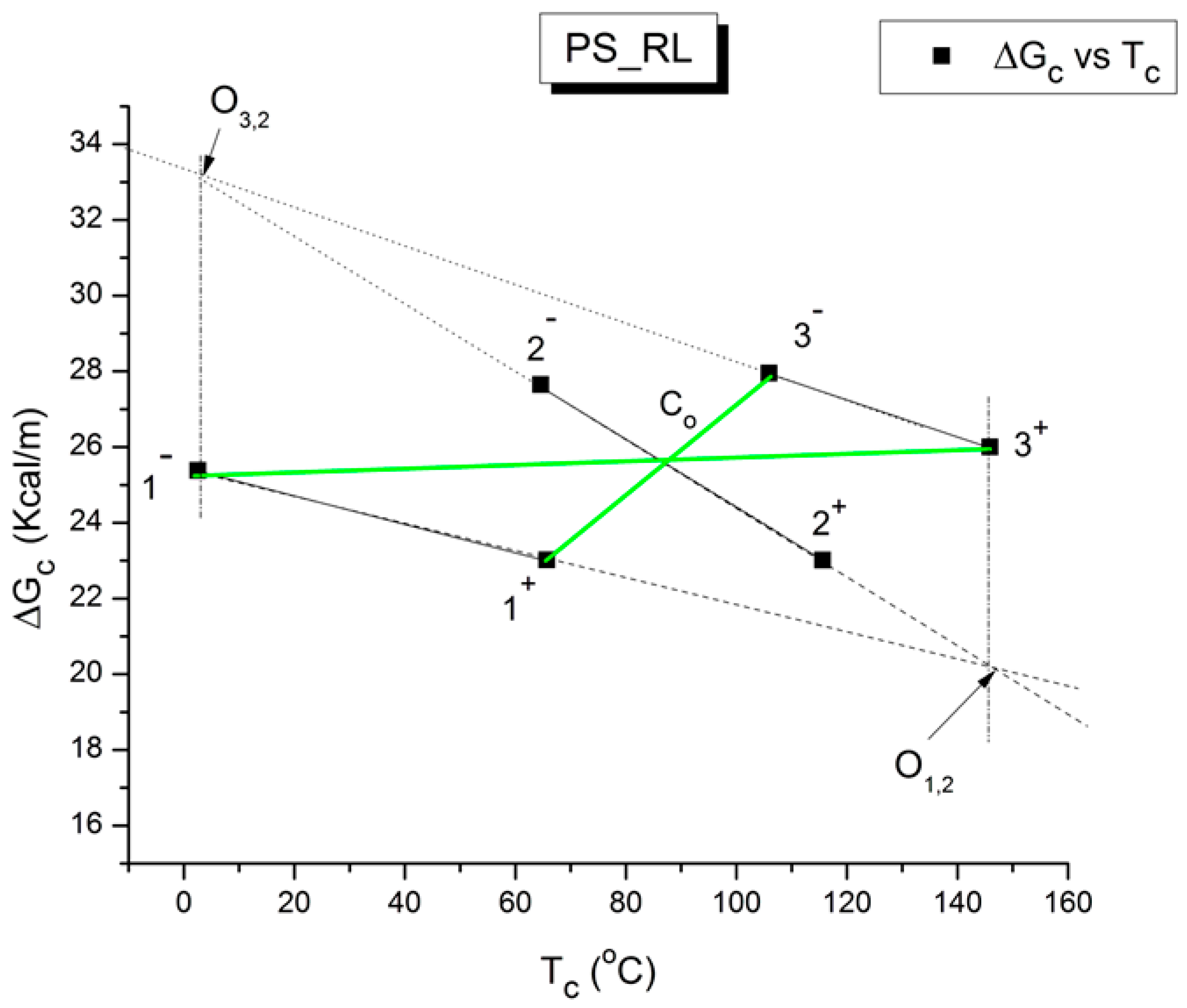
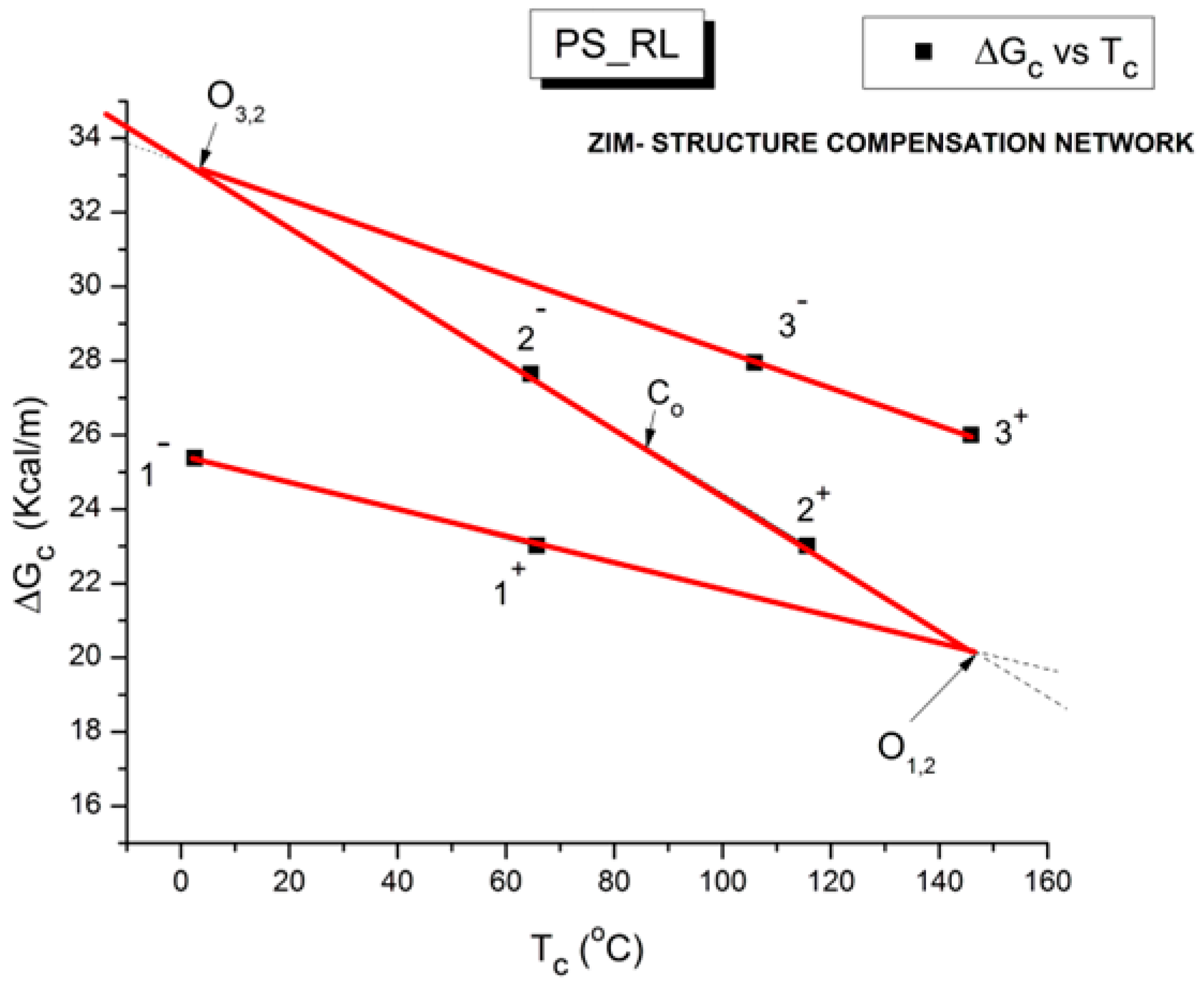
Another important particularity of the network of compensation points that is of great significance for the properties of the amorphous state of polymers is its ability to determine the T
g of the amorphous phases in polymers. We will refer to both
Figure 52 and
Figure 53 to explain this important finding. In
Figure 52, we redrew the same graph that was used in a couple of previous figures but removed all the unnecessary details in the previous graph to reveal the presence of the so-called “Z structure” of the network of compensation points. The illustrated graph is actually a Z looked at in a mirror, which is the reason we called it the ZIM structure. A similar “Z structure” is already observed at the scale of the elementary Debye relaxations to characterize a T
g transition (
Figure 11), but in
Figure 52, we are working at a different scale, the scale of the compensation lines, not the elementary relaxations. Nevertheless, we observe a Z structure associated with the three transitions characterizing phase 1, phase 2, and say phase 3; in a way,
Figure 52 is the second higher stage of a Russian doll assembly that repeats itself at different scales.
Figure 52.
Z-structure (ZIM) compensation network for PS_RL.
Figure 52.
Z-structure (ZIM) compensation network for PS_RL.
Figure 53 is a plot of ΔS
