
Molecular-Level Insights into Meta-Phenylenediamine and Sulfonated Zinc Phthalocyanine Interactions for Enhanced Polyamide Membranes: A DFT and TD-DFT Study



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Computational Environment
2.2. Geometry Optimization
2.3. TD-DFT and UV–Vis Spectral Simulation
2.4. FT-IR Vibrational Analysis
2.5. Solvent Effects Using PCM
2.6. Frontier Molecular Orbitals and Global Reactivity
2.7. Conformational Analysis via Dihedral Scan
3. Results
3.1. Geometry of Molecular Optimization of the MPD, Zn(SO2−)4Pc and MPD/Zn(SO2−)4Pc
3.2. Conformational Energy and Electronic Structure of MPD/Zn(SO2−)4Pc
3.2.1. Conformational Energy and Stability
3.2.2. Effect on Electronic Structure: HOMO–LUMO Gap
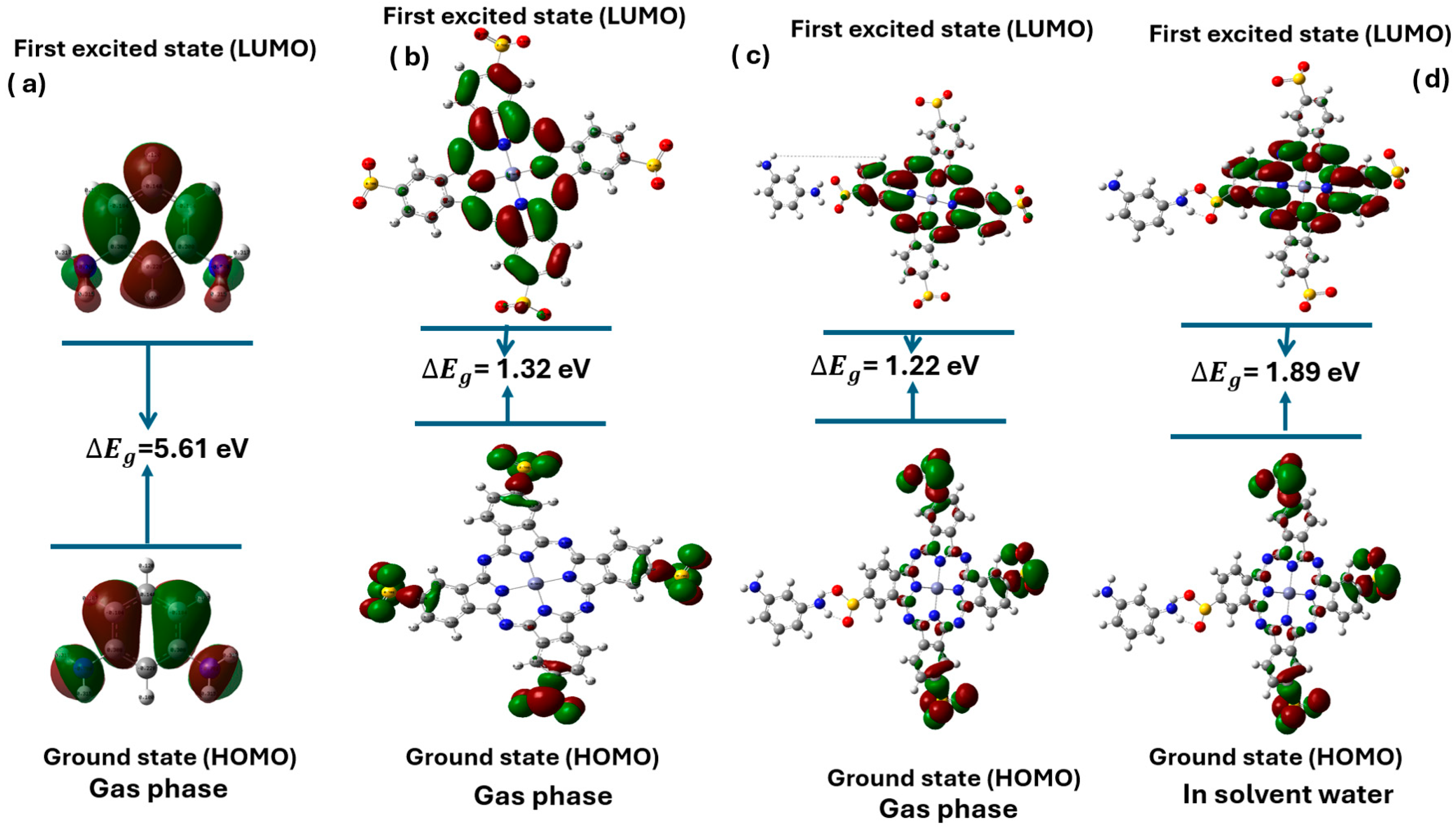
3.3. Analysis and Comparison of HOMO-LUMO Gap Energies
3.4. Global Reactivity Descriptors
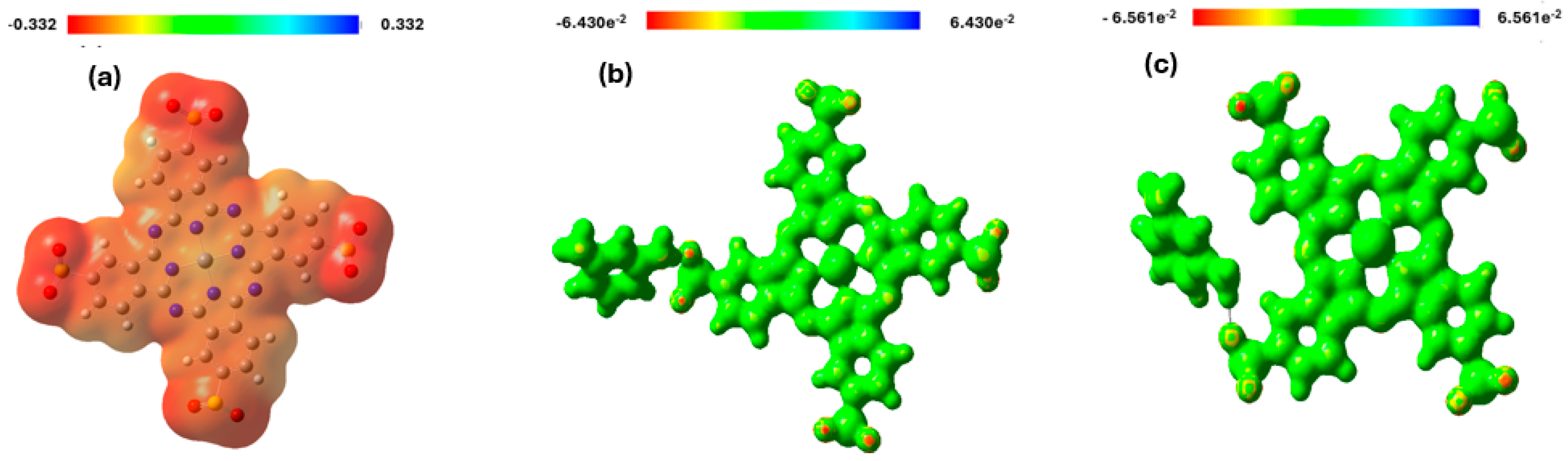
3.5. Molecular Electrostatic Potential (MEP) Analysis
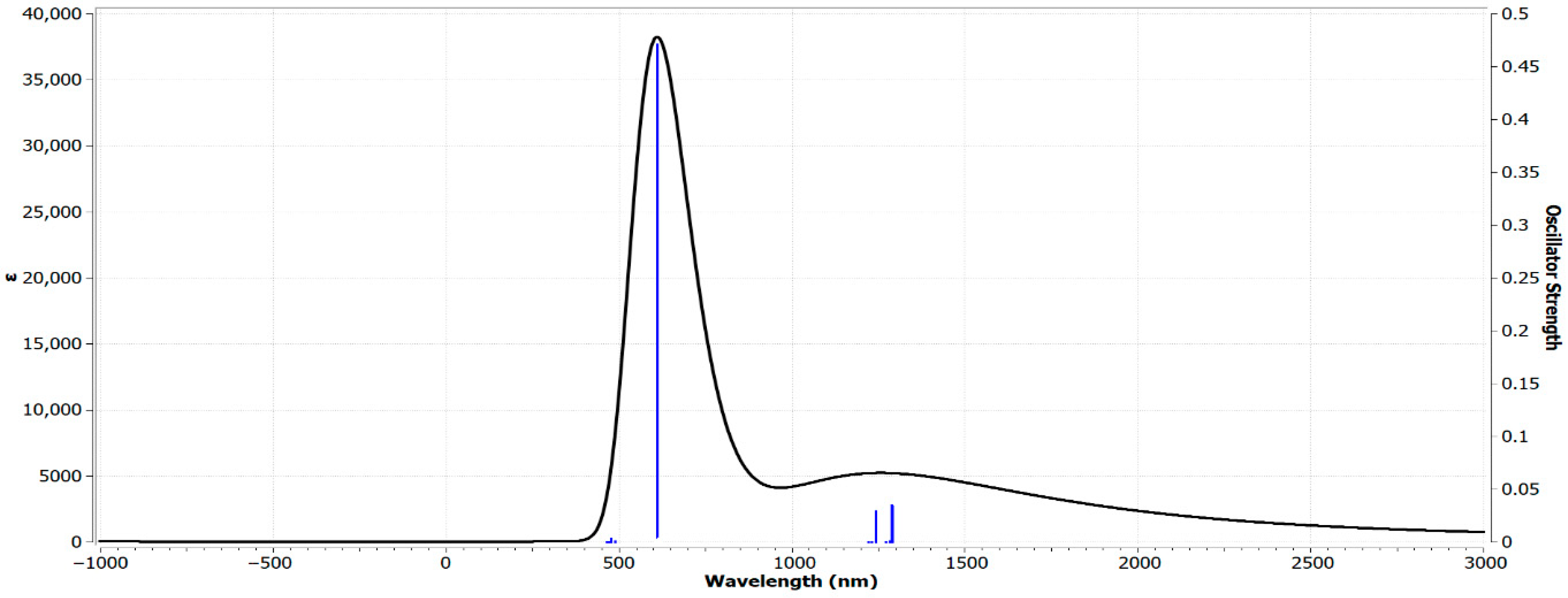
3.6. Analysis of the UV–Vis Spectrum of Zn(SO2−)4Pc Using TD-DFT at the 6-31G(d) Basis Set
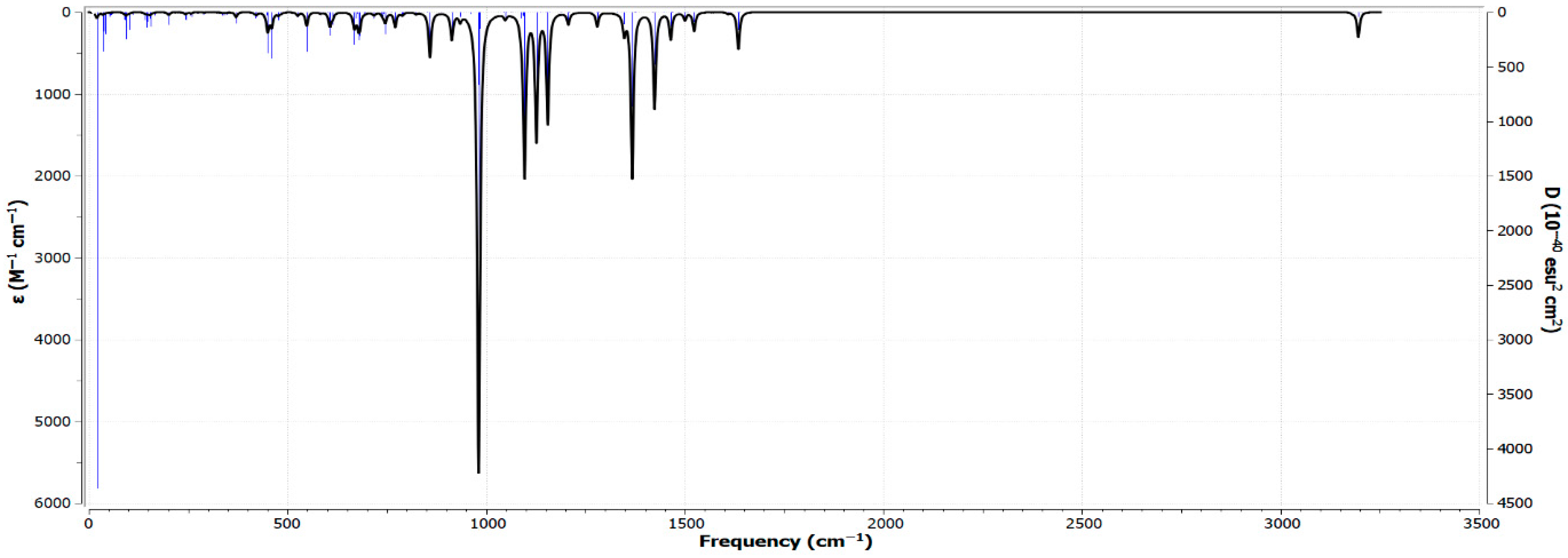
3.7. Analysis of the FT-IR Spectrum and Vibrational Modes of Zn(SO2−)4Pc in the Gas Phase Using RB3LYP/6-31G(d)
3.8. Cross-Correlation Between DFT Predictions and Membrane Properties
3.8.1. Porosity (ε)
- is the bulk density of the membrane.
- is the polymer density.
3.8.2. Hydrophilicity Mechanism
- γSV is the solid–vapor interfacial tension.
- γSL is the solid–liquid interfacial tension.
- γLV is the liquid–vapor interfacial tension.
3.8.3. Surface Roughness and π–π Interactions
3.8.4. Electronic Properties and Reactivity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MPD | Meta-phenylenediamine |
| Zn(SO2−)4Pc | Sulfonated zinc phthalocyanine |
| IPol | Interfacial polymerization |
| MEP | Molecular electrostatic potential |
| DFT | Density Functional Theory |
