

Three-Dimensional Printing of Personalized Carbamazepine Tablets Using Hydrophilic Polymers: An Investigation of Correlation Between Dissolution Kinetics and Printing Parameters

Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Process Development of 3D-Printed Tablets
HME
Three-Dimensional Printing
Process Development Using Design of Experiments (DoE)
2.2.2. Characterization of Raw Materials, Intermediate Extrudates, and Printed Tablets
Thermalgravimetric Analysis (TGA)
Differential Scanning Calorimetry (DSC)
Powder X-Ray Diffraction (PXRD)
Hot-Stage Polarized Light Microscopy (PLM)
Qualification and Quantification of CBZ Using HPLC
2.2.3. In Vitro Drug Release Study
3. Results and Discussion
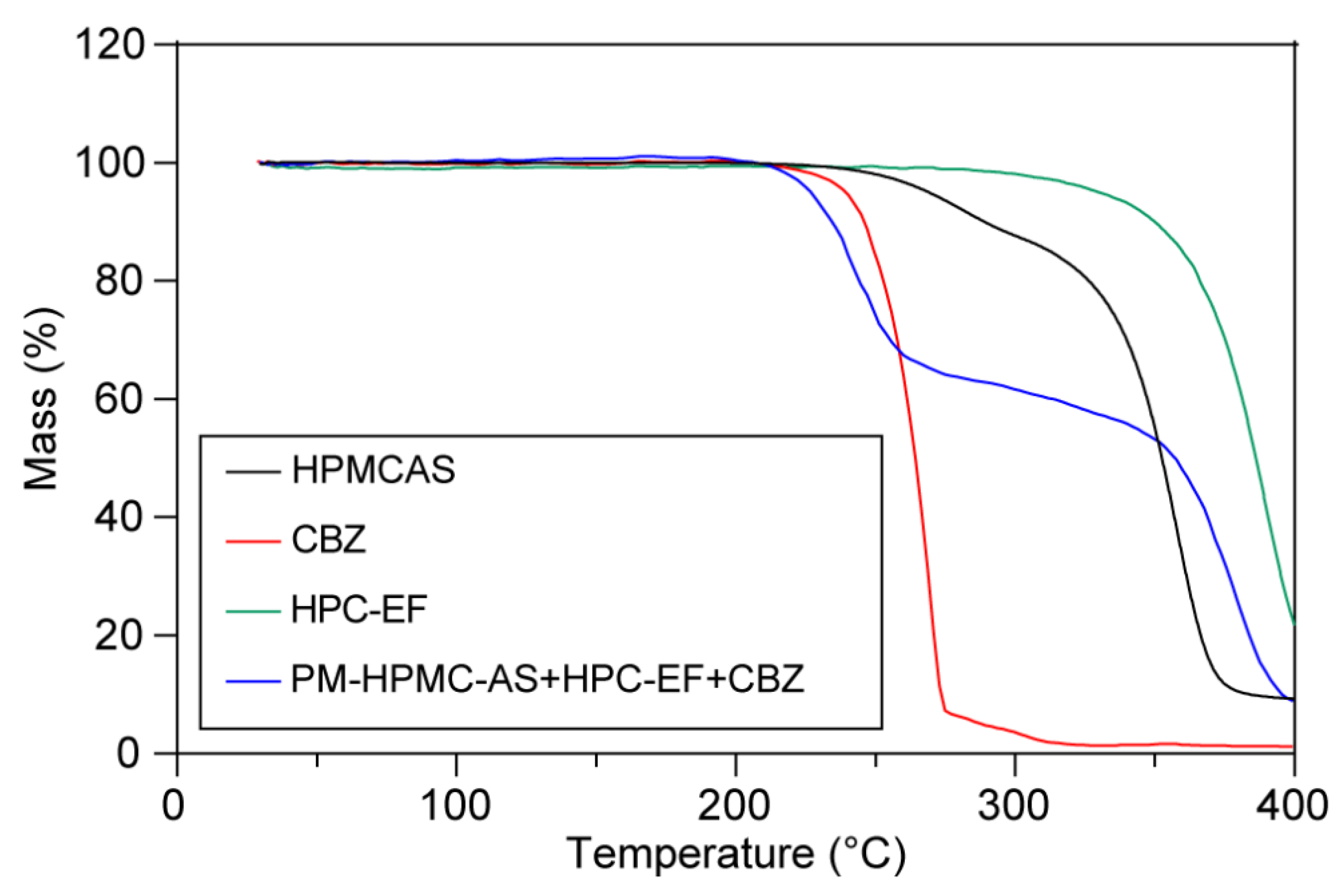
3.1. Thermal Stability
3.2. Preparation of 3D Printable Filaments
3.3. Solid-State Analysis
3.3.1. Characterization of Thermal Behavior of Raw Materials and Filaments
3.3.2. Hot-Staged PLM of Raw Materials and Filaments
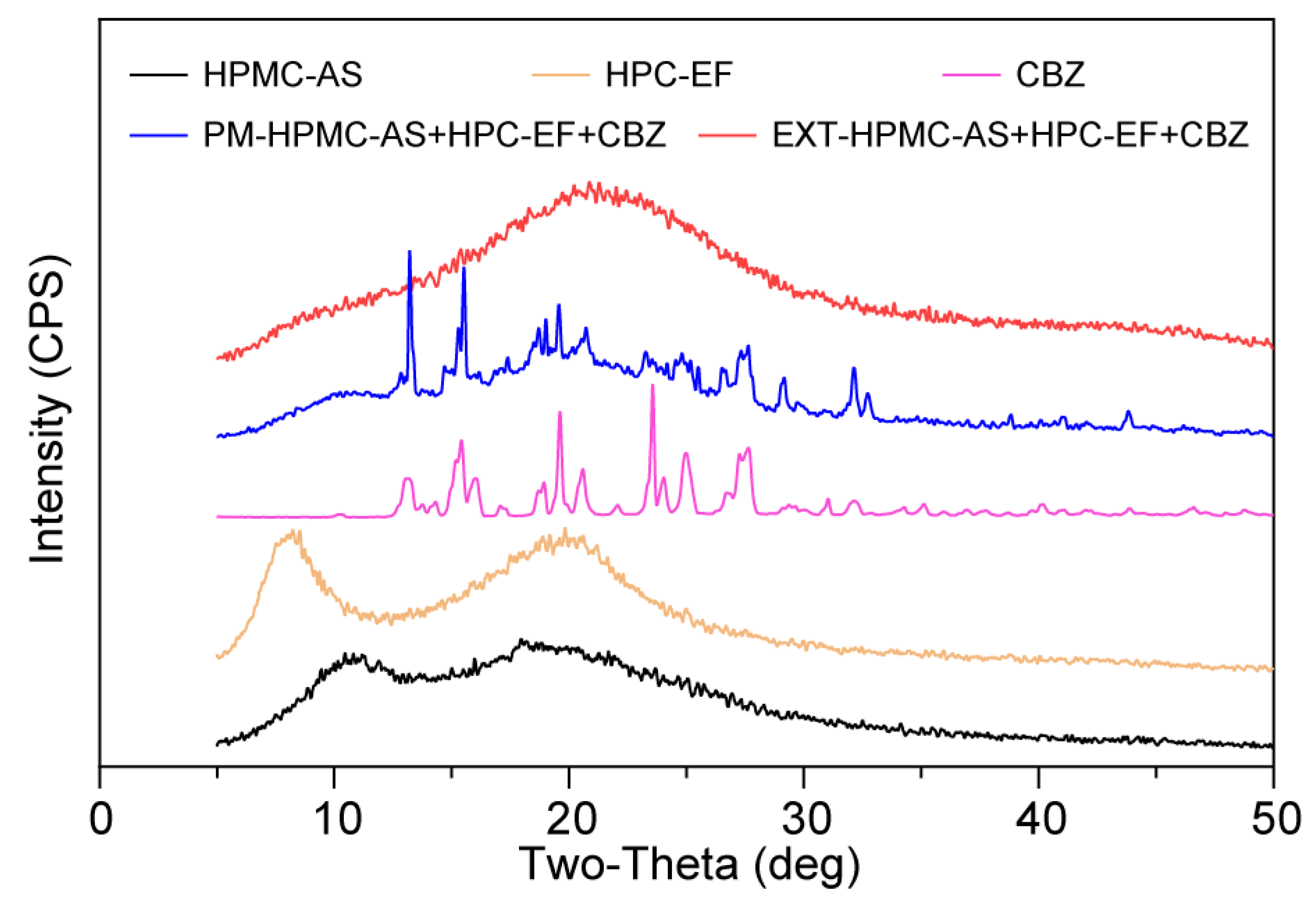
3.3.3. Characterization of Crystalline States of Raw Materials and Filaments
3.4. DoE Studies
3.4.1. Physical Properties of 3D-Printed Tablets
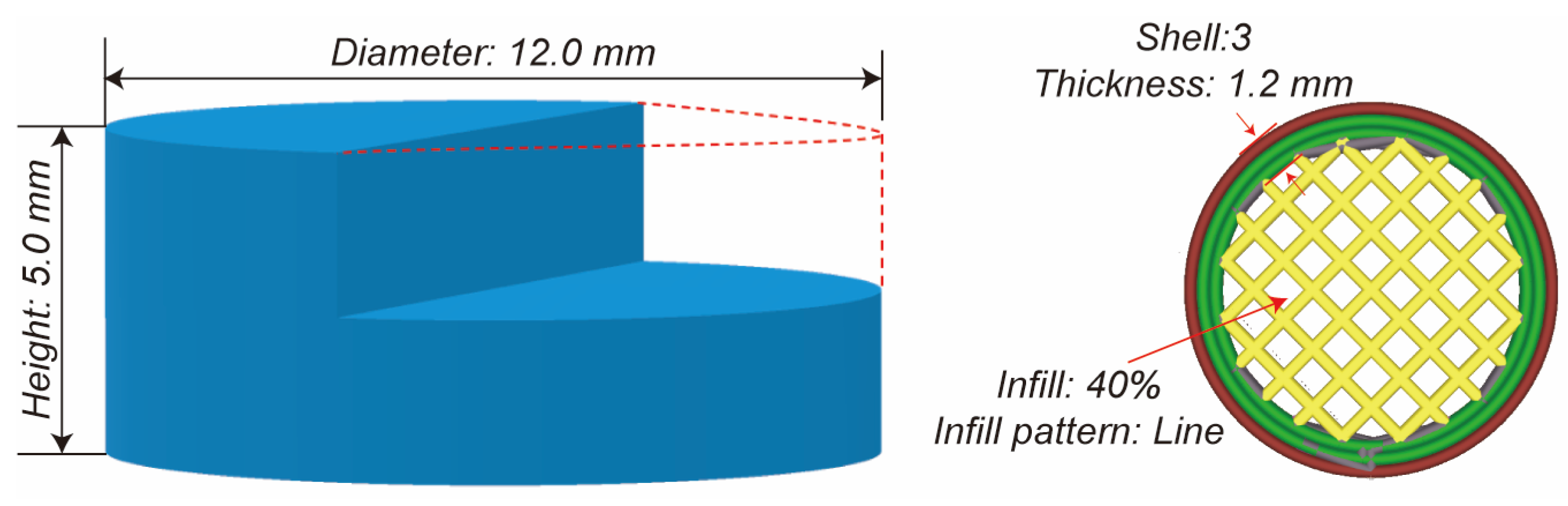
Morphology Study
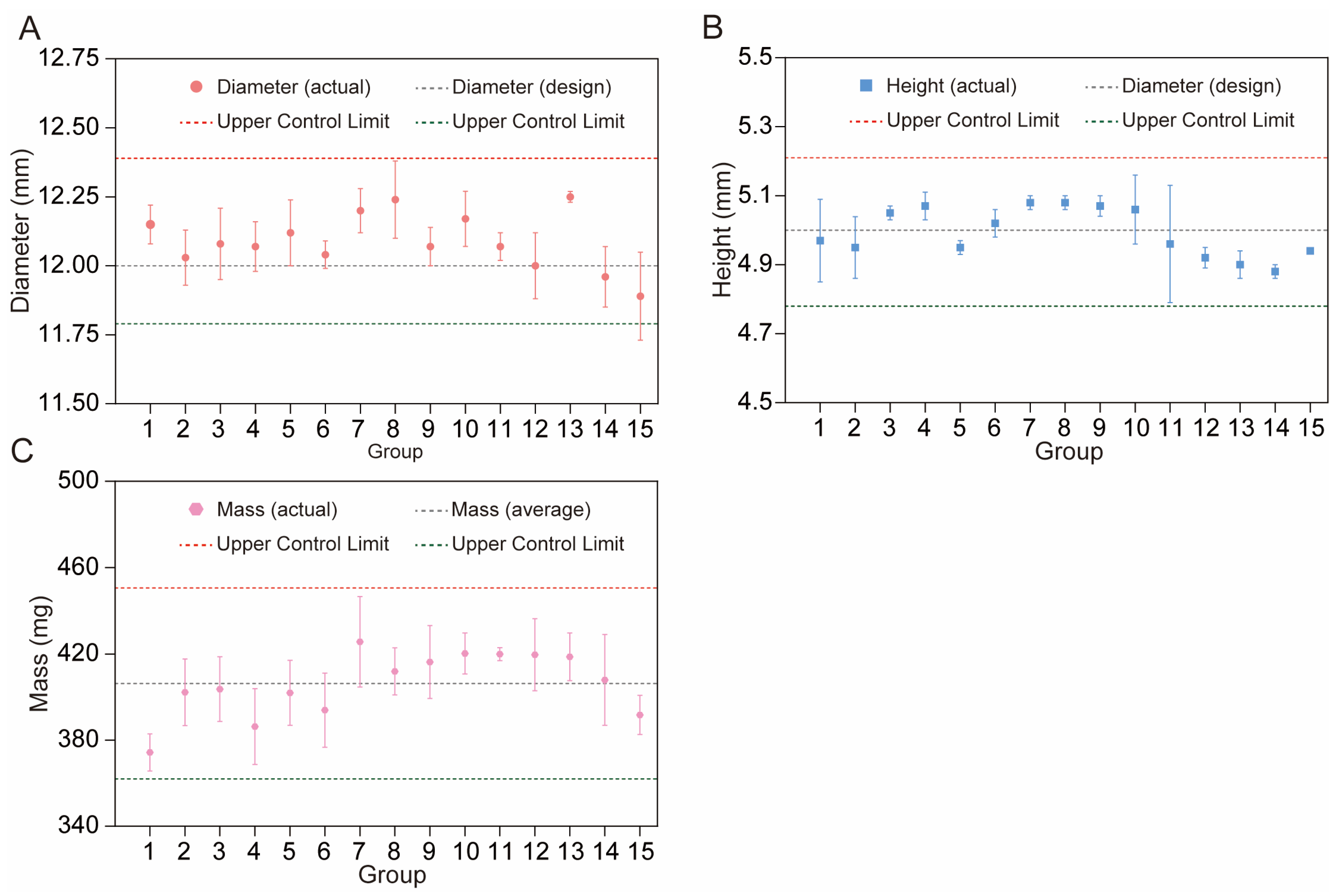
Height and Diameter
Mass
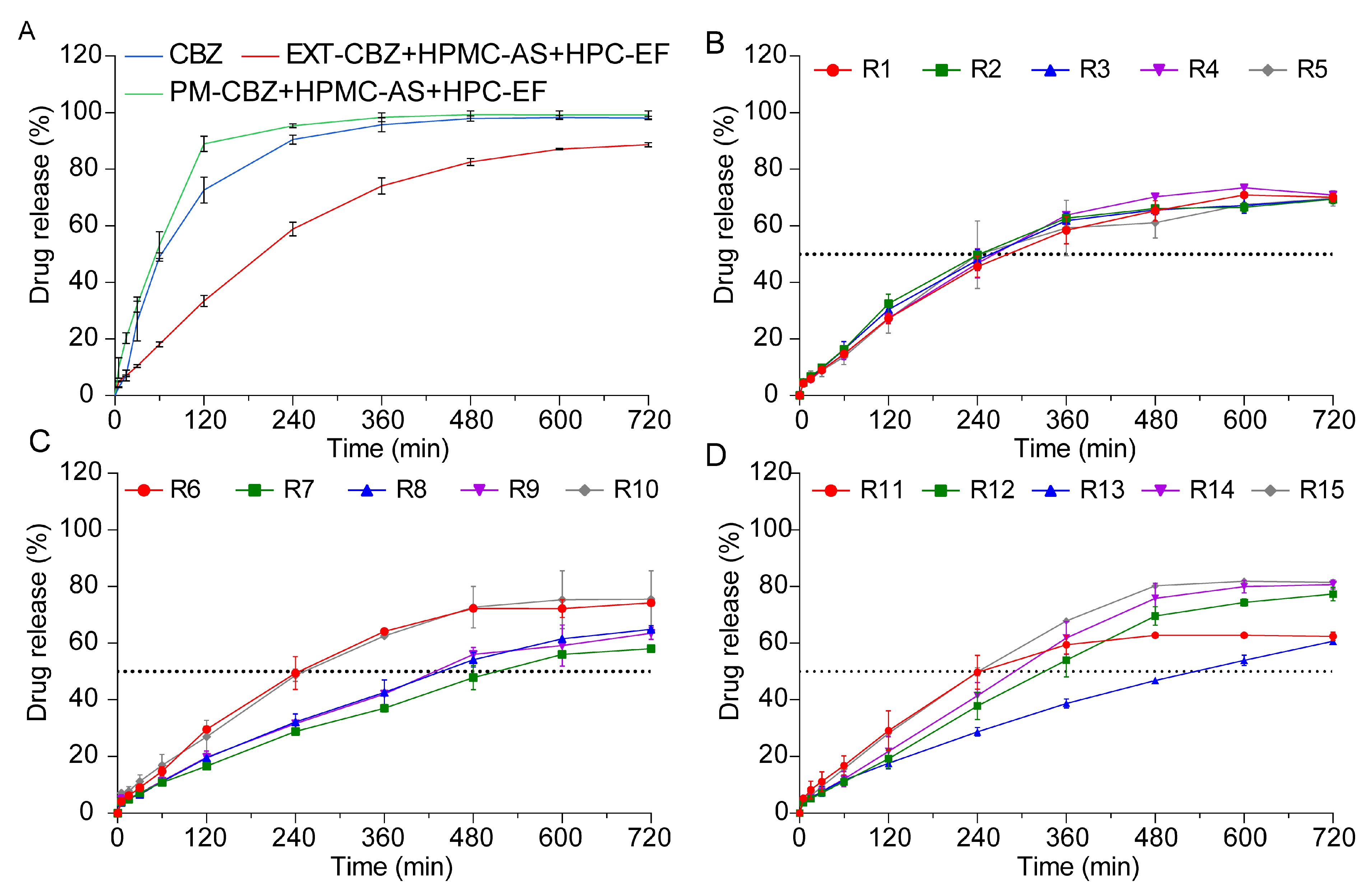
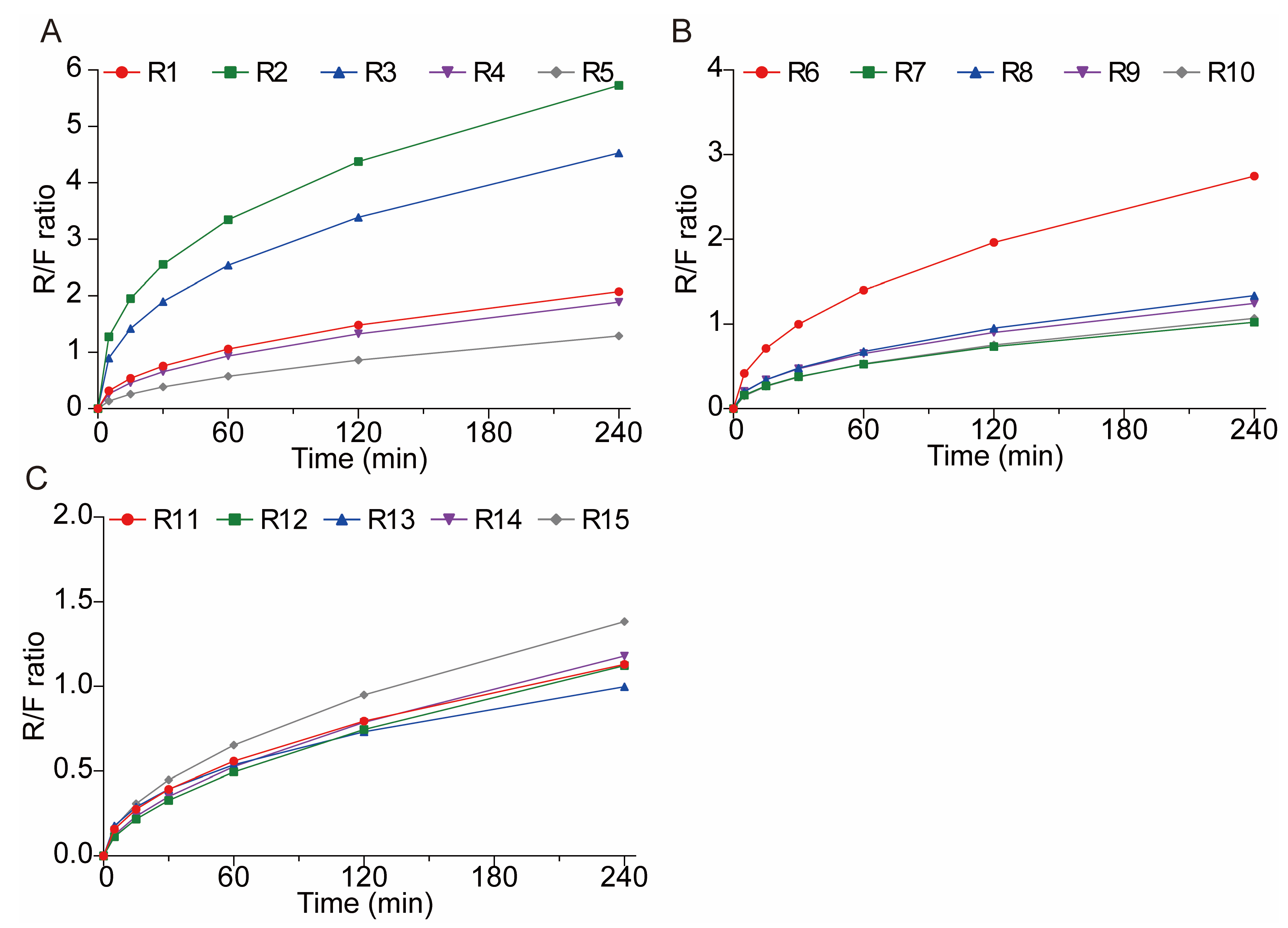
3.4.2. In Vitro Drug Release Studies
Quality and Quantity of the CBZ
In Vitro Drug Release Studies of 3D-Printed Tablets
Analysis of Q4h and t50%
Response Surface Methodology
The Release Kinetics
3.5. Reproducibility
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Elemento, O. The Future of Precision Medicine: Towards a More Predictive Personalized Medicine. Emerg. Top. Life Sci. 2020, 4, 175–177. [Google Scholar] [CrossRef]
- Adepu, S.; Ramakrishna, S. Controlled Drug Delivery Systems: Current Status and Future Directions. Molecules 2021, 26, 5905. [Google Scholar] [CrossRef]
- Thacharodi, A.; Singh, P.; Meenatchi, R.; Ahmed, Z.H.T.; Kumar, R.R.S.; Neha, V.; Kavish, S.; Maqbool, M.; Hassan, S. Revolutionizing Healthcare and Medicine: The Impact of Modern Technologies for a Healthier Future—A Comprehensive Review. Health Care Sci. 2024, 3, 329–349. [Google Scholar] [CrossRef]
- Bayda, S.; Adeel, M.; Tuccinardi, T.; Cordani, M.; Rizzolio, F. The History of Nanoscience and Nanotechnology: From Chemical–Physical Applications to Nanomedicine. Molecules 2019, 25, 112. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, Q.; Nasif, K.F.A.; Xie, Y.; Deng, B.; Niu, S.; Pouriyeh, S.; Dai, Z.; Chen, J.; Xie, C.Y. AI-Driven Deep Learning Techniques in Protein Structure Prediction. Int. J. Mol. Sci. 2024, 25, 8426. [Google Scholar] [CrossRef] [PubMed]
- Koppad, S.; Annappa, B.; Gkoutos, G.V.; Acharjee, A. Cloud Computing Enabled Big Multi-Omics Data Analytics. Bioinform. Biol. Insights 2021, 15, 11779322211035921. [Google Scholar] [CrossRef]
- Sun, C.; Wang, A.; Zhou, Y.; Chen, P.; Wang, X.; Huang, J.; Gao, J.; Wang, X.; Shu, L.; Lu, J.; et al. Spatially Resolved Multi-Omics Highlights Cell-Specific Metabolic Remodeling and Interactions in Gastric Cancer. Nat. Commun. 2023, 14, 2692. [Google Scholar] [CrossRef]
- Zhu, T.; Yang, M.; Wang, Y.; Xu, J.; Dai, H. Towards Precision Medicine: Leading with Big Data and Multidisciplinary Collaboration. Precis. Medicat. 2024, 1, 100003. [Google Scholar] [CrossRef]
