
Molecular Dynamics Simulation of Hydrogen Permeation Behavior in Epoxy Resin Systems





Abstract
1. Introduction
2. Materials and Methods
2.1. Molecular Dynamics Simulation
