
Development of a Continuous Extrusion Process for Alginate Biopolymer Films for Sustainable Applications


Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
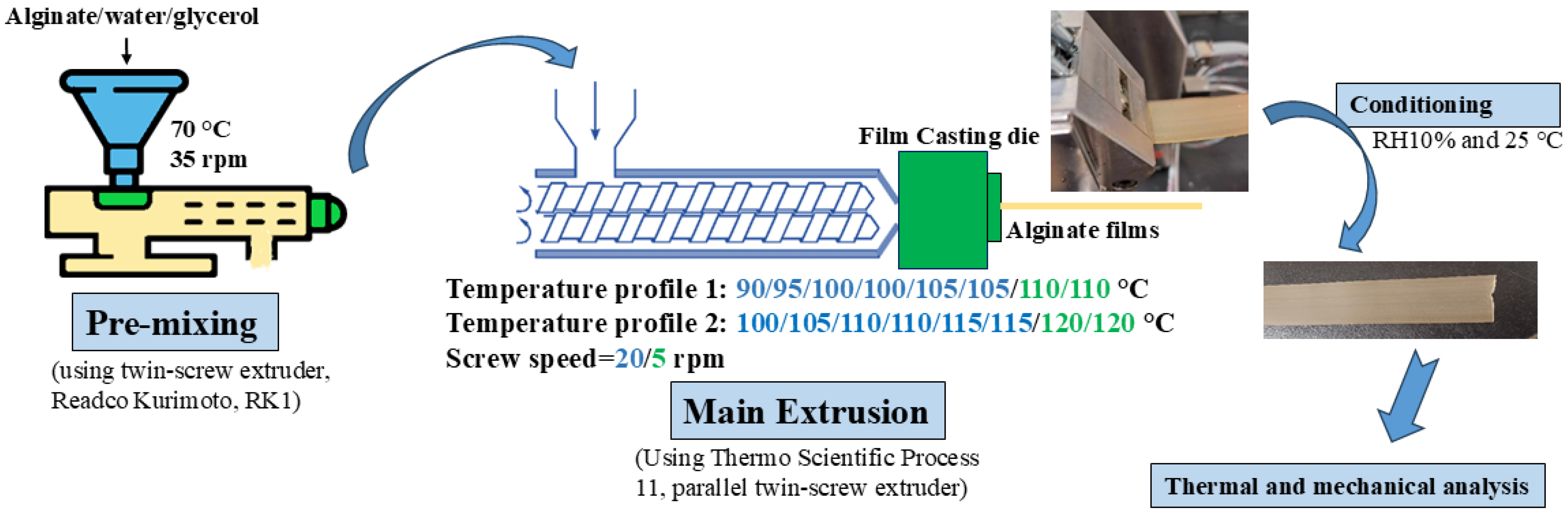
2.2. Film-Forming Methods
2.3. Characterization Methods
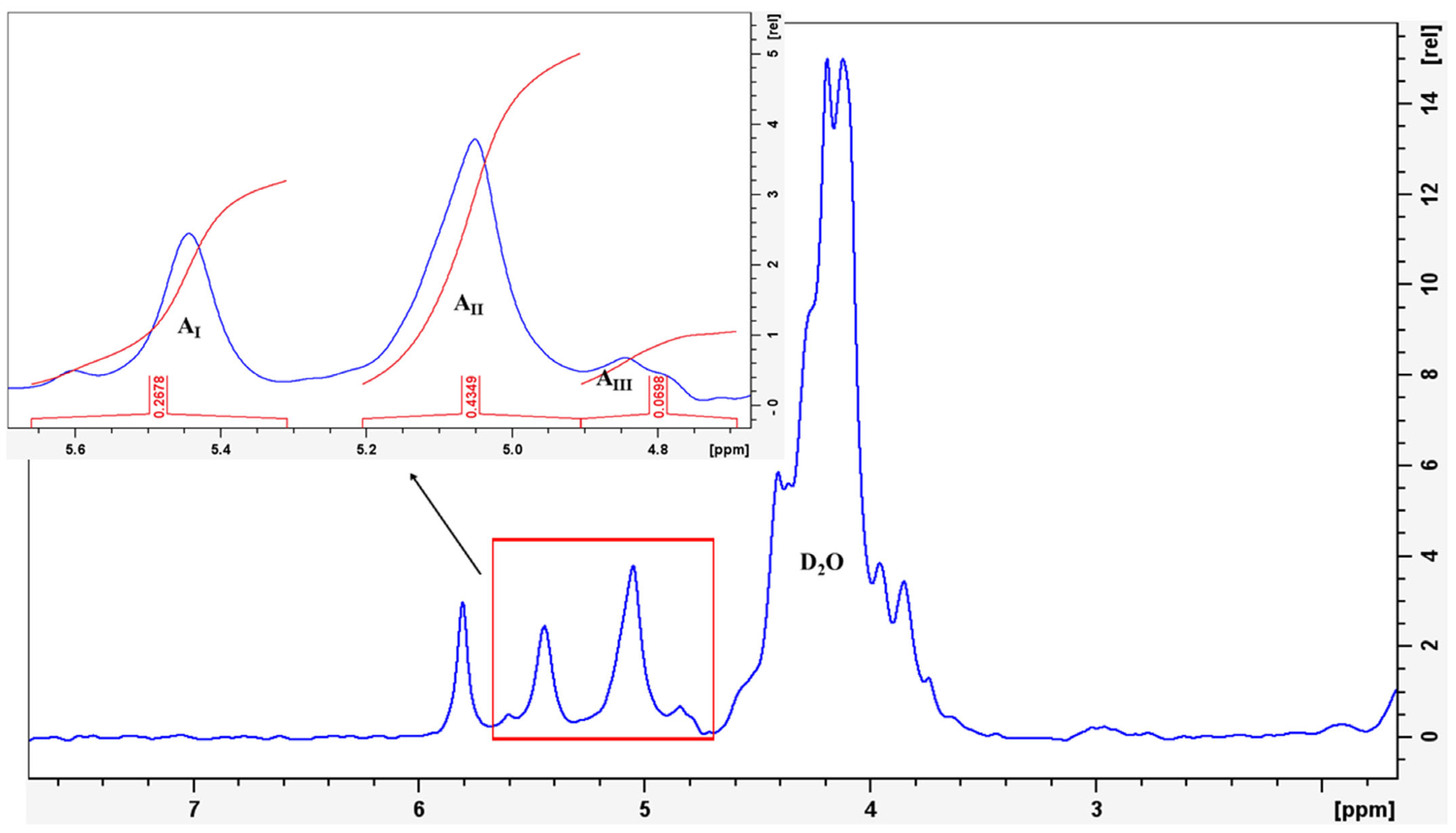
2.3.1. 1H NMR
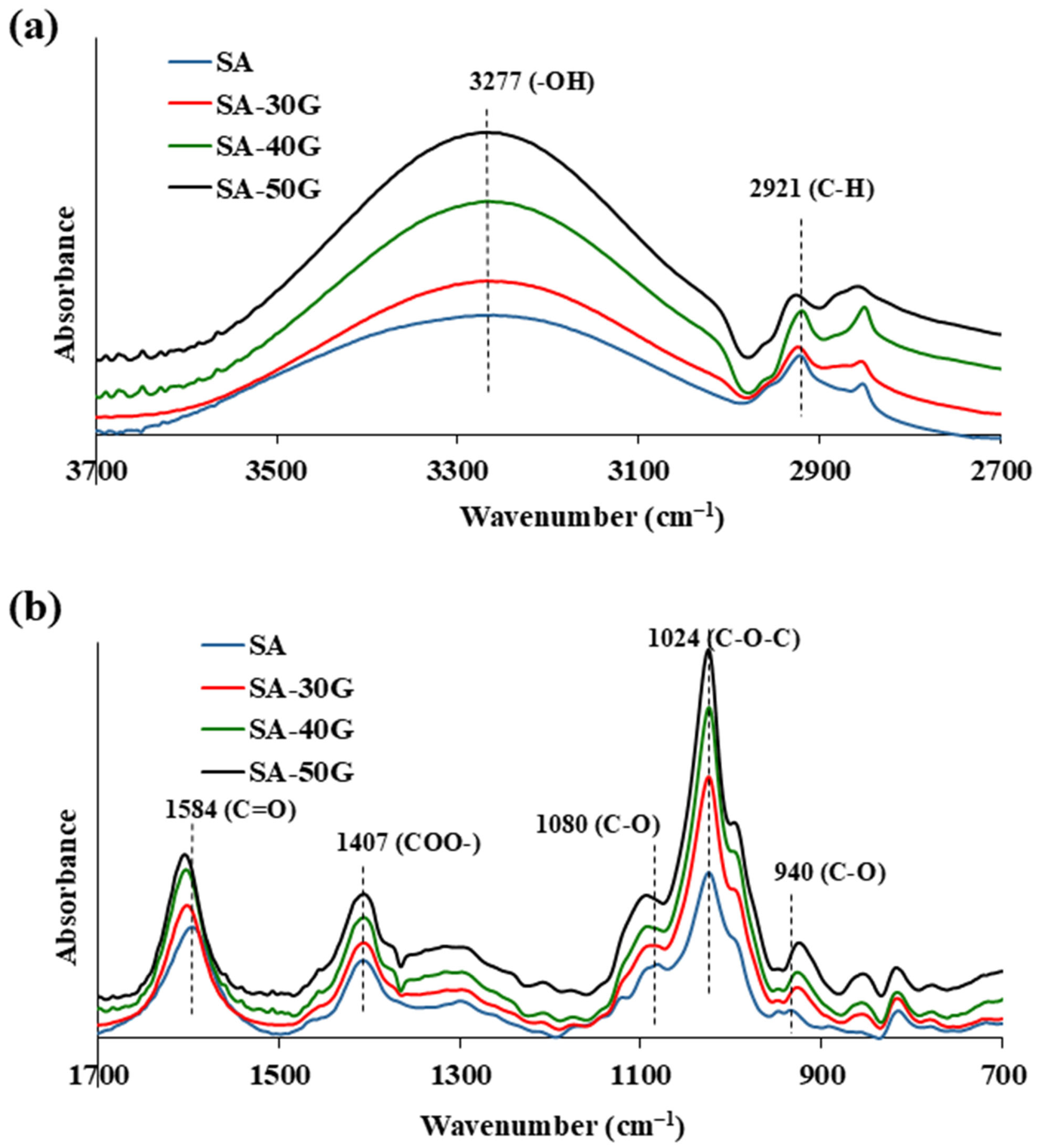
2.3.2. FT-IR
2.3.3. DSC (Differential Scanning Calorimeter)
2.3.4. DMA (Dynamical Mechanical Analysis)
2.3.5. Tensile Test
3. Results
3.1. The Effect of Plasticizer Concentration on the Structure and Thermal and Mechanical Properties of Alginate
3.1.1. Chemical Structure
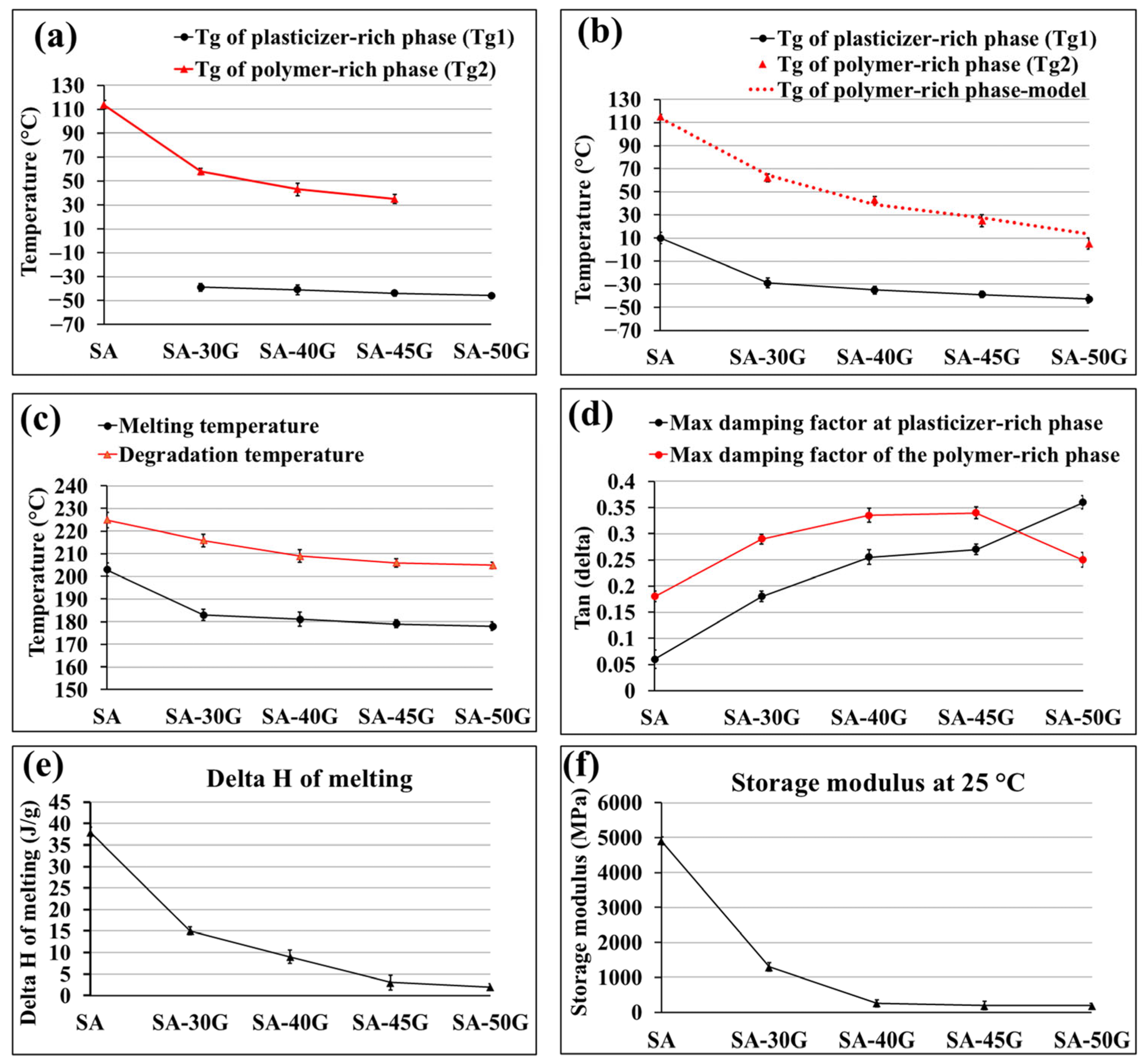
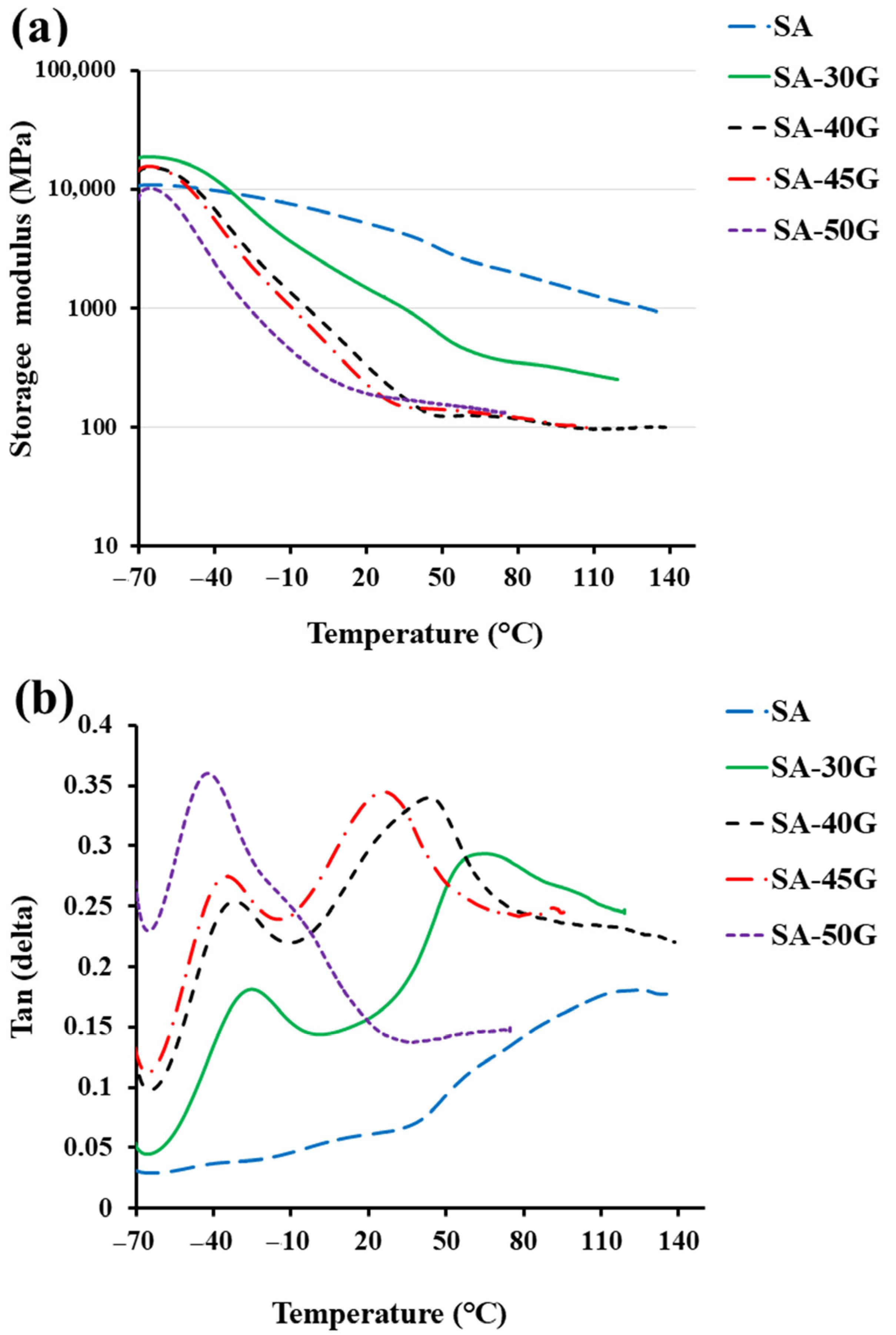
3.1.2. Thermal Properties
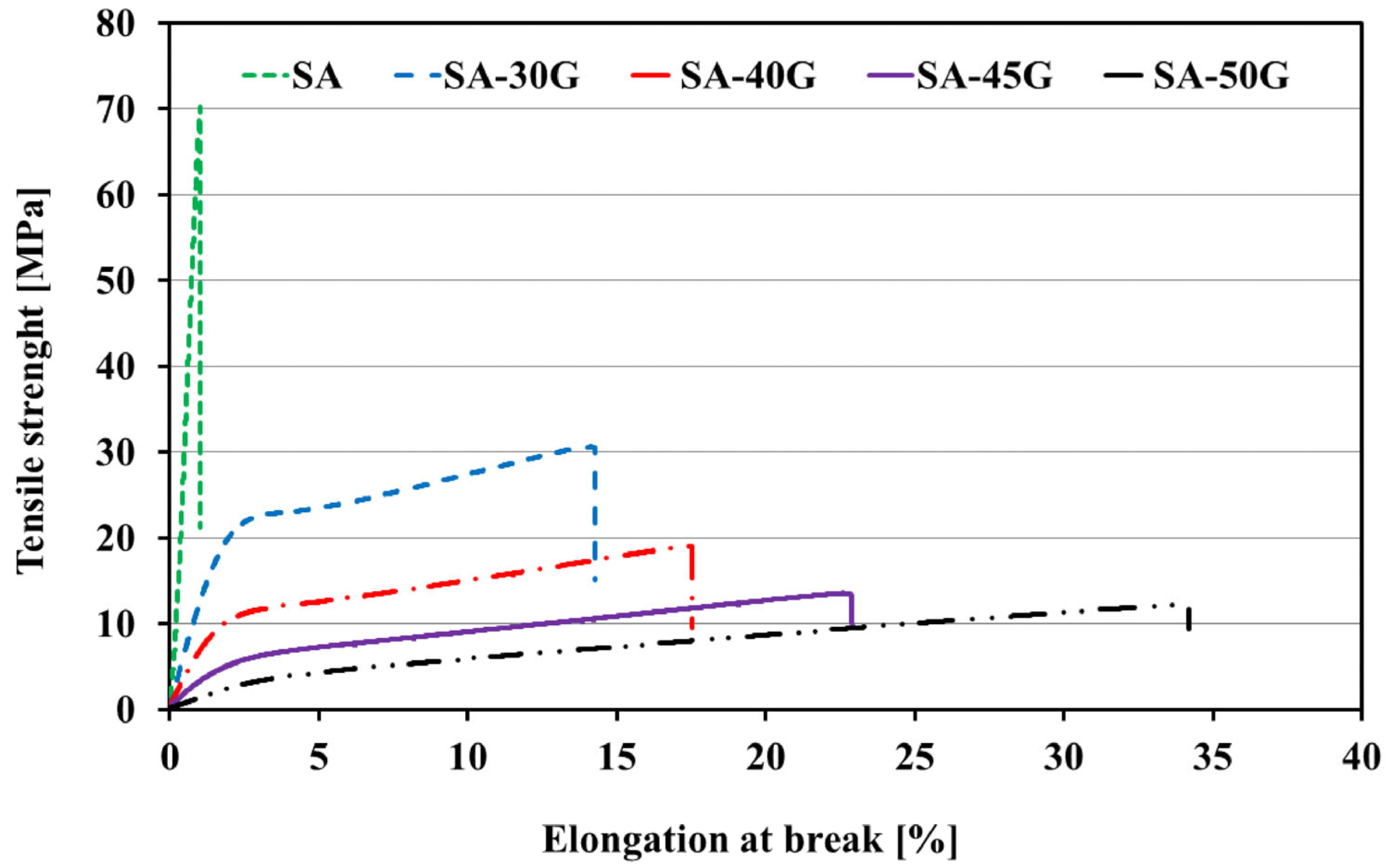
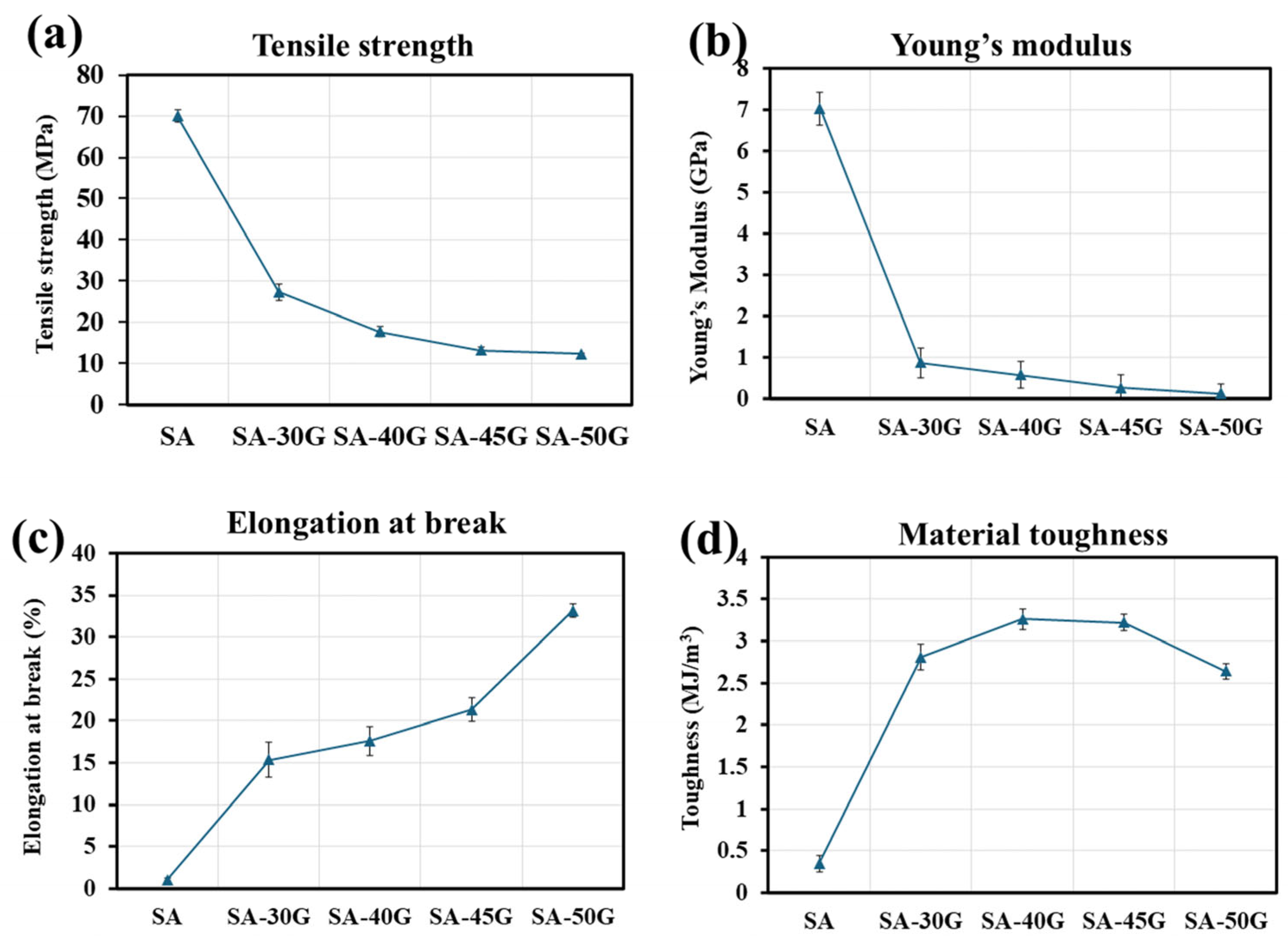
3.1.3. Mechanical Properties
3.2. The Effect of Processing Temperature on Thermal and Mechanical Properties of Alginate
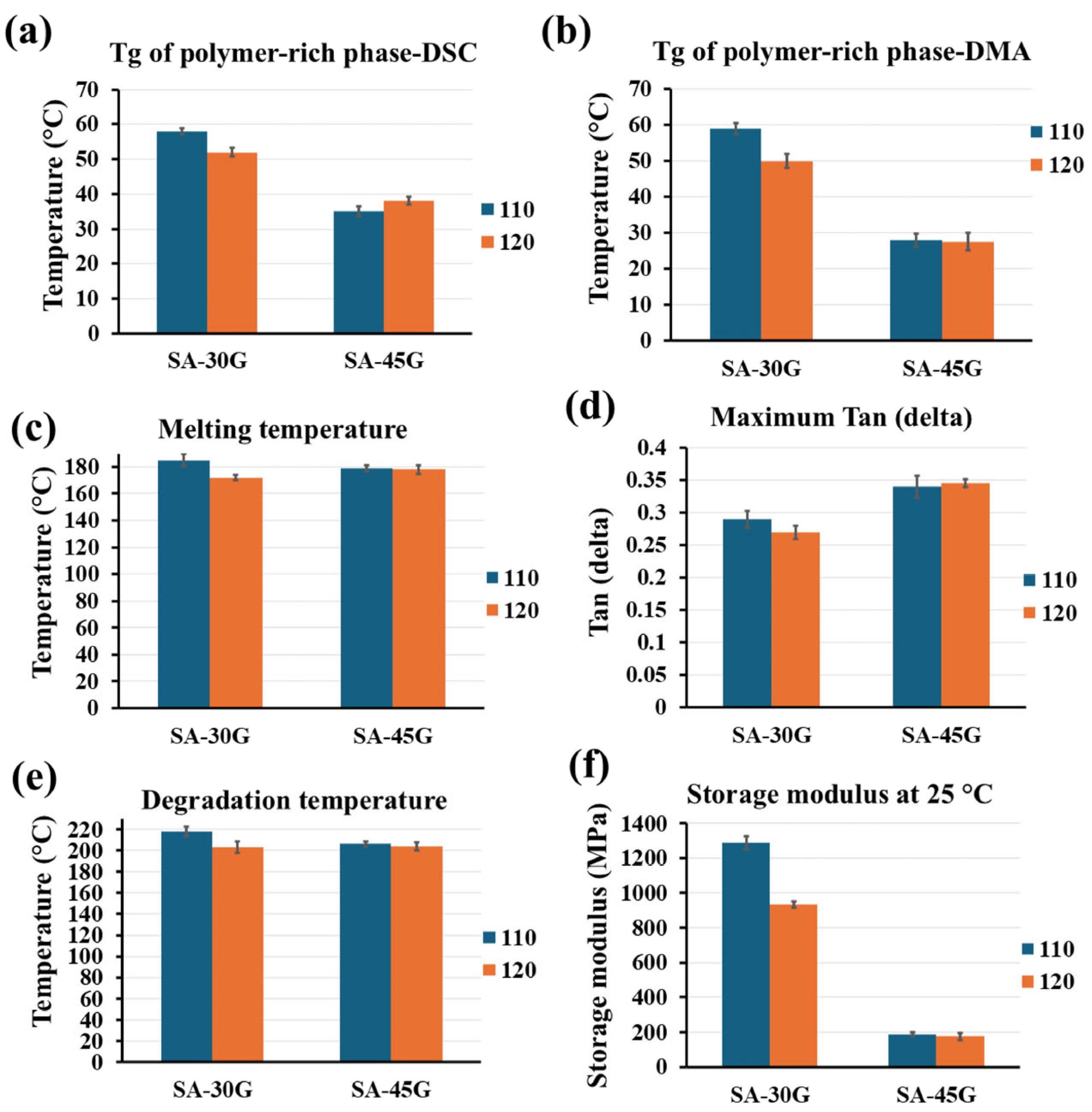
3.2.1. Thermal Properties
3.2.2. Mechanical Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abdul Khalil, H.P.S.; Banerjee, A.; Saurabh, C.K.; Tye, Y.Y.; Suriani, A.B.; Mohamed, A.; Karim, A.A.; Rizal, S.; Paridah, M.T. Biodegradable Films for Fruits and Vegetables Packaging Application: Preparation and Properties. Food Eng. Rev. 2018, 10, 139–153. [Google Scholar] [CrossRef]
- Abdullah, N.A.; Mohamad, Z.; Khan, Z.I.; Jusoh, M.; Zakaria, Z.Y.; Ngadi, N. Alginate Based Sustainable Films and Composites for Packaging: A Review. Chem. Eng. Trans. 2021, 83, 271–276. [Google Scholar] [CrossRef]
- MacHugh, D.J.; McHugh, D.J. (Eds.) Production and Utilization of Products from Commercial Seaweeds; FAO fisheries technical paper; Food and Agriculture Organization of the United Nations: Rome, Italy, 1987; ISBN 978-92-5-102612-0. [Google Scholar]
- Moore, A. (Ed.) Alginic Acid: Chemical Structure, Uses and Health Benefits; Chemistry research and applications; Nova Publishers: New York, NY, USA, 2015; ISBN 978-1-63463-224-9. [Google Scholar]
- Abourehab, M.A.S.; Rajendran, R.R.; Singh, A.; Pramanik, S.; Shrivastav, P.; Ansari, M.J.; Manne, R.; Amaral, L.S.; Deepak, A. Alginate as a Promising Biopolymer in Drug Delivery and Wound Healing: A Review of the State-of-the-Art. Int. J. Mol. Sci. 2022, 23, 9035. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Jiang, J.; Zhao, L.; Zhang, J.; Wang, F. Applications of Alginate as a Functional Food Ingredient. In Biopolymers for Food Design; Elsevier: Amsterdam, The Netherlands, 2018; pp. 409–429. ISBN 978-0-12-811449-0. [Google Scholar]