- Beg, S.; Almalki, W.H.; Malik, A.; Farhan, M.; Aatif, M.; Rahman, Z.; Alruwaili, N.K.; Alrobaian, M.; Tarique, M.; Rahman, M. 3D Printing for Drug Delivery and Biomedical Applications. Drug Discov. Today 2020, 25, 1668–1681. [Google Scholar] [CrossRef]
- Lee, M.-S.; Yao, D.-J.; Hsu, W. An Implantable Drug-Delivery System on a Chip. Curr. Top. Med. Chem. 2015, 15, 1516–1524. [Google Scholar] [CrossRef]
- Cobo, A.; Sheybani, R.; Meng, E. MEMS: Enabled Drug Delivery Systems. Adv. Health Mater. 2015, 4, 969–982. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kang, W.; Wang, W. Photocleavage-based Photoresponsive Drug Delivery. Photochem. Photobiol. 2022, 98, 288–302. [Google Scholar] [CrossRef]
- Guzzi, E.A.; Tibbitt, M.W. Additive Manufacturing of Precision Biomaterials. Adv. Mater. 2020, 32, e1901994. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, F.; Xu, H.; Fuenmayor, E.; Major, I. Material Compatibility and Processing Challenges in Droplet Deposition Modelling Additive Manufacturing: A Study on Pharmaceutical Excipients Polyvinylpyrrolidone/Vinyl Acetate (PVP/VA) and Polycaprolactone (PCL). Eur. J. Pharm. Sci. 2024, 200, 106850. [Google Scholar] [CrossRef]
- Ervasti, T.; Simonaho, S.-P.; Ketolainen, J.; Forsberg, P.; Fransson, M.; Wikström, H.; Folestad, S.; Lakio, S.; Tajarobi, P.; Abrahmsén-Alami, S. Continuous Manufacturing of Extended Release Tablets via Powder Mixing and Direct Compression. Int. J. Pharm. 2015, 495, 290–301. [Google Scholar] [CrossRef]
- Croitoru-Sadger, T.; Yogev, S.; Shabtay-Orbach, A.; Mizrahi, B. Two-Component Cross-Linkable Gels for Fabrication of Solid Oral Dosage Forms. J. Control. Release 2019, 303, 274–280. [Google Scholar] [CrossRef]
- Tan, D.K.; Maniruzzaman, M.; Nokhodchi, A. Advanced Pharmaceutical Applications of Hot-Melt Extrusion Coupled with Fused Deposition Modelling (FDM) 3D Printing for Personalised Drug Delivery. Pharmaceutics 2018, 10, 203. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, J.; Li, Y.; Yang, W.; Ni, W.; Jia, Y.; Yu, M.; Zhang, J. Improve Solubility and Develop Personalized Itraconazole Dosages via Forming Amorphous Solid Dispersions with Hydrophilic Polymers Utilizing HME and 3D Printing Technologies. Polymers 2024, 16, 3302. [Google Scholar] [CrossRef]
- Huang, L.; Yang, W.; Bu, Y.; Yu, M.; Xu, M.; Guo, J.; Ni, W.; Jia, Y.; Zhang, J. Patient-Focused Programable Release Indomethacin Tablets Prepared via Conjugation of Hot Melt Extrusion (HME) and Fused Depositional Modeling (FDM) -3D Printing Technologies. J. Drug Deliv. Sci. Technol. 2024, 97, 105797. [Google Scholar] [CrossRef]
- Goole, J.; Amighi, K. 3D Printing in Pharmaceutics: A New Tool for Designing Customized Drug Delivery Systems. Int. J. Pharm. 2016, 499, 376–394. [Google Scholar] [CrossRef]
- Dumpa, N.; Butreddy, A.; Wang, H.; Komanduri, N.; Bandari, S.; Repka, M.A. 3D Printing in Personalized Drug Delivery: An Overview of Hot-Melt Extrusion-Based Fused Deposition Modeling. Int. J. Pharm. 2021, 600, 120501. [Google Scholar] [CrossRef]
- Martinez, P.R.; Goyanes, A.; Basit, A.W.; Gaisford, S. Influence of Geometry on the Drug Release Profiles of Stereolithographic (SLA) 3D-Printed Tablets. AAPS Pharmscitech 2018, 19, 3355–3361. [Google Scholar] [CrossRef]