p vs. ΔH
p limited to the positive and negative compensations C
2+ and C
2−, with red and blue lines passing through the positive or negative data points, respectively. It can be shown [
16] that the intersection of these compensations corresponds to the T
g state of the amorphous state relaxing via the process of TWD. We visually see that the intersection occurs approximately at ΔH
p = 60 Kcal/m and ΔS
p = 90 cal/m-K. These values are the enthalpy and entropy of activation of the mode of relaxation triggered by t polarization at T
p = T
g: we designate them ΔH
g and ΔS
g. In previous communications (p. 73 of ref. [
5] and ref. [
16]), we have also explained how to find the value of T
g from the thermo-kinetics values of the amorphous phases of the positive and negative compensations. The first method is based on the assumption that at T
g: ΔG
g= (ΔG
c+ + ΔG
c−)/2 and since for all T
p, then ΔG = ΔH
p − T ΔS
p for T
p = T
g, we have ΔG
g = ΔH
g – T
g ΔS
g. In other words, we have the following:
Equation (13) summarizes the first method to find the value of Tg from TWD to characterize an amorphous phase. It can be used in conjunction with the values of ΔG
c+ and ΔG
c− from
Figure 52, applied to compensations 1, 2, and 3, and the values of ΔH
g and ΔS
g, which are calculated similarly to the way explained for phase 2 in
Figure 53 to find the T
g for phases 1 and 3 besides phase 2.