- Vijayalakshmi, K.; Latha, S.; Rose, M. Industrial Applications of Alginate. In Industrial Applications of Marine Biopolymers; Sudha, P., Ed.; CRC Press: Boca Raton, FL, USA, 2017; pp. 545–575. ISBN 978-1-4987-3148-5. [Google Scholar]
- Zhang, K.; Zhang, W.; Ruan, P.; Zhou, Y.; Yao, B.; Wang, Y.; Wang, Z. Fabrication and Characterization of Truxillic Alginate Bioplastic with Decent Antibacterial, Antioxidant and Degradability Properties in Food Packaging. Mater. Today Commun. 2024, 41, 111022. [Google Scholar] [CrossRef]
- Majhi, S.; Tyagi, A.; Mishra, A. Bio-Polymeric Packaging Material for Packaging of Raw Food. In Encyclopedia of Renewable and Sustainable Materials; Elsevier: Amsterdam, The Netherlands, 2020; pp. 44–51. ISBN 978-0-12-813196-1. [Google Scholar]
- Avella, M.; Pace, E.D.; Immirzi, B.; Impallomeni, G.; Malinconico, M.; Santagata, G. Addition of Glycerol Plasticizer to Seaweeds Derived Alginates: Influence of Microstructure on Chemical–Physical Properties. Carbohydr. Polym. 2007, 69, 503–511. [Google Scholar] [CrossRef]
- Jost, V.; Kobsik, K.; Schmid, M.; Noller, K. Influence of Plasticiser on the Barrier, Mechanical and Grease Resistance Properties of Alginate Cast Films. Carbohydr. Polym. 2014, 110, 309–319. [Google Scholar] [CrossRef]
- Gao, C.; Pollet, E.; Avérous, L. Properties of Glycerol-Plasticized Alginate Films Obtained by Thermo-Mechanical Mixing. Food Hydrocoll. 2017, 63, 414–420. [Google Scholar] [CrossRef]
- Jeya Jeevahan, J.; Chandrasekaran, M.; Venkatesan, S.P.; Sriram, V.; Britto Joseph, G.; Mageshwaran, G.; Durairaj, R.B. Scaling up Difficulties and Commercial Aspects of Edible Films for Food Packaging: A Review. Trends Food Sci. Technol. 2020, 100, 210–222. [Google Scholar] [CrossRef]
- Mortalò, C.; Russo, P.; Miorin, E.; Zin, V.; Paradisi, E.; Leonelli, C. Extruded Composite Films Based on Polylactic Acid and Sodium Alginate. Polymer 2023, 282, 126162. [Google Scholar] [CrossRef]