- Haring, A.P.; Tong, Y.; Halper, J.; Johnson, B.N. Programming of Multicomponent Temporal Release Profiles in 3D Printed Polypills via Core–Shell, Multilayer, and Gradient Concentration Profiles. Adv. Health Mater. 2018, 7, e1800213. [Google Scholar] [CrossRef]
- Pavan Kalyan, B.; Kumar, L. 3D Printing: Applications in Tissue Engineering, Medical Devices, and Drug Delivery. AAPS PharmSciTech 2022, 23, 92. [Google Scholar] [CrossRef]
- Dey, M.; Ozbolat, I.T. 3D Bioprinting of Cells, Tissues and Organs. Sci. Rep. 2020, 10, 14023. [Google Scholar] [CrossRef]
- Acierno, D.; Patti, A. Fused Deposition Modelling (FDM) of Thermoplastic-Based Filaments: Process and Rheological Properties—An Overview. Materials 2023, 16, 7664. [Google Scholar] [CrossRef]
- Li, X.; Liu, B.; Pei, B.; Chen, J.; Zhou, D.; Peng, J.; Zhang, X.; Jia, W.; Xu, T. Inkjet Bioprinting of Biomaterials. Chem. Rev. 2020, 120, 10793–10833. [Google Scholar] [CrossRef] [PubMed]
- Doddapaneni, V.V.K.; Lee, K.; Aysal, H.E.; Paul, B.K.; Pasebani, S.; Sierros, K.A.; Okwudire, C.E.; Chang, C. A Review on Progress, Challenges, and Prospects of Material Jetting of Copper and Tungsten. Nanomaterials 2023, 13, 2303. [Google Scholar] [CrossRef] [PubMed]
- Lamm, M.E.; Wang, L.; Kishore, V.; Tekinalp, H.; Kunc, V.; Wang, J.; Gardner, D.J.; Ozcan, S. Material Extrusion Additive Manufacturing of Wood and Lignocellulosic Filled Composites. Polymers 2020, 12, 2115. [Google Scholar] [CrossRef] [PubMed]
- Bulina, N.V.; Baev, S.G.; Makarova, S.V.; Vorobyev, A.M.; Titkov, A.I.; Bessmeltsev, V.P.; Lyakhov, N.Z. Selective Laser Melting of Hydroxyapatite: Perspectives for 3D Printing of Bioresorbable Ceramic Implants. Materials 2021, 14, 5425. [Google Scholar] [CrossRef]
- Charoo, N.A.; Barakh Ali, S.F.; Mohamed, E.M.; Kuttolamadom, M.A.; Ozkan, T.; Khan, M.A.; Rahman, Z. Selective Laser Sintering 3D Printing—An Overview of the Technology and Pharmaceutical Applications. Drug Dev. Ind. Pharm. 2020, 46, 869–877. [Google Scholar] [CrossRef]
- Sierra-Vega, N.O.; Rostom, S.; Annaji, M.; Kamal, N.; Ashraf, M.; O’Connor, T.; Zidan, A. Personalization of Intravaginal Rings by Droplet Deposition Modeling Based 3D Printing Technology. Int. J. Pharm. 2024, 665, 124754. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.; Belton, P.; Qi, S. Direct Granule Feeding of Thermal Droplet Deposition 3D Printing of Porous Pharmaceutical Solid Dosage Forms Free of Plasticisers. Pharm. Res. 2022, 39, 599–610. [Google Scholar] [CrossRef]
- Turker, B. Redesigning FDM Platforms for Bio-Printing Applications. Micromachines 2025, 16, 226. [Google Scholar] [CrossRef]
- Horváth, Z.M.; Grundšteins, K.; Radziņš, O.; Kons, A.; Bērziņš, A.; Viter, R.; Lamprou, D.A.; Mohylyuk, V. FDM 3D-Printed Oral Dosage Form of Prednisolone—Improvement of Printability and Influencing Drug Release. Int. J. Pharm. 2025, 673, 125391. [Google Scholar] [CrossRef] [PubMed]
- Kocabas, L.I.; Ayyoubi, S.; Tajqurishi, M.; Quodbach, J.; Vermonden, T.; Kok, R.J. 3D-Printed Prednisolone Phosphate Suppositories with Tunable Dose and Rapid Release for the Treatment of Inflammatory Bowel Disease. Int. J. Pharm. 2024, 649, 123639. [Google Scholar] [CrossRef] [PubMed]