| TD-DFT | Time-Dependent DFT |
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
References
- Shen, S.L.; Wang, Y.; Wang, J.X.; Li, Y.H.; Zhang, J.F.; Chen, L. Fabrication of Thin-Film Composite Membranes with High Perm selectivity via Interfacial Polymerization. J. Membr. Sci. 2022, 647, 120301. [Google Scholar]
- Tang, L.; Guo, F.; He, L.; Zhao, H.; Yang, W.; Wang, X. Advances in Thin-Film Composite Membrane Design for Water Treatment. Desalination 2021, 520, 115362. [Google Scholar]
- Li, Z.; Ding, H.J.; Yang, Y.; Sun, L.; Xu, R.; Chen, Y. Mitigating Chlorine Degradation in RO Membranes: A Comprehensive Review. J. Water Process Eng. 2023, 52, 103354. [Google Scholar]
- Rahimpour, A.; Jahanshahi, M.; Rajaeian, S.M.; Tien, H.N.; Matsuura, T.; Ismail, A.F. Nanomaterial-Based Modifications of Polyamide Membranes for Enhanced Performance. J. Membr. Sci. 2020, 602, 117962. [Google Scholar]
- Szekeres, M.; Arthanareeswaran, K.; Asatekin, J.; Chen, C.; Gupta, S. Phthalocyanine-Based Membranes for Water Purification and Selective Separation. J. Appl. Polym. Sci. 2023, 140, 5312. [Google Scholar]
- Bottari, G.; Herranz, M.Á.; Wibmer, L.; Volland, L.; Laura Rodríguez-Pérez, L.; Guldi, D.M.; Hirsch, A.; Torres, T. Chemical functionalization and characterization of graphene-based materials. Chem. Soc. Rev. 2017, 46, 4464–4500. [Google Scholar] [CrossRef]
- Şahin, G.; Harmandar, K.; Jamoussi, B.; Durmuş, M. The new water-soluble zinc (II) phthalocyanines substituted with morpholine groups: Synthesis and optical properties. J. Photochem. Photobiol. A Chem. 2020, 401, 112736. [Google Scholar]
- Urbani, M.; Grätzel, M.; Nazeeruddin, M.K.; Martín, N.; D’Souza, F.; Torres, T. Molecular Engineering of Phthalocyanines for Dye-Sensitized Solar Cells. Chem. Rev. 2014, 114, 12330–12396. [Google Scholar] [CrossRef]
- Shabatina, T.A.; Vernigor, A.A.; Borisova, N.A.; Konovalov, A.I. Coordination Chemistry of Phthalocyanines: Functionalization Strategies and Applications. Coord. Chem. Rev. 2019, 398, 213016. [Google Scholar]