- Rech, A.; Siamos, E.; Nicholas, P.; Daugaard, A.E. Recyclable Extrudable Biopolymer Composites from Alginate and Lignocellulosic Biomass Waste. ACS Sustain. Chem. Eng. 2023, 11, 8939–8947. [Google Scholar] [CrossRef]
- Gao, C.; Pollet, E.; Avérous, L. Innovative Plasticized Alginate Obtained by Thermo-Mechanical Mixing: Effect of Different Biobased Polyols Systems. Carbohydr. Polym. 2017, 157, 669–676. [Google Scholar] [CrossRef]
- Pongjanyakul, T.; Puttipipatkhachorn, S. Alginate-Magnesium Aluminum Silicate Films: Effect of Plasticizers on Film Properties, Drug Permeation and Drug Release from Coated Tablets. Int. J. Pharm. 2007, 333, 34–44. [Google Scholar] [CrossRef]
- Olivas, G.I.; Barbosa-Cánovas, G.V. Alginate–Calcium Films: Water Vapor Permeability and Mechanical Properties as Affected by Plasticizer and Relative Humidity. LWT Food Sci. Technol. 2008, 41, 359–366. [Google Scholar] [CrossRef]
- Paixão, L.C.; Lopes, I.A.; Barros Filho, A.K.D.; Santana, A.A. Alginate Biofilms Plasticized with Hydrophilic and Hydrophobic Plasticizers for Application in Food Packaging. J. Appl. Polym. Sci. 2019, 136, 48263. [Google Scholar] [CrossRef]
- Sharmin, N.; Sone, I.; Walsh, J.L.; Sivertsvik, M.; Fernández, E.N. Effect of Citric Acid and Plasma Activated Water on the Functional Properties of Sodium Alginate for Potential Food Packaging Applications. Food Packag. Shelf Life 2021, 29, 100733. [Google Scholar] [CrossRef]
- Falguera, V.; Quintero, J.P.; Jiménez, A.; Muñoz, J.A.; Ibarz, A. Edible Films and Coatings: Structures, Active Functions and Trends in Their Use. Trends Food Sci. Technol. 2011, 22, 292–303. [Google Scholar] [CrossRef]
- Eslami, Z.; Elkoun, S.; Robert, M.; Adjallé, K. A Review of the Effect of Plasticizers on the Physical and Mechanical Properties of Alginate-Based Films. Molecules 2023, 28, 6637. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.T.; Taconi, K.A. The Glycerin Glut: Options for the Value-added Conversion of Crude Glycerol Resulting from Biodiesel Production. Environ. Prog. 2007, 26, 338–348. [Google Scholar] [CrossRef]
- Pagliaro, M.; Rossi, M. The Future of Glycerol: New Uses of a Versatile Raw Material; RSC Green Chemistry Series; Royal Society of Chemistry: Cambridge, UK, 2008; ISBN 978-0-85404-124-4. [Google Scholar]