- Fanous, M.; Bitar, M.; Gold, S.; Sobczuk, A.; Hirsch, S.; Ogorka, J.; Imanidis, G. Development of Immediate Release 3D-Printed Dosage Forms for a Poorly Water-Soluble Drug by Fused Deposition Modeling: Study of Morphology, Solid State and Dissolution. Int. J. Pharm. 2021, 599, 120417. [Google Scholar] [CrossRef]
- Chen, Y.; Traore, Y.L.; Walker, L.; Yang, S.; Ho, E.A. Fused Deposition Modeling Three-Dimensional Printing of Flexible Polyurethane Intravaginal Rings with Controlled Tunable Release Profiles for Multiple Active Drugs. Drug Deliv. Transl. Res. 2022, 12, 906–924. [Google Scholar] [CrossRef]
- Obeid, S.; Madžarević, M.; Ibrić, S. Tailoring Amlodipine Release from 3D Printed Tablets: Influence of Infill Patterns and Wall Thickness. Int. J. Pharm. 2021, 610, 121261. [Google Scholar] [CrossRef]
- Alms, J.; Hopmann, C.; Wang, J.; Hohlweck, T. Non-Isothermal Crystallisation Kinetics of Polypropylene at High Cooling Rates and Comparison to the Continuous Two-Domain PvT Model. Polymers 2020, 12, 1515. [Google Scholar] [CrossRef]
- Vanaei, H.R.; Raissi, K.; Deligant, M.; Shirinbayan, M.; Fitoussi, J.; Khelladi, S.; Tcharkhtchi, A. Toward the Understanding of Temperature Effect on Bonding Strength, Dimensions and Geometry of 3D-Printed Parts. J. Mater. Sci. 2020, 55, 14677–14689. [Google Scholar] [CrossRef]
- Miyanaji, H.; Momenzadeh, N.; Yang, L. Effect of Printing Speed on Quality of Printed Parts in Binder Jetting Process. Addit. Manuf. 2018, 20, 1–10. [Google Scholar] [CrossRef]
- Gündoğdu, T.K.; Deniz, I.; Çalışkan, G.; Şahin, E.S.; Azbar, N. Experimental Design Methods for Bioengineering Applications. Crit. Rev. Biotechnol. 2016, 36, 368–388. [Google Scholar] [CrossRef]
- Munnangi, S.R.; Youssef, A.A.A.; Narala, N.; Lakkala, P.; Vemula, S.K.; Alluri, R.; Zhang, F.; Repka, M.A. Continuous Manufacturing of Solvent-Free Cyclodextrin Inclusion Complexes for Enhanced Drug Solubility via Hot-Melt Extrusion: A Quality by Design Approach. Pharmaceutics 2023, 15, 2203. [Google Scholar] [CrossRef]
- Silva, L.A.D.; Almeida, S.L.; Alonso, E.C.P.; Rocha, P.B.R.; Martins, F.T.; Freitas, L.A.P.; Taveira, S.F.; Cunha-Filho, M.S.S.; Marreto, R.N. Preparation of a Solid Self-Microemulsifying Drug Delivery System by Hot-Melt Extrusion. Int. J. Pharm. 2018, 541, 1–10. [Google Scholar] [CrossRef]
- Zhang, J.; Thakkar, R.; Zhang, Y.; Maniruzzaman, M. Structure-Function Correlation and Personalized 3D Printed Tablets Using a Quality by Design (QbD) Approach. Int. J. Pharm. 2020, 590, 119945. [Google Scholar] [CrossRef] [PubMed]
- Sá Filho, A.; Martins, J.L.R.; Costa, R.F.; Pedrino, G.R.; Duarte, V.S.; Silva, O.N.; Napolitano, H.B.; Fajemiroye, J.O. Polymorphism and Pharmacological Assessment of Carbamazepine. Int. J. Mol. Sci. 2024, 25, 9835. [Google Scholar] [CrossRef]
- Czernicki, W.; Baranska, M. Carbamazepine Polymorphs: Theoretical and Experimental Vibrational Spectroscopy Studies. Vib. Spectrosc. 2013, 65, 12–23. [Google Scholar] [CrossRef]
- Huang, L.; Ni, W.; Jia, Y.; Zhu, M.; Yang, T.; Yu, M.; Zhang, J. Process Development for the Continuous Manufacturing of Carbamazepine-Nicotinamide Co-Crystals Utilizing Hot-Melt Extrusion Technology. Pharmaceutics 2025, 17, 568. [Google Scholar] [CrossRef] [PubMed]