- Jiang, J.; Wang, S.; Lin, J.; Zhang, W.; Chen, H.; Liu, Z. Tuning the Optoelectronic Properties of Phthalocyanines for Organic Electronic Devices. Adv. Funct. Mater. 2021, 31, 2105894. [Google Scholar]
- Fu, X.; Lin, J.; Liang, Z.; Yao, R.; Wu, W.; Fang, Z.; Zou, W.; Wu, Z.; Ning, H.; Peng, J. Graphene oxide as a promising nanofiller for polymer composite. Surf. Interfaces 2023, 37, 102747. [Google Scholar] [CrossRef]
- Tawfik, A.; Eraky, M.; Khalil, M.N.; Osman, A.I.; Rooney, D.W. Sulfonated graphene nanomaterials for membrane antifouling, pollutant removal, and production of chemicals from biomass: A review. Environ. Chem. Lett. 2022, 21, 1093–1116. [Google Scholar] [CrossRef]
- Rath, R.; Mohanty, S.; Kumar, P.; Nayak, S.K.; Unnikrishnan, L. Synergistic effect of silica-covered graphene oxide (In-Situ) hybrid nanocomposites for use as a polymer electrolyte membrane for fuel cell applications. Surf. Interfaces 2023, 38, 102761. [Google Scholar] [CrossRef]
- Ma, L.; Zhao, D.; Zheng, J. Construction of electrostatic and π–π interaction to enhance interfacial adhesion between carbon nanoparticles and polymer matrix. J. Appl. Polym. Sci. 2019, 137, 48633. [Google Scholar] [CrossRef]
- Liu, Y.; Xiong, H.; Yu, X.; Huang, H.; Li, L.; Ji, J.; Huang, Y.; Xu, M. Interfacial fabrication of polypyrrole/sulfonated reduced graphene oxide nanocomposites for electrochemical capacitors. Polym. Compos. 2017, 39, E378–E385. [Google Scholar] [CrossRef]
- Sun, Y.; Tian, L.; Qiao, Z.; Geng, C.; Guo, X.; Zhong, C. Surface modification of bilayer structure on metal-organic frameworks towards mixed matrix membranes for efficient propylene/propane separation. J. Membr. Sci. 2022, 648, 120350. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, Z.; Li, W.; Huang, B.; Wang, J.; Sun, P. DFT Study on Polyamide Membrane Functionalization for Improved Water Filtration. Theor. Chem. Acc. 2022, 141, 1085. [Google Scholar]