Figure 53.
The two compensation lines, “positive” (points 3 to 9 at the top) and “negative” ((9,10,11) at the bottom) define the behavior of amorphous phase “2”. Their intersection occurs at Tg, providing ΔHg and ΔSg.
Figure 53.
The two compensation lines, “positive” (points 3 to 9 at the top) and “negative” ((9,10,11) at the bottom) define the behavior of amorphous phase “2”. Their intersection occurs at Tg, providing ΔHg and ΔSg.
The second method to determine T
g from the results in a relaxation map is explained with regard to
Figure 54 and
Figure 55.
Figure 54 is a plot of ΔG
p (Kcal/m) at various T
p values, where ΔG
p is simply the calculated value of ΔG = ΔH
p – T ΔS
p for T = T
p. ΔG(T) is the description of the relaxation of a single Debye relaxation in the ΔG plane during the heating step in
Figure 3. Notice the difference between ΔG
p and ΔG
c, the latter being the value of ΔG extrapolated at the point of compensation between several relaxations. For a polymer without any singular treatment to process it, the ΔG
p vs. T
p behavior is typically linear and almost invariably simple: ΔG
p ~ 0.07 T
p (K) in Kcal/m. This reference behavior, ΔG
pe, is represented by the red dashed line in
Figure 54; subscript “e” in ΔG
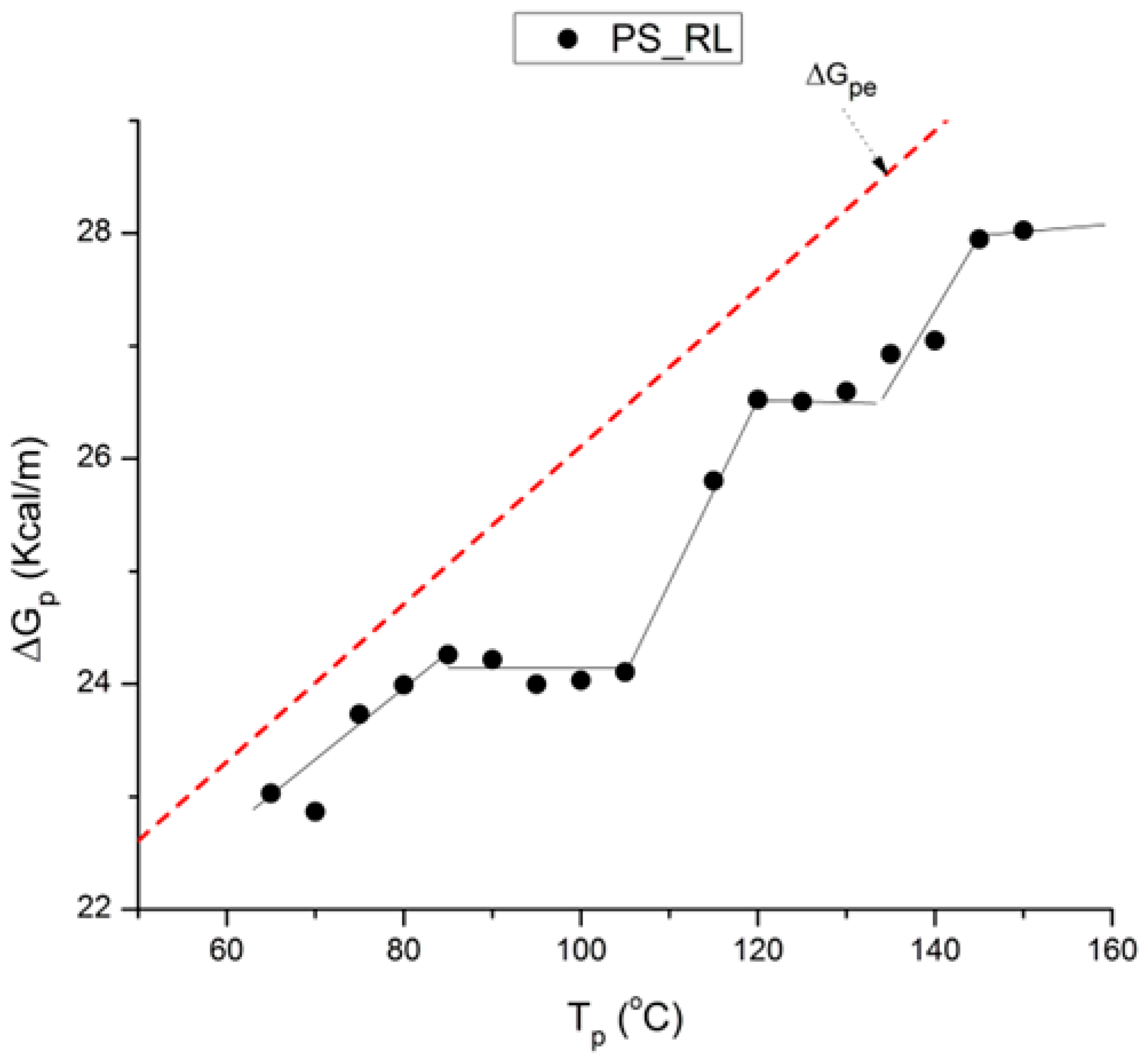
pe refers to the pure dielectric origin of the activation of the dipoles, resulting in a local dis-equilibrium, and of its return to equilibrium during the depolarization process. The PS_RL rheomolded sample in
Figure 54 (the black squares) was purposely brought out of equilibrium to reveal the complex mechanisms of internal motions during the TWD process when the samples return to their equilibrium state during their heating or thermal annealing relaxation stages. The three stages of the relaxation process are indeed visible in
Figure 54. It is clear that the sample is “sustaining” its initial non-equilibrium state to a persisting degree if we compare the evolution of the squares to the dashed reference line representing the equilibrium state. Yet, opening a short parenthesis, we should note that the highest temperature for T