- Macedo, J.; Vanhoorne, V.; Vervaet, C.; Pinto, J.F. Influence of Formulation Variables on the Processability and Properties of Tablets Manufactured by Fused Deposition Modelling. Int. J. Pharm. 2023, 637, 122854. [Google Scholar] [CrossRef]
- Yu, D.; Li, J.; Wang, H.; Pan, H.; Li, T.; Bu, T.; Zhou, W.; Zhang, X. Role of Polymers in the Physical and Chemical Stability of Amorphous Solid Dispersion: A Case Study of Carbamazepine. Eur. J. Pharm. Sci. 2022, 169, 106086. [Google Scholar] [CrossRef] [PubMed]
- McNamara, D.P.; Whitney, K.M.; Goss, S.L. Use of a Physiologic Bicarbonate Buffer System for Dissolution Characterization of Ionizable Drugs. Pharm. Res. 2003, 20, 1641–1646. [Google Scholar] [CrossRef]
- Liu, X.; Lu, M.; Guo, Z.; Huang, L.; Feng, X.; Wu, C. Improving the Chemical Stability of Amorphous Solid Dispersion with Cocrystal Technique by Hot Melt Extrusion. Pharm. Res. 2012, 29, 806–817. [Google Scholar] [CrossRef]
- Grzesiak, A.L.; Lang, M.; Kim, K.; Matzger, A.J. Comparison of the Four Anhydrous Polymorphs of Carbamazepine and the Crystal Structure of Form I**Supplementary Material: X-ray Crystallographic Information File (CIF) of Triclinic CBZ (Form I) Is Available. J. Pharm. Sci. 2003, 92, 2260–2271. [Google Scholar] [CrossRef]
- Sahoo, A.; Suryanarayanan, R.; Siegel, R.A. Stabilization of Amorphous Drugs by Polymers: The Role of Overlap Concentration (C*). Mol. Pharm. 2020, 17, 4401–4406. [Google Scholar] [CrossRef]
- Rustichelli, C.; Gamberini, G.; Ferioli, V.; Gamberini, M.C.; Ficarra, R.; Tommasini, S. Solid-State Study of Polymorphic Drugs: Carbamazepine. J. Pharm. Biomed. Anal. 2000, 23, 41–54. [Google Scholar] [CrossRef]
- Mawazi, S.M.; Doolaanea, A.A.; Hadi, H.A.; Chatterjee, B. The Impact of Carbamazepine Crystallinity on Carbamazepine-Loaded Microparticle Formulations. Int. J. Pharm. 2021, 602, 120638. [Google Scholar] [CrossRef] [PubMed]
- Huskić, I.; Christopherson, J.-C.; Užarević, K.; Friščić, T. In Situ Monitoring of Vapour-Induced Assembly of Pharmaceutical Cocrystals Using a Benchtop Powder X-Ray Diffractometer. Chem. Commun. 2016, 52, 5120–5123. [Google Scholar] [CrossRef]
- Hofer, J.D.; Rauk, A.P. Use of Bayesian Methods to Analyze and Visualize Content Uniformity Capability Versus United States Pharmacopeia and ASTM Standards. J. Pharm. Sci. 2017, 106, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Ciavarella, A.B.; Khan, M.A.; Gupta, A.; Faustino, P.J. Dose Uniformity of Scored and Unscored Tablets: Application of the FDA Tablet Scoring Guidance for Industry. PDA J. Pharm. Sci. Technol. 2016, 70, 523–532. [Google Scholar] [CrossRef]
- Srividya, B.; Ghosh, A. Mechanistic Insights into Amorphous Solid Dispersions: Bridging Theory and Practice in Drug Delivery. Pharm. Res. 2025, 42, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Yun, T.; Lee, S.; Yun, S.; Cho, D.; Bang, K.; Kim, K. Investigation of Stabilized Amorphous Solid Dispersions to Improve Oral Olaparib Absorption. Pharmaceutics 2024, 16, 958. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, G.N.; Romanini, M.; Hauptmann, A.; Loerting, T.; Shalaev, E.; Tamarit, J.L.; Pardo, L.C.; Macovez, R. Genuine Antiplasticizing Effect of Water on a Glass-Former Drug. Sci. Rep. 2017, 7, 7470. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, M.; Mukherjee, S.; Kumar, P. Analysis and Optimization of Geometry of 3D Printer Part Cooling Fan Duct. Mater. Today Proc. 2022, 50, 2482–2487. [Google Scholar] [CrossRef]