- Hanack, M.; Schneider, T.; Eastwood, D.E. Electronic Properties of Metal Phthalocyanines: A Computational Perspective. J. Porphyr. Phthalocyanines 1998, 2, 293–305. [Google Scholar]
- Thomas, K.R.; Lin, J.T.; Hsu, Y.C.; Ho, K.C. Organic dyes containing thienylfluorene conjugation for solar cells. Chem. Commun. 2005, 32, 4098–4100. [Google Scholar] [CrossRef]
- Odobel, F.; Blart, E.; Lagree, M.; Pleux, C. Based Photosensitizers: Applications in Light-Harvesting and Solar Energy Conversion. Coord. Chem. Rev. 2010, 254, 2585–2593. [Google Scholar]
- Jamoussi, B.; Al-Sharif, M.N.M.; Gzara, L.; Organji, H.; Almeelbi, T.B.; Chakroun, R.; Al-Mur, B.A.; Al Makishah, N.H.M.; Madkour, M.H.F.; Aloufi, F.A. Hybrid Zinc Phthalocyanine/PVDF-HFP System for Reducing Biofouling in Water Desalination: DFT Theoretical and MolDock Investigations. Polymers 2024, 16, 1738. [Google Scholar] [CrossRef]
- Sasa, N.; Okada, K.; Nakamura, K.; Okada, S. Synthesis, structural and conformational analysis and chemical properties of phthalocyaninatometal complexes. J. Mol. Struct. 1998, 446, 163–178. [Google Scholar] [CrossRef]
- Gajda, Ł.; Kupka, T.; Broda, M.A. Solvent impact on the planarity and aromaticity of free and monohydrated zinc phthalocyanine: A theoretical study. Struct. Chem. 2017, 29, 667–679. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J. GaussView, version 6.0; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Solğun, D.G.; Yıldıko, Ü.; Ağırtaş, M.S. Synthesis, DFT calculations, photophysical, photochemical properties of peripherally metallophthalocyanines bearing (2-(benzo[d] [1,3] dioxol-5-ylmethoxy) phenoxy) substituents. In Research Square; Springer: Berlin/Heidelberg, Germany, 2021. [Google Scholar] [CrossRef]