- Sanyang, M.; Sapuan, S.; Jawaid, M.; Ishak, M.; Sahari, J. Effect of Plasticizer Type and Concentration on Tensile, Thermal and Barrier Properties of Biodegradable Films Based on Sugar Palm (Arenga pinnata) Starch. Polymers 2015, 7, 1106–1124. [Google Scholar] [CrossRef]
- Rhim, J.-W. Physical and Mechanical Properties of Water Resistant Sodium Alginate Films. LWT Food Sci. Technol. 2004, 37, 323–330. [Google Scholar] [CrossRef]
- Giz, A.S.; Berberoglu, M.; Bener, S.; Aydelik-Ayazoglu, S.; Bayraktar, H.; Alaca, B.E.; Catalgil-Giz, H. A Detailed Investigation of the Effect of Calcium Crosslinking and Glycerol Plasticizing on the Physical Properties of Alginate Films. Int. J. Biol. Macromol. 2020, 148, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Janik, W.; Nowotarski, M.; Ledniowska, K.; Shyntum, D.Y.; Krukiewicz, K.; Turczyn, R.; Sabura, E.; Furgoł, S.; Kudła, S.; Dudek, G. Modulation of Physicochemical Properties and Antimicrobial Activity of Sodium Alginate Films Through the Use of Chestnut Extract and Plasticizers. Sci. Rep. 2023, 13, 11530. [Google Scholar] [CrossRef]
- Epure, V.; Griffon, M.; Pollet, E.; Avérous, L. Structure and Properties of Glycerol-Plasticized Chitosan Obtained by Mechanical Kneading. Carbohydr. Polym. 2011, 83, 947–952. [Google Scholar] [CrossRef]
- Matet, M.; Heuzey, M.-C.; Pollet, E.; Ajji, A.; Avérous, L. Innovative Thermoplastic Chitosan Obtained by Thermo-Mechanical Mixing with Polyol Plasticizers. Carbohydr. Polym. 2013, 95, 241–251. [Google Scholar] [CrossRef]
- Xie, F.; Gao, C.; Avérous, L. Alginate-Based Materials: Enhancing Properties through Multiphase Formulation Design and Processing Innovation. Mater. Sci. Eng. R Rep. 2024, 159, 100799. [Google Scholar] [CrossRef]
- Tiefenbacher, K.F. Glossary of Terms in Wafers, Waffles and Adjuncts. In The Technology of Wafers and Waffles II; Elsevier: Amsterdam, The Netherlands, 2019; pp. 325–411. ISBN 978-0-12-809437-2. [Google Scholar]
- Fishman, M.L.; Coffin, D.R.; Konstance, R.P.; Onwulata, C.I. Extrusion of Pectin/Starch Blends Plasticized with Glycerol. Carbohydr. Polym. 2000, 41, 317–325. [Google Scholar] [CrossRef]
- Paluch, M.; Ostrowska, J.; Tyński, P.; Sadurski, W.; Konkol, M. Structural and Thermal Properties of Starch Plasticized with Glycerol/Urea Mixture. J. Polym. Environ. 2022, 30, 728–740. [Google Scholar] [CrossRef]