p in
Figure 54 does not reach the T
LL value of the sample, which increases beyond 160 °C (the classical value for PS) by the mechanical treatment during cooling [
10,
11,
12].
Figure 54.
ΔGp vs. Tp for PS_RL. The dashed red line is the expected behavior for a stable sample (with no internal stress before polarization). ΔGp = ΔHp – Tp(K)ΔSp.
Figure 54.
ΔGp vs. Tp for PS_RL. The dashed red line is the expected behavior for a stable sample (with no internal stress before polarization). ΔGp = ΔHp – Tp(K)ΔSp.
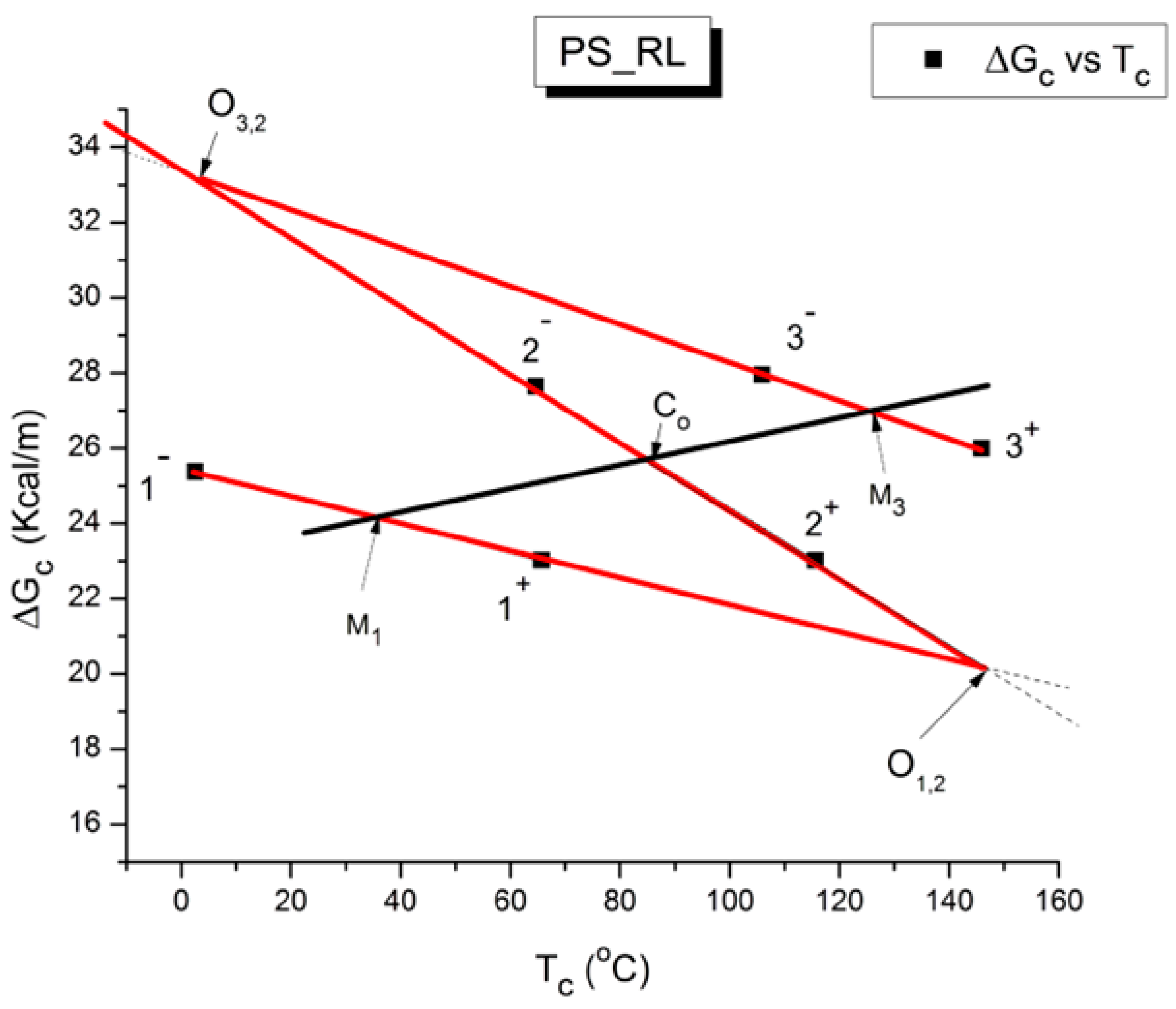
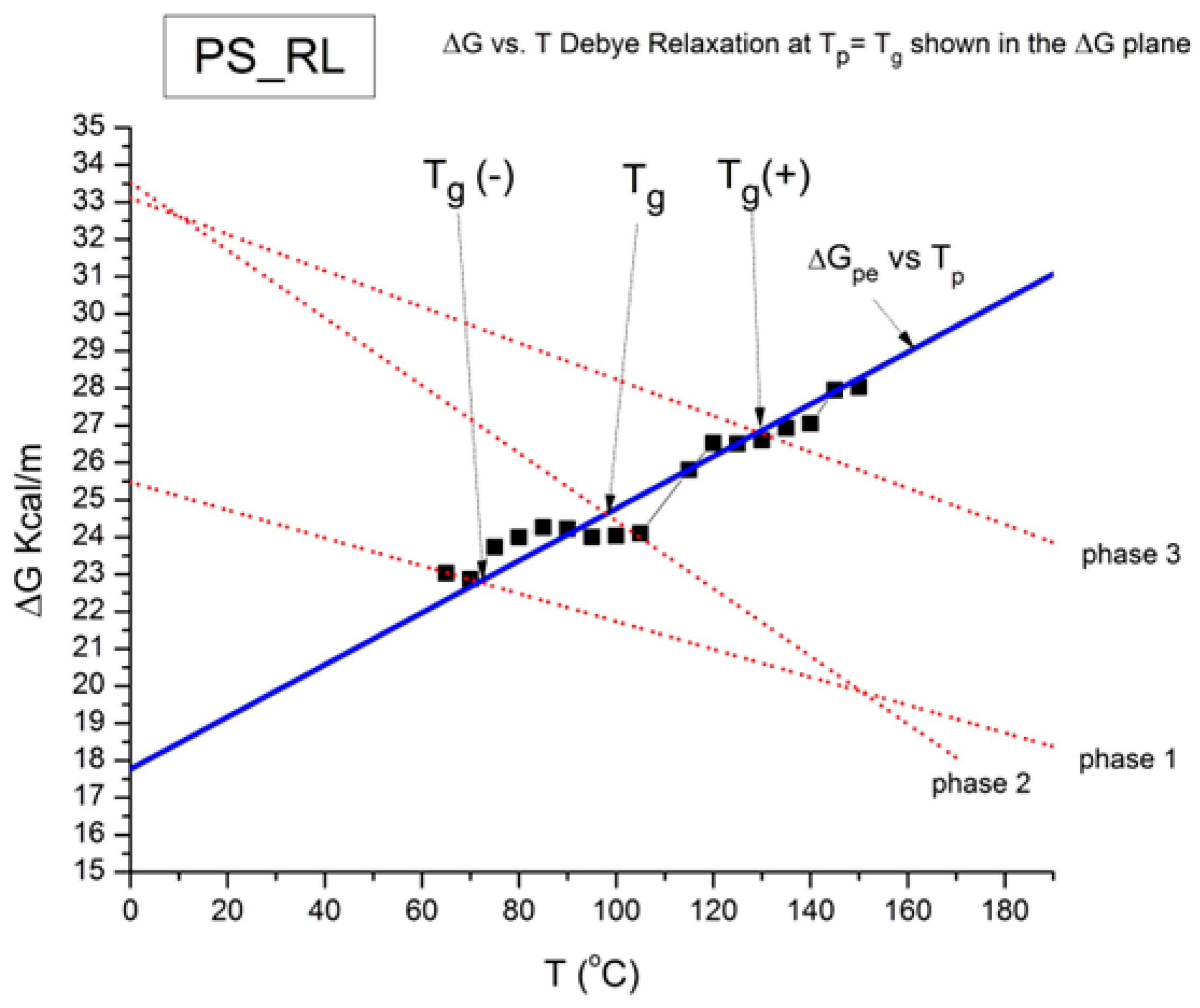
Figure 55 shows the superposition of the graphs in
Figure 52 and
Figure 54. The ZIM structure of
Figure 52 is here visible as red dashed lines. The blue line has the same slope as the dashed line of
Figure 54 but is shifted by regression to fit the square data points. In other words, the blue line is obtained by linear regression of the squares forcing the value of the slope to equal 0.07 Kcal/m-K.