- Csonka, G.I.; French, A.D.; Johnson, G.P. Density Functional Theory: A Powerful Tool for Predicting and Interpreting Vibrational Spectra. Molecules 2021, 26, 3012. [Google Scholar]
- Laurent, A.D.; Jacquemin, D. TD-DFT benchmarks: A review. Int. J. Quantum Chem. 2013, 113, 2019–2039. [Google Scholar] [CrossRef]
- Scheiner, S. Molecular Electrostatic Potential Topology Analysis of Noncovalent Interactions. Acc. Chem. Res. 2023, 56, 1532–1541. [Google Scholar]
- Pechlaner, M.; van Gunsteren, W.F. Algorithms to apply dihedral-angle constraints in molecular or stochastic dynamics simulations. J. Chem. Phys. 2020, 152, 024109. [Google Scholar] [CrossRef]
- Martins, J.B.L.; Cabral, B.J.C. Electron binding energies of SO2 at the surface of a water cluster. J. Chem. Phys. 2023, 159, 234301. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Pearson, R.G. Absolute electronegativity and hardness: Applications to organic chemistry. J. Org. Chem. 1989, 54, 1423–1430. [Google Scholar] [CrossRef]
- Geerlings, P.; Proft, F.D.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Padmanabhan, J.; Parthasarathi, R.; Subramanian, V.; Chattaraj, P.K. Electrophilicity-based charge transfer descriptor. J. Phys. Chem. 2007, 111, 1358–1361. [Google Scholar] [CrossRef] [PubMed]
- Villemin, D.; Abbaz, T.; Bendjeddou, A. Molecular structure, HOMO, LUMO, MEP, natural bond orbital analysis of benzo and anthraquinodimethane derivatives. Pharm. Biol. Eval. 2018, 5, 27. [Google Scholar]
- Basha, F.; Khan, F.L.; Muthu, S.; Raja, M. Computational evaluation on molecular structure (Monomer, Dimer), RDG, ELF, electronic (HOMO-LUMO, MEP) properties, and spectroscopic profiling of 8-Quinolinesulfonamide with molecular docking studies. Comput. Theor. Chem. 2021, 1198, 113169. [Google Scholar] [CrossRef]
- Palewska, K.; Sworakowski, J.; Lipiński, J.; Nešpůrek, S. Effect of electric permittivity of the solvent on aggregation process of the water-soluble sulfonated metal phthalocyanines. J. Photochem. Photobiol. A Chem. 2011, 223, 149–156. [Google Scholar] [CrossRef]