- Yan, Q.; Hou, H.; Guo, P.; Dong, H. Effects of Extrusion and Glycerol Content on Properties of Oxidized and Acetylated Corn Starch-Based Films. Carbohydr. Polym. 2012, 87, 707–712. [Google Scholar] [CrossRef]
- Souza, R.C.R.; Andrade, C.T. Processing and Properties of Thermoplastic Starch and Its Blends with Sodium Alginate. J. Appl. Polym. Sci. 2001, 81, 412–420. [Google Scholar] [CrossRef]
- López, O.V.; Ninago, M.D.; Lencina, M.M.S.; García, M.A.; Andreucetti, N.A.; Ciolino, A.E.; Villar, M.A. Thermoplastic Starch Plasticized with Alginate–Glycerol Mixtures: Melt-Processing Evaluation and Film Properties. Carbohydr. Polym. 2015, 126, 83–90. [Google Scholar] [CrossRef]
- Córdoba, A.; Cuéllar, N.; González, M.; Medina, J. The Plasticizing Effect of Alginate on the Thermoplastic Starch/Glycerin Blends. Carbohydr. Polym. 2008, 73, 409–416. [Google Scholar] [CrossRef]
- Eslami, Z.; Elkoun, S.; Ahyaya, L.; Robert, M. Influence of M/G Ratio and Molecular Weight on Water Sorption and Glass Transition Behavior of Alginate Films. Int. J. Biol. Macromol. 2025, 315, 144639. [Google Scholar] [CrossRef]
- Gordon, M.; Taylor, J.S. Ideal Copolymers and the Second-order Transitions of Synthetic Rubbers. i. Non-crystalline Copolymers. J. Appl. Chem. 1952, 2, 493–500. [Google Scholar] [CrossRef]
- Sablani, S.S.; Kasapis, S.; Rahman, M.S. Evaluating Water Activity and Glass Transition Concepts for Food Stability. J. Food Eng. 2007, 78, 266–271. [Google Scholar] [CrossRef]
- Shah, B.N.; Schall, C.A. Measurement and Modeling of the Glass Transition Temperatures of Multi-Component Solutions. Thermochim. Acta 2006, 443, 78–86. [Google Scholar] [CrossRef]
- ASTM D638-22; Test Method for Tensile Properties of Plastics. D20 Committee ASTM International: West Conshohocken, PA, USA, 2022. [CrossRef]
- Grasdalen, H.; Larsen, B.; Smidsrød, O. A p.m.r. Study of the Composition and Sequence of Uronate Residues in Alginates. Carbohydr. Res. 1979, 68, 23–31. [Google Scholar] [CrossRef]
- Draget, K.I.; Smidsrød, O.; Skjåk-Bræk, G. Alginates from Algae. In Biopolymers Online; Steinbüchel, A., Ed.; Wiley: Hoboken, NJ, USA, 2002; ISBN 978-3-527-30290-1. [Google Scholar]
- Haug, A.; Larsen, B.; Smidsrød, O. Uronic Acid Sequence in Alginate from Different Sources. Carbohydr. Res. 1974, 32, 217–225. [Google Scholar] [CrossRef]