Figure 55.
This figure illustrates the superposition of the graphs in
Figure 52 and
Figure 54. The ZIM structure of
Figure 52 is here visible as red dashed lines. The blue line has the same slope as the dashed line of
Figure 54 but is shifted by regression to fit the square data points. See the text.
Figure 55.
This figure illustrates the superposition of the graphs in
Figure 52 and
Figure 54. The ZIM structure of
Figure 52 is here visible as red dashed lines. The blue line has the same slope as the dashed line of
Figure 54 but is shifted by regression to fit the square data points. See the text.
According to the second method for the determination of a T
g transition from a TWD relaxation map [
16], the value of T
g is at the cross-section of the compensation line of the Z structure pertinent to the relaxation process analyzed with the ΔG
p vs. T
p straight line.
Figure 55 illustrates the method to determine T
g for compensation 1 (amorphous phase 1 with T
g(−)), compensation 2 (amorphous phase 2 with T
g), and compensation 3 (amorphous phase 3 with T
g(+)). Note that, contrary to what we suggested in a previous communication (p. 495 of ref. [
5]), we are affirming in this paper that T
g(−) is not associated with T
β transition, nor is T
g(+) associated with T
LL: the visible breaks are parented to a true fragmentation of T
g, or actually T
α, the mechanical manifestation of T
g, due to the initial conditions at the beginning of the thermal–mechanical treatment. In the Discussion (
Section 3.2), we will show that simulations with the dual-phase model of the interactions succeed in generating positive and negative compensations, multi-compensations, super-compensations, and fragmented behavior at T
g. The results below summarize our findings to describe T
g(−), T
g, and T
g(+) in
Figure 53, Equation (13), and
Figure 55:
| ΔS−g = 37.42 cal/m-K, T−g = 34.2 °C (method 1) or 72.9 °C (method 2); |
| ΔH−g = 35.70 Kcal/m; |
| ΔSg = 90.93, Tg = 90.2 °C (method 1) or 100.0 °C (method 2); |
| ΔHg = 58.37; |
| ΔS+g = 48.72, T+g = 126.0 °C (method 1) or 129.0 °C (method 2); |
| ΔH+g = 46.42. |
The only noticeable discrepancy between the two methods to calculate T
g is for T
g(−), and we have no explanation as to why. The two methods are compatible at a 0.5-degree error for samples in a state of equilibrium [
4], an empirical rule that does not appear to apply for samples out of equilibrium due to their processing history.
We propose that these “break transitions” of T
g, revealed by multi-compensations structuring as a Z network, correspond to different dissipative states of the interactions between the dual-conformers of the amorphous matter. The T
β transition in PS, and in other amorphous polymers, is indeed a local property of the dual-conformers, corresponding to the lower horizontal branch of the Z structure at the scale of the elementary relaxations (lower scale). The T
g(−) break is different because it is the lower branch of a Z structure of compensations of the dual-conformers (higher scale), not of the dual-conformers themselves (lower scale). The same reasoning applies to the case of T
LL and T
g(+): they are equivalent but not the same because working at different scales. The T
LL transition is observed in a relaxation map at the T
p temperature that corresponds to the upper horizontal branch of a Z structure involving a lower scale. The T
g(+) break is the upper branch of a Z structure, apparently the equivalent of T
LL but working at the higher scale level of the compensation network.
Section 3.2 and
Section 4 will explain what we really mean by “a different scale” in a dual-phase dissipative context [
7].
Finally, we present a plot of ΔS
p versus T
p for PS_RL in
Figure 56. For a sample in a state of internal equilibrium, there is only one T
g, and the positive and negative compensations that cross at T
g (like in
Figure 53) are transposed, in a ΔS
p vs. T
p plot, to two hyperbolic functions on both sides of T
g sharing the same infinity axis at T
p = T
g [
16].
Figure 56 applies to a sample in non-equilibrium, showing the fragmentation of T
g in three T
gs values and the conversion of the hyperbolic functions to linear ones. There are no distinctions in behavior for either T
g(−), T
g, or T
g(+). This confirms what was hinted before, namely the fragmented transitions perceived below and above T
g have the same relaxation origin as T
g itself (see
Section 3.2 and
Section 4). Another observation in
Figure 56 is the sharp discontinuity observed between the fragmented transitions in a way rarely observed in physics except at critical transitions generated by chaotic behavior. This comment should be remembered when a dual-phase explanation of the fragmentation of T
g in the TWD experiments is provided in the Discussion (Figure 68).