- Xia, D.; Li, W.; Wang, H.; Zheng, X.; Guo, Y.; Yang, X. Study on Spectra Properties of Novel Octa-Substituted Phthalocyanines. Spectrosc. Spectr. Anal. 2011, 31, 2171–2175. [Google Scholar]
- Tackley, D.R.; Dent, G.; Ewen Smith, W. Phthalocyanines: Structure and vibrations. Phys. Chem. Chem. Phys. 2001, 3, 1419–1426. [Google Scholar] [CrossRef]
- Napier, A.; Collins, R.A. FTIR characteristics of halogenated phthalocyanines exhibiting polymorphism. Thin Solid Film 1994, 248, 166–177. [Google Scholar] [CrossRef]
- Han, L.; Fan, C.; Liu, Y.; Yang, Y.; Li, H.; Tang, L.; Li, H.; Liu, Y.; Wu, H.; Jiang, Z. Engineering Solute–Solvent Interactions for the Synthesis of Covalent Organic Polymer Nanosheets. Small 2025, 21, e202506441. [Google Scholar] [CrossRef]
- Politzer, P.; Lane, P.; Murray, J.S. Electrostatic Potentials, Intralattice Attractive Forces and Crystal Densities of Nitrogen-Rich C,H,N,O Salts. Crystals 2016, 6, 7. [Google Scholar] [CrossRef]
- Zhao, D.; Chen, L.; Peng, M.; Xue, B.; Yao, Z.; Huang, W.; Wang, Z.; Liu, J. The complex influence of membrane roughness on colloidal fouling: A dialectical perspective. J. Membr. Sci. 2025, 725, 124014. [Google Scholar] [CrossRef]
- Ma, X.; Yang, Z.; Yao, Z.; Guo, H.; Xu, Z.; Tang, C.Y. Tuning roughness features of thin film composite polyamide membranes for simultaneously enhanced permeability, selectivity and anti-fouling performance. J. Colloid. Interface Sci. 2019, 540, 382–388. [Google Scholar] [CrossRef] [PubMed]
- Bhalani, D.V.; Trivedi, J.S.; Jewrajka, S.K. Selective grafting of morphologically modified poly(vinylidene fluoride) ultrafiltration membrane by poly(acrylic acid) for inducing antifouling property. Appl. Surf. Sci. 2021, 544, 148905. [Google Scholar] [CrossRef]
- Yankova, R.; Yotova, T.S.; Avramov, M. DFT Investigation of Electronic Structure, Reactivity, and Molecular Interactions in a Selenate–Selenite System. Russ. J. Gen. Chem. 2025, 95, 1543–1553. [Google Scholar] [CrossRef]