- Belattmania, Z.; Kaidi, S.; El Atouani, S.; Katif, C.; Bentiss, F.; Jama, C.; Reani, A.; Sabour, B.; Vasconcelos, V. Isolation and FTIR-ATR and 1H NMR Characterization of Alginates from the Main Alginophyte Species of the Atlantic Coast of Morocco. Molecules 2020, 25, 4335. [Google Scholar] [CrossRef]
- Flórez-Fernández, N.; Domínguez, H.; Torres, M.D. A Green Approach for Alginate Extraction from Sargassum Muticum Brown Seaweed Using Ultrasound-Assisted Technique. Int. J. Biol. Macromol. 2019, 124, 451–459. [Google Scholar] [CrossRef]
- Sartori, C.; Finch, D.S.; Ralph, B.; Gilding, K. Determination of the Cation Content of Alginate Thin Films by FTi.r. Spectroscopy. Polymer 1997, 38, 43–51. [Google Scholar] [CrossRef]
- Chan, H.M.; Nyam, K.L.; Yusof, Y.A.; Pui, L.P. Investigation of Properties of Polysaccharide-Basededible Film Incorporated with Functional Melastoma Malabathricum Extract. Carpathian J. Food Sci. Technol. 2020, 12, 120–134. [Google Scholar] [CrossRef]
- Dai, Y.-N.; Li, P.; Zhang, J.-P.; Wang, A.-Q.; Wei, Q. A Novel pH sensitiveN-Succinyl Chitosan/Alginate Hydrogel Bead for Nifedipine Delivery. Biopharm. Drug Dispos. 2008, 29, 173–184. [Google Scholar] [CrossRef]
- Azucena Castro-Yobal, M.; Contreras-Oliva, A.; Saucedo-Rivalcoba, V.; Rivera-Armenta, J.L.; Hernández-Ramírez, G.; Salinas-Ruiz, J.; Herrera-Corredor, A. Evaluation of Physicochemical Properties of Film-Based Alginate for Food Packing Applications. e-Polymers 2021, 21, 82–95. [Google Scholar] [CrossRef]
- Ghosal, K.; Das, A.; Das, S.K.; Mahmood, S.; Ramadan, M.A.M.; Thomas, S. Synthesis and Characterization of Interpenetrating Polymeric Networks Based Bio-Composite Alginate Film: A Well-Designed Drug Delivery Platform. Int. J. Biol. Macromol. 2019, 130, 645–654. [Google Scholar] [CrossRef]
- Pérez, A.; Sandoval, A.J.; Cova, A.; Müller, A.J. Glass Transitions and Physical Aging of Cassava Starch—Corn Oil Blends. Carbohydr. Polym. 2014, 105, 244–252. [Google Scholar] [CrossRef]
- Liu, H.; Xie, F.; Yu, L.; Chen, L.; Li, L. Thermal Processing of Starch-Based Polymers. Prog. Polym. Sci. 2009, 34, 1348–1368. [Google Scholar] [CrossRef]
- Russo, R.; Malinconico, M.; Santagata, G. Effect of Cross-Linking with Calcium Ions on the Physical Properties of Alginate Films. Biomacromolecules 2007, 8, 3193–3197. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.; Abbate, M.; Malinconico, M.; Santagata, G. Effect of Polyglycerol and the Crosslinking on the Physical Properties of a Blend Alginate-Hydroxyethylcellulose. Carbohydr. Polym. 2010, 82, 1061–1067. [Google Scholar] [CrossRef]