- Barba, D.; Arias, A.; Garcia-Gonzalez, D. Temperature and Strain Rate Dependences on Hardening and Softening Behaviours in Semi-Crystalline Polymers: Application to PEEK. Int. J. Solids Struct. 2020, 182–183, 205–217. [Google Scholar] [CrossRef]
- Bhardwaj, S.P.; Arora, K.K.; Kwong, E.; Templeton, A.; Clas, S.-D.; Suryanarayanan, R. Mechanism of Amorphous Itraconazole Stabilization in Polymer Solid Dispersions: Role of Molecular Mobility. Mol. Pharm. 2014, 11, 4228–4237. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | F | T | S | Diameter | RSD 1 | RSD 2 | Height | RSD 1 | RSD 2 | Mass | RSD 1 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| (%) | (°C) | (mm/s) | (mm) | % | (mm) | % | (mg) | % | |||
| R1 | 20 | 40 | 60 | 12.15 ± 0.07 | 0.58 | 1.25 | 4.97 ± 0.12 | 2.41 | −0.60 | 374.3 ± 8.6 | 2.30 |
| R2 | 60 | 60 | 60 | 12.03 ± 0.10 | 0.83 | 0.25 | 4.95 ± 0.09 | 1.82 | −1.00 | 402.3 ± 15.5 | 3.85 |
| R3 | 60 | 60 | 60 | 12.08 ± 0.13 | 1.08 | 0.67 | 5.05 ± 0.02 | 0.40 | 1.00 | 403.7 ± 15.0 | 3.72 |
| R4 | 100 | 80 | 60 | 12.07 ± 0.09 | 0.75 | 0.58 | 5.07 ± 0.04 | 0.79 | 1.40 | 386.3 ± 17.6 | 4.56 |
| R5 | 60 | 60 | 60 | 12.12 ± 0.12 | 0.99 | 1.00 | 4.95 ± 0.02 | 0.40 | −1.00 | 402.0 ± 15.1 | 3.76 |
| R6 | 60 | 80 | 100 | 12.04 ± 0.05 | 0.42 | 0.33 | 5.02 ± 0.04 | 0.80 | 0.40 | 394.0 ± 17.3 | 4.39 |
| R7 | 100 | 60 | 20 | 12.20 ± 0.08 | 0.66 | 1.67 | 5.08 ± 0.02 | 0.39 | 1.60 | 425.7 ± 21.0 | 4.93 |
| R8 | 60 | 80 | 20 | 12.24 ± 0.14 | 1.14 | 2.00 | 5.08 ± 0.02 | 0.39 | 1.60 | 412.0 ± 11.0 | 2.67 |
| R9 | 20 | 60 | 20 | 12.07 ± 0.07 | 0.58 | 0.58 | 5.07 ± 0.03 | 0.59 | 1.40 | 416.3 ± 17.0 | 4.08 |
| R10 | 20 | 80 | 60 | 12.17 ± 0.10 | 0.82 | 1.42 | 5.06 ± 0.10 | 1.98 | 1.20 | 420.3 ± 9.5 | 2.26 |
| R11 | 100 | 40 | 60 | 12.07 ± 0.05 | 0.41 | 0.58 | 4.96 ± 0.17 | 3.43 | −0.80 | 420.0 ± 3.0 | 0.71 |
| R12 | 60 | 40 | 100 | 12.00 ± 0.12 | 1.00 | 0.00 | 4.92 ± 0.03 | 0.61 | −1.60 | 419.7 ± 16.8 | 4.00 |
| R13 | 60 | 40 | 20 | 12.25 ± 0.02 | 0.16 | 2.08 | 4.90 ± 0.04 | 0.82 | −2.00 | 418.7 ± 11.1 | 2.65 |
| R14 | 20 | 60 | 100 | 11.96 ± 0.11 | 0.92 | −0.33 | 4.88 ± 0.02 | 0.41 | −2.40 | 408.0 ± 21.1 | 5.17 |
| R15 | 100 | 60 | 100 | 11.89 ± 0.16 | 1.35 | −0.92 | 4.94 ± 0.01 | 0.20 | −1.20 | 391.7 ± 9.1 | 2.32 |
| Average | 12.09 | 4.99 | 406.33 | ||||||||
| LCL | 11.79 | 4.78 | 361.99 | ||||||||
| UCL | 12.39 | 5.21 | 450.67 | ||||||||
| Group | F (%) | T (°C) | S (mm/s) | Q4h (%) | t50% (min) |
|---|---|---|---|---|---|
| R1 | 20 | 40 | 60 | 45.49 | 245 |
| R2 | 60 | 60 | 60 | 49.63 | 240 |
| R3 | 60 | 60 | 60 | 47.77 | 255 |
| R4 | 100 | 80 | 60 | 46.59 | 260 |
| R5 | 60 | 60 | 60 | 48.36 | 250 |
| R6 | 60 | 80 | 100 | 49.17 | 245 |
| R7 | 100 | 60 | 20 | 28.72 | 510 |
| R8 | 60 | 80 | 20 | 32.18 | 430 |
| R9 | 20 | 60 | 20 | 31.35 | 435 |
| R10 | 20 | 80 | 60 | 48.36 | 250 |
| R11 | 100 | 40 | 60 | 26.96 | 245 |
| R12 | 60 | 40 | 100 | 37.18 | 335 |
| R13 | 60 | 40 | 20 | 28.62 | 530 |
| R14 | 20 | 60 | 100 | 40.86 | 290 |
| R15 | 100 | 60 | 100 | 49.82 | 240 |
| F | T | S | F × T | F × S | T × S | F2 | T2 | S2 | |
|---|---|---|---|---|---|---|---|---|---|
| p-value | 0.8234 | 0.0608 | 0.0002 | 0.5857 | 0.1749 | 0.8744 | 1.0000 | 0.6001 | 0.0005 |