Let us now go back to the analysis of the slowly cooled sample PS_VA (
Figure 41). We said earlier that two negative compensations are clearly visible (
Figure 41) and that point 5 (T
p = 95 °C) was “lost”. In view of what we learned from the analysis of PS_RL, we can now reexamine the situation (
Figure 57). Knowing that positive and negative compensations alternate to define the state of an amorphous phase with a T
g transition at their crossing, point 5 is in fact located on a positive compensation that linearly lines up points 4, 5, and 6. The positive compensation line is not defined with certainty in this case since we only have one intermediate point between points 4 and 6 to determine its slope and intercept. Yet, the fact that point 5 is exactly located on the line joining points 4 and 6 gives a good probability that our assumption is correct.
We have learned how to find the values of the compensation points from the analysis of PS_RL above and will apply the same method below. We calculated the following parameters:
The coordinates of the compensation points T
c and ΔG
c, expressed in the ΔG plane, are found from the equation of the EE curves (above) and are compiled in
Table 5.
Notice that, in the above description of PS_VA, we named the compensations comp(1)
−, comp(2)
+, and comp(3)
−”, since the positive compensation was added to our initial count of 2 in
Figure 41 after we realized that point 5 was not “lost” and that a new compensation should be added to the set of the two negative compensations. Additionally, to match the annotations used for PS_RL, comp(3)
− is actually the negative compensation line of comp(2), so its name should be changed to comp(2)
−. Hence, In our new understanding of the TWD results of PS_VA, let us change the annotations to match the ones used that will reflect the differences between PS_VA and PS_RL appear more clearly:
Figure 58 is a plot of ΔG
c vs. T
c, using the data from
Table 5 while considering the new understanding of the compensation points. The first difference observed between
Figure 58 for PS_VA and
Figure 48 for PS_RL is the reversal in the vertical position of the negative and positive compensation points: the negative compensations C
1− and C
2− are below C
2+ for PS_VA, and the opposite is seen for PS_RL. This is a major difference apparently only due to their different thermo-mechanical histories. The comparison of the network of compensations for these two treatments makes it easier to realize that three additional compensation points are missing for PS_VA and would have probably become visible if we had extended the T
p range of the TWD Debye relaxations at both ends: polarizing at lower values than point 1 (T
p = 60 °C) to make C
1+ visible and to higher values than point 8 (T
p = 125 °C) to make C
3+ and C
3− visible. Using the three existing compensation points (
Figure 58) and assuming that the geometrical rules established for PS_RL remain valid for PS_VA, we can construct what the compensation network could look like for PS_VA. This extrapolated network construction is shown in
Figure 59. Amazingly, provided our assumptions are verified by new experiments with a broader range of T
p values, the network for the slowly cooled sample becomes a simple parallelogram tilted upwardly, with the three positive compensation points aligned (red line on top), the three negative compensation points also aligned (blue line at the bottom), and the parallelism of the lines passing through the positive and negative compensation points. When the three compensation points are aligned, it means that their corresponding compensation lines compensate at a single point: this is a super-compensation of the single Eyring relaxations. This same situation is observed in
Figure 45 and
Figure 56 for PS_RL. Yet, for PS_RL, the coordinates of the super-compensation points of the negative and positive super-compensations are different for both axes. The fact that the network of compensations is a parallelogram for PS_VA (
Figure 58) and only a quadrilateral for PS_RL (
Figure 52) is significant and represents a characteristic of the state of non-equilibrium of the amorphous phase that can be correlated to specific thermal–mechanical treatments.
The ΔH
g and ΔS
g values for the visible transition (T
g) at the middle of C
2+ and C
2− can be calculated from the intersection of the compensation lines (4,5,6) and (6,7,8): ΔH
g = 68.0 Kcal/m and ΔS
g = 112.4 cal/m-K. The value of T
g from Equation (13) is determined as 92 °C. The vertex of the pencil of the line is C
o (T
co = 91.78 °C, ΔG
co = 26.95 Kcal/m), and therefore T
co = T
g, ΔG
co = ΔG
g. For the PS_VA sample, with no internal stress, T
g is exactly located at the vertex of the network of compensations. This is not true for samples that are not at equilibrium and the analysis presented in this section allows for the characterization of the differences.
Figure 60 below illustrates ΔS
p vs. T
p for PS_VA, which should be compared to
Figure 56 for PS_RL.