- Treenate, P.; Monvisade, P.; Yamaguchi, M. The Effect of Glycerol/Water and Sorbitol/Water on the Plasticization of Hydroxyethylacryl Chitosan/Sodium Alginate Films. MATEC Web Conf. 2015, 30, 02006. [Google Scholar] [CrossRef]
- Madrigal, L.; Sandoval, A.J.; Müller, A.J. Effects of Corn Oil on Glass Transition Temperatures of Cassava Starch. Carbohydr. Polym. 2011, 85, 875–884. [Google Scholar] [CrossRef]
- Espíndola, S.P.; Norder, B.; Koper, G.J.M.; Picken, S.J. The Glass Transition Temperature of Heterogeneous Biopolymer Systems. Biomacromolecules 2023, 24, 1627–1637. [Google Scholar] [CrossRef] [PubMed]
- Çaykara, T.; Demirci, S.; Eroğlu, M.S.; Güven, O. Poly(Ethylene Oxide) and Its Blends with Sodium Alginate. Polymer 2005, 46, 10750–10757. [Google Scholar] [CrossRef]
- Farah, S.; Anderson, D.G.; Langer, R. Physical and Mechanical Properties of PLA, and Their Functions in Widespread Applications—A Comprehensive Review. Adv. Drug Deliv. Rev. 2016, 107, 367–392. [Google Scholar] [CrossRef]
- Van Soest, J.J.G.; Benes, K.; De Wit, D.; Vliegenthart, J.F.G. The Influence of Starch Molecular Mass on the Properties of Extruded Thermoplastic Starch. Polymer 1996, 37, 3543–3552. [Google Scholar] [CrossRef]
- Oh, G. A Simplified Toughness Estimation Method Based on Standard Tensile Data. Int. J. Press. Vessel. Pip. 2022, 199, 104733. [Google Scholar] [CrossRef]
- Ciccia, N.R.; Shi, J.X.; Pal, S.; Hua, M.; Malollari, K.G.; Lizandara-Pueyo, C.; Risto, E.; Ernst, M.; Helms, B.A.; Messersmith, P.B.; et al. Diverse Functional Polyethylenes by Catalytic Amination. Science 2023, 381, 1433–1440. [Google Scholar] [CrossRef]
- Ma, Y.; Liu, H.; Zhou, L.; Yu, Y.; Gong, J. Tough, Degradable Bioplastics Enabling by Noncovalent Assembly of Polysaccharides and Inorganic Ionic Oligomers. ACS Appl. Polym. Mater. 2025, 7, 1896–1908. [Google Scholar] [CrossRef]
- Chen, Z.; Li, P.; Ji, Q.; Xing, Y.; Ma, X.; Xia, Y. All-Polysaccharide Composite Films Based on Calcium Alginate Reinforced Synergistically by Multidimensional Cellulose and Hemicellulose Fractionated from Corn Husks. Mater. Today Commun. 2023, 34, 105090. [Google Scholar] [CrossRef]