| Significance | NS | NS | *** | NS | NS | NS | NS | NS | *** |
| Code Equation | t50% = 0.21761 − 0.011099 × S + 0.00041 × S2 | ||||||||
| F | T | S | F × T | F × S | T × S | F2 | T2 | S2 | |
|---|---|---|---|---|---|---|---|---|---|
| p-value | 0.5282 | 0.0935 | 0.0005 | 0.4435 | 0.0911 | 0.3680 | 0.9990 | 0.5901 | 0.0005 |
| Significance | NS | NS | *** | NS | NS | NS | NS | NS | *** |
| Code Equation | 1/Sqrt(Q4h) = 692.34375 − 64.86486 × S + 2.29410 × S2 | ||||||||
| # | Zero-Order | First-Order | Higuchi | Korsmeyer–Peppas | Peppas–Sahlin | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| K0 | R2 | k | R2 | k | R2 | kkp | n | R2 | k1 | k2 | m | R2 | |
| CBZ | 0.187 | 0.8436 | 0.010 | 0.9982 | 4.470 | 0.9423 | 9.396 | 0.377 | 0.9588 | 3.153 | −0.024 | 0.675 | 0.9911 |
| PM | 0.192 | 0.8036 | 0.014 | 0.9973 | 4.666 | 0.9182 | 14.387 | 0.313 | 0.9516 | 5.524 | −0.073 | 0.595 | 0.9884 |
| EXT | 0.154 | 0.9555 | 0.004 | 0.9989 | 3.514 | 0.9896 | 2.303 | 0.570 | 0.9891 | 0.532 | −0.001 | 0.895 | 0.9994 |
| R1 | 0.122 | 0.9579 | 0.002 | 0.9938 | 2.797 | 0.9908 | 1.860 | 0.567 | 0.9905 | 0.458 | −0.001 | 0.880 | 0.9995 |
| R2 | 0.123 | 0.9341 | 0.002 | 0.9840 | 2.841 | 0.9842 | 2.713 | 0.508 | 0.9839 | 0.683 | −0.002 | 0.829 | 0.9983 |
| R3 | 0.122 | 0.9420 | 0.002 | 0.9874 | 2.817 | 0.9868 | 2.456 | 0.523 | 0.9863 | 0.616 | −0.001 | 0.841 | 0.9988 |
| R4 | 0.127 | 0.9493 | 0.002 | 0.9915 | 2.916 | 0.9850 | 1.933 | 0.568 | 0.9844 | 0.321 | 0.000 | 0.956 | 0.9993 |
| R5 | 0.120 | 0.9483 | 0.002 | 0.9880 | 2.753 | 0.9869 | 2.082 | 0.546 | 0.9863 | 0.549 | −0.001 | 0.850 | 0.9966 |
| R6 | 0.130 | 0.9493 | 0.003 | 0.9928 | 2.984 | 0.9866 | 2.073 | 0.560 | 0.9859 | 0.419 | −0.001 | 0.913 | 0.9990 |
| R7 | 0.092 | 0.9889 | 0.001 | 0.9978 | 2.073 | 0.9908 | 0.707 | 0.676 | 0.9976 | 0.499 | 0.475 | 0.362 | 0.9977 |
| R8 | 0.103 | 0.9874 | 0.002 | 0.9988 | 2.320 | 0.9918 | 0.775 | 0.680 | 0.9980 | −0.699 | 1.100 | 0.319 | 0.9980 |
| R9 | 0.102 | 0.9844 | 0002 | 0.9972 | 2.295 | 0.9908 | 0.868 | 0.660 | 0.9961 | −0.406 | 1.062 | 0.318 | 0.9961 |
| R10 | 0.132 | 0.9592 | 0.003 | 0.9953 | 3.018 | 0.9895 | 2.125 | 0.558 | 0.9896 | 0.515 | −0.001 | 0.872 | 0.9980 |
| R11 | 0.115 | 0.9217 | 0.002 | 0.9739 | 2.675 | 0.9787 | 3.073 | 0.477 | 0.9796 | 0.695 | −0.002 | 0.824 | 0.9982 |
| R12 | 0.126 | 0.9834 | 0.002 | 0.9966 | 2.811 | 0.9857 | 0.746 | 0.717 | 0.9933 | −2.620 | 1.893 | 0.303 | 0.9938 |
| R13 | 0.093 | 0.9909 | 0.001 | 0.9986 | 2.093 | 0.9933 | 0.772 | 0.664 | 0.9996 | 1.004 | 0.282 | 0.391 | 0.9997 |
| R14 | 0.136 | 0.9757 | 0.003 | 0.9954 | 3.043 | 0.9847 | 0.979 | 0.686 | 0.9897 | −5.017 | 3.366 | 0.270 | 0.9907 |
| R15 | 0.143 | 0.9605 | 0.003 | 0.9965 | 3.241 | 0.9871 | 1.764 | 0.600 | 0.9875 | 0.296 | 0.000 | 0.980 | 0.9991 |
| # | Zero-Order | First-Order | Higuchi | Korsmeyer–Peppas | Peppas–Sahlin | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| K0 | R2 | k | R2 | k | R2 | kkp | n | R2 | k1 | k2 | m | R2 | |
| R1 | 0.201 | 0.9944 | 0.003 | 0.9982 | 2.552 | 0.9790 | 0.704 | 0.760 | 0.9984 | 1.039 | 0.151 | 0.485 | 0.9988 |
| R2 | 0.224 | 0.9893 | 0.003 | 0.9970 | 2.856 | 0.9812 | 0.900 | 0.734 | 0.9968 | 0.452 | 0.662 | 0.388 | 0.9968 |
| R3 | 0.215 | 0.9909 | 0.003 | 0.9976 | 2.744 | 0.9821 | 0.879 | 0.731 | 0.9977 | 0.886 | 0.406 | 0.418 | 0.9979 |