The only certainty about
Figure 60 is that the last three points on the left (points 6, 7, and 8 in
Table 1) can possibly be fitted by a hyperbola since they belong to the negative branch of the T
g transition of an amorphous phase at equilibrium [
5,
16]. A hyperbolic fit is always feasible with three points, yet the interest is that, for the x-asymptote, the fit gives the vertical red line corresponding to T
p = 373 K, clearly what we expect to find for polystyrene. This is not, however, what we found for T
g using Equation (13): we found 92 °C. On the other hand, the two points on the left side of the red line belong to a hyperbola fitting the behavior of a positive branch of a T
g transition (the asymptote for that left branch is T
g = 373 K). Hence, points 3, 4, 5, 6, and 7 are correctly positioned with respect to a T
g transitional behavior for a sample at equilibrium. The other points (1–4) are not on that hyperbolic branch, and there are possibly two reasons for this deviation. First, these points may still not be at equilibrium despite the long cooling time because the cooling sample became a glass ever since it reached 100 °C, resulting in a drastic reduction in the rate to reach equilibrium. The other possibility is that points 1–4 belong to the negative branch of the T
g(−) transition, which may exist at low T
p.
Figure 61 below, showing ΔG
p vs. T
p for PS_VA, may help solve this uncertainty when it is compared to
Figure 54. The first four points of
Figure 54 are shifted vertically to position themselves around the “equilibrium” red straight dashed line in
Figure 61 and, therefore, these four points belong to the negative hyperbola of T
g(−) in
Figure 60, corresponding to the compensation point C1
− of
Figure 59.
Another comparison of PS_RL and PS_VA relates to the super-compensation coordinates. For PS_VA in
Figure 59, we saw that the red and blue lines joining the positive and the negative compensation points, respectively, were parallel to each other, separated by a distance (δΔG
c = 3.494 Kcal/m and δT
c = 31.1 °C (31.276). The equation of the positive super-compensation line is ΔG
c = 27.62 + 3.036 10
−3 T
c(K), and for the blue super-compensation line, it is ΔG
c = 24.12 + 3.036 10
−3 T
c(K). The intercept and slope of the super-compensation line provide the coordinates of the super-compensation points “of the lower scale correlation map”: ΔH
c = intercept and ΔS
c = −slope. Hence, the super-compensation coordinates for the positive and negative compensations of PS_VA can be compared to the ones shown in
Figure 45 and
Figure 46 for PS_RL, respectively.
| PS_VA | PS_RL |
| ΔHsc+ = 27.62 Kcal/m | ΔHsc+ = 23.0 Kcal/m |
| ΔSsc+ = −3.04 cal/m-K | ΔSsc+ = 0 cal/m-K |
| |
| |
| ΔHsc− = 24.12 Kcal/m | ΔHsc− = 17.5 Kcal/m |
| ΔSsc− = −3.04 cal/m-K | ΔSsc− = −23.0 cal/m-K |
One sees that the thermo-mechanical treatment essentially lowers the enthalpy of the super-compensations, for both positive and negative compensations, with the effect being stronger for the negative compensations (6.67 vs. 4.62 Kcal/m). The network of compensations becomes asymmetrical pursuant to the thermo-mechanical treatment. When there was no mechanical treatment, and the sample was cooled very slowly in the mold to induce a near-equilibrium state for the molded sample, the network of compensations appeared symmetrical; yet, the structure of the polarity of the network of compensations was reversed (upside down). Note that the confirmation of these conclusions is pending a repeat of the TWD testing of the slowly cooled samples using a broader span of T
p values to validate the construction of the compensations’ network. Our analysis of the PS_VA results was performed with only three compensation points and using the empirical rules established from analyzing samples in non-equilibrium amorphous states. We have only presented the details of our analysis for one treated ample, PS_R, but have presented elsewhere (II.4 of [
5]) the influence of several other thermal–mechanical treatments validating the generalization of the results presented in this review: the presence of three scales to consider the interactions, namely the scale of their Debye relaxation, the scale of their compensation, and the scale of the compensation of the compensations, i.e., of their super-compensation, with each scale interactively coupled to the next one forming a general network of compensations.
In summary of this
Section 2.3, it should be concluded that the thermal-windowing depolarization (TWD) procedure applied to the rheomolded PS samples (compression-molded under vibration) allows for the quantification of the influence of the thermal–mechanical history on the “internal stress” incurred by the rheomolding variables. In this
Section 2.3, we also showed the power of the TWD analysis to gain insights into the interactive coupling relaxation mechanisms between the local molecular “tags”, i.e., the dipoles, activated dielectrically. The relaxation map of these rheomolded samples indicates that the deconvoluted dipole relaxations belong to a highly ordered network of compensation lines themselves, compensating to a single point of the relaxation map. This “super-compensation” behavior appears to indicate a unique simple kinetic origin to the various electrical relaxation mechanisms found below T
g, at T
g, and above T
g. The phenomenon of compensation of the compensation lines of the Arrhenius relaxations generated at various T
p values seems to be a key revelator of the fundamental mechanism responsible for this apparently diversified and complex behavior [
5,
16]. As we said in our objectives of this review, we will attempt to understand, in the
Section 3 below, the dual-phase gears capable of generating such apparent simplicity and how the interactions in the amorphous phase are modulated by the presence of compensations at different scales.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}