- Li, M.; Liu, P.; Zou, W.; Yu, L.; Xie, F.; Pu, H.; Liu, H.; Chen, L. Extrusion Processing and Characterization of Edible Starch Films with Different Amylose Contents. J. Food Eng. 2011, 106, 95–101. [Google Scholar] [CrossRef]
- Thunwall, M.; Boldizar, A.; Rigdahl, M. Compression Molding and Tensile Properties of Thermoplastic Potato Starch Materials. Biomacromolecules 2006, 7, 981–986. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Alginate/Glycerol (w/w) | Alginate/Water (w/w) | Temperature Profile | Mass Fraction of the Components for the Samples Conditioned at the Relative Humidity of 10% and 25 °C | ||
|---|---|---|---|---|---|---|
| xA * | xG * | xW * | ||||
| SA | 100/0 | 65/35 | 1 | 0.930 | 0 | 0.067 |
| SA-30G | 70/30 | 70/30 | 1&2 | 0.672 | 0.282 | 0.046 |
| SA-40G | 60/40 | 75/25 | 1 | 0.564 | 0.380 | 0.056 |
| SA-45G | 55/45 | 80/20 | 1&2 | 0.520 | 0.422 | 0.058 |
| SA-50G | 50/50 | 85/15 | 1 | 0.470 | 0.470 | 0.060 |
| Composition | Value |
|---|---|
| FG | 0.53 ± 0.06 |
| FM | 0.47 ± 0.03 |
| FGG | 0.144 ± 0.009 |
| FMG = FGM | 0.386 ± 0.02 |
| FMM | 0.084 ± 0.05 |
| M/G | 0.89 ± 0.12 |
| η | 1.55 ± 0.01 |
| Gordon-Taylor Parameters | KAG | KAW | TgA (°C) | E |
|---|---|---|---|---|
| Value | 0.9 | 2.3 | 156 | 3.85 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eslami, Z.; Elkoun, S.; Mirzapour, M.; Robert, M. Development of a Continuous Extrusion Process for Alginate Biopolymer Films for Sustainable Applications. Polymers 2025, 17, 1818. https://doi.org/10.3390/polym17131818
Eslami Z, Elkoun S, Mirzapour M, Robert M. Development of a Continuous Extrusion Process for Alginate Biopolymer Films for Sustainable Applications. Polymers. 2025; 17(13):1818. https://doi.org/10.3390/polym17131818
Chicago/Turabian StyleEslami, Zahra, Saïd Elkoun, Miraidin Mirzapour, and Mathieu Robert. 2025. "Development of a Continuous Extrusion Process for Alginate Biopolymer Films for Sustainable Applications" Polymers 17, no. 13: 1818. https://doi.org/10.3390/polym17131818
APA StyleEslami, Z., Elkoun, S., Mirzapour, M., & Robert, M. (2025). Development of a Continuous Extrusion Process for Alginate Biopolymer Films for Sustainable Applications. Polymers, 17(13), 1818. https://doi.org/10.3390/polym17131818