- Qi, Y.; Gong, W.; Yan, Q. Bridging deep learning force fields and electronic structures with a physics-informed approach. Npj Comput. Mater. 2025, 11, 177. [Google Scholar] [CrossRef]
- Chahmana, S.; Benghanem, F.; Fellah, M.; Aityoucef, H.; Bennaadja, S.; Foudia, M.; Djili, A.; Ghedjati, S.; El-Hiti, G.A. Integrated exploration of molecular structure, quantum chemical properties, molecular docking, and antioxidant activity of 4-(2-hydroxyanilino)pent-3-en-2-one. Results Chem. 2024, 8, 101622. [Google Scholar] [CrossRef]
- Owen, A.E.; Anyambula, I.A.; Benson, C.U.; Ojumola, F.O.; Alawa, J.A.; Benjamin, I.; Iyam, S.O.; Ogar, C.U.; Ojong, M.A.; Ojong, R.; et al. Exploration of semi-carbazone derivatives as promising agents against cholera: Insights from spectroscopic analysis, reactivity studies (ELF, HOMO-LUMO, NBO), solvation effects, and molecular docking investigations. Chem. Phys. Impact 2024, 8, 100438. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | Phase | Minimum Total Energy (Hartree) | Dipole (Debye) |
|---|---|---|---|
| MPD | Gas phase | −342.954877 | 2.603122 |
| Zn(SO2−)4Pc | Gas phase | −5638.225264 | 2.554394 |
| MPD/Zn(SO2−)4Pc | Gas phase | −5981.218660 | 19.817231 |
| MPD/Zn (SO2−)4Pc | In water | −5981.823923 | 23.18453 |
| Dihedral Angle | Energy (Hartree) | Dipole Moment (Debye) |
|---|---|---|
| 60° | −5981.218297 | 18.23 |
| 90° | −5981.214242 | 14.79 |
| 120° | −5981.218660 | 19.82 |
| 180° | −5981.218660 | 19.26 |
| Dihedral Angle | HOMO (eV) | LUMO (eV) | ΔE (eV) |
|---|---|---|---|
| 60° | 3.19625 | −4.421036158 | 1.224240886 |
| 90° | 3.21856 | −4.427022666 | 1.208458274 |
| 120° | 3.19326 | −4.416138106 | 1.222880316 |
| 180° | 3.20605 | −4.440628366 | 1.234581218 |
| Molecule/System | Phase | ΔEg (eV) |
|---|---|---|
| (a) MPD | Gas phase | 5.61 eV |