| R4 | 0.205 | 0.9957 | 0.003 | 0.9982 | 2.589 | 0.9764 | 0.635 | 0.784 | 0.9985 | 0.999 | 0.116 | 0.509 | 0.9990 |
| R5 | 0.214 | 0.9972 | 0.003 | 0.9949 | 2.674 | 0.9666 | 0.501 | 0.837 | 0.9967 | 0.894 | 0.048 | 0.580 | 0.9980 |
| R6 | 0.217 | 0.9952 | 0.003 | 0.9978 | 2.734 | 0.9748 | 0.634 | 0.795 | 0.9979 | 0.925 | 0.177 | 0.486 | 0.9982 |
| R7 | 0.218 | 0.9896 | 0.002 | 0.9924 | 1.653 | 0.9843 | 0.745 | 0.662 | 0.9954 | 1.013 | 0.075 | 0.479 | 0.9974 |
| R8 | 0.143 | 0.9921 | 0.002 | 0.9957 | 1.831 | 0.9814 | 0.637 | 0.714 | 0.9970 | 0.931 | 0.084 | 0.492 | 0.9981 |
| R9 | 0.142 | 0.9884 | 0.002 | 0.9926 | 1.831 | 0.9824 | 0.790 | 0.671 | 0.9946 | 1.084 | 0.105 | 0.466 | 0.9961 |
| R10 | 0.214 | 0.9918 | 0.003 | 0.9923 | 2.735 | 0.9769 | 1.010 | 0.702 | 0.9932 | 1.452 | 0.098 | 0.504 | 0.9957 |
| R11 | 0.220 | 0.9931 | 0.003 | 0.9963 | 2.810 | 0.9818 | 0.968 | 0.716 | 0.9975 | 1.426 | 0.099 | 0.509 | 0.9988 |
| R12 | 0.161 | 0.9965 | 0.002 | 0.9944 | 2.018 | 0.9652 | 0.396 | 0.828 | 0.9952 | 0.686 | 0.030 | 0.592 | 0.9974 |
| R13 | 0.130 | 0.9847 | 0.002 | 0.9899 | 1.697 | 0.9915 | 0.951 | 0.618 | 0.9978 | 1.220 | 0.105 | 0.447 | 0.9991 |
| R14 | 0.177 | 0.9970 | 0.002 | 0.9953 | 2.222 | 0.9666 | 0.439 | 0.826 | 0.9961 | 0.766 | 0.037 | 0.583 | 0.9979 |
| R15 | 0.217 | 0.9964 | 0.003 | 0.9970 | 2.739 | 0.9738 | 0.649 | 0.790 | 0.9977 | 1.072 | 0.076 | 0.542 | 0.9987 |
| # | F | T | S | Diameter | Height | Mass | Q4h | t50% |
|---|---|---|---|---|---|---|---|---|
| (%) | (°C) | (mm/s) | (mm) | (mm) | (mg) | (%) | (min) | |
| R2 | 60 | 12.03 ± 0.10 | 4.95 ± 0.09 | 402.3 ± 15.5 | 49.63 | 240 | ||
| R3 | 60 | 60 | 12.08 ± 0.13 | 5.05 ± 0.02 | 403.7 ± 15.0 | 47.77 | 255 | |
| R5 | 12.12 ± 0.12 | 4.95 ± 0.02 | 402.0 ± 15.1 | 48.36 | 250 | |||
| Average | 12.08 | 4.98 | 402.67 | 48.59 | 248.3 | |||
| S.D. | 0.05 | 0.06 | 0.91 | 0.95 | 7.6 | |||
| RSD 1 | 0.37 | 1.16 | 0.23 | 1.96 | 3.06 | |||
| RSD 2 | 0.38 | 1.15 | NA | NA | NA | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, L.; Zhang, X.; Huang, Q.; Zhu, M.; Yang, T.; Zhang, J. Three-Dimensional Printing of Personalized Carbamazepine Tablets Using Hydrophilic Polymers: An Investigation of Correlation Between Dissolution Kinetics and Printing Parameters. Polymers 2025, 17, 2126. https://doi.org/10.3390/polym17152126
Huang L, Zhang X, Huang Q, Zhu M, Yang T, Zhang J. Three-Dimensional Printing of Personalized Carbamazepine Tablets Using Hydrophilic Polymers: An Investigation of Correlation Between Dissolution Kinetics and Printing Parameters. Polymers. 2025; 17(15):2126. https://doi.org/10.3390/polym17152126
Chicago/Turabian StyleHuang, Lianghao, Xingyue Zhang, Qichen Huang, Minqing Zhu, Tiantian Yang, and Jiaxiang Zhang. 2025. "Three-Dimensional Printing of Personalized Carbamazepine Tablets Using Hydrophilic Polymers: An Investigation of Correlation Between Dissolution Kinetics and Printing Parameters" Polymers 17, no. 15: 2126. https://doi.org/10.3390/polym17152126
APA StyleHuang, L., Zhang, X., Huang, Q., Zhu, M., Yang, T., & Zhang, J. (2025). Three-Dimensional Printing of Personalized Carbamazepine Tablets Using Hydrophilic Polymers: An Investigation of Correlation Between Dissolution Kinetics and Printing Parameters. Polymers, 17(15), 2126. https://doi.org/10.3390/polym17152126