| (b) Zn(SO2−)4Pc | Gas phase | 1.32 eV |
| (c) MPD/Zn(SO2−)4Pc | Gas phase | 1.22eV |
| (d) MPD/Zn(SO2−)4Pc | In solvent water | 1.89 eV |
| Molecules | MPD | Zn(SO2−)4Pc | MPD/Zn(SO2−)4Pc (Gas Phase) | MPD/Zn(SO2−)4Pc (Water) |
|---|---|---|---|---|
| Ionization potential (IP = −EHOMO) | 0.18457 | −0.12296 | −0.11735 | 0.16822 |
| Electron affinity (EA = −ELUMO) | −0.02158 | −0.17139 | −0.16229 | 0.09879 |
| Chemical hardness (η = (IP − EA)/2) | 0.103075 | 0.02421 | 0.02247 | 0.03471 |
| Chemical softness (s = 1/2η) | 4.850836 | 20.64835 | 22.25189 | 14.40299 |
| Chemical potential (μ = (IP + EA)/2) | −0.081495 | −0.147175 | −0.13982 | −0.13350 |
| Electronegativity (χ = (1 + EA)/2) | 0.48921 | 0.147175 | 0.51123 | 0.51735 |
| Electrophilicity index (ω = μ2/2η) | 0.032216 | 0.4472533 | 0.435016 | 0.25671 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gargouri, A.; Jamoussi, B. Molecular-Level Insights into Meta-Phenylenediamine and Sulfonated Zinc Phthalocyanine Interactions for Enhanced Polyamide Membranes: A DFT and TD-DFT Study. Polymers 2025, 17, 2019. https://doi.org/10.3390/polym17152019
Gargouri A, Jamoussi B. Molecular-Level Insights into Meta-Phenylenediamine and Sulfonated Zinc Phthalocyanine Interactions for Enhanced Polyamide Membranes: A DFT and TD-DFT Study. Polymers. 2025; 17(15):2019. https://doi.org/10.3390/polym17152019
Chicago/Turabian StyleGargouri, Ameni, and Bassem Jamoussi. 2025. "Molecular-Level Insights into Meta-Phenylenediamine and Sulfonated Zinc Phthalocyanine Interactions for Enhanced Polyamide Membranes: A DFT and TD-DFT Study" Polymers 17, no. 15: 2019. https://doi.org/10.3390/polym17152019
APA StyleGargouri, A., & Jamoussi, B. (2025). Molecular-Level Insights into Meta-Phenylenediamine and Sulfonated Zinc Phthalocyanine Interactions for Enhanced Polyamide Membranes: A DFT and TD-DFT Study. Polymers, 17(15), 2019. https://doi.org/10.3390/polym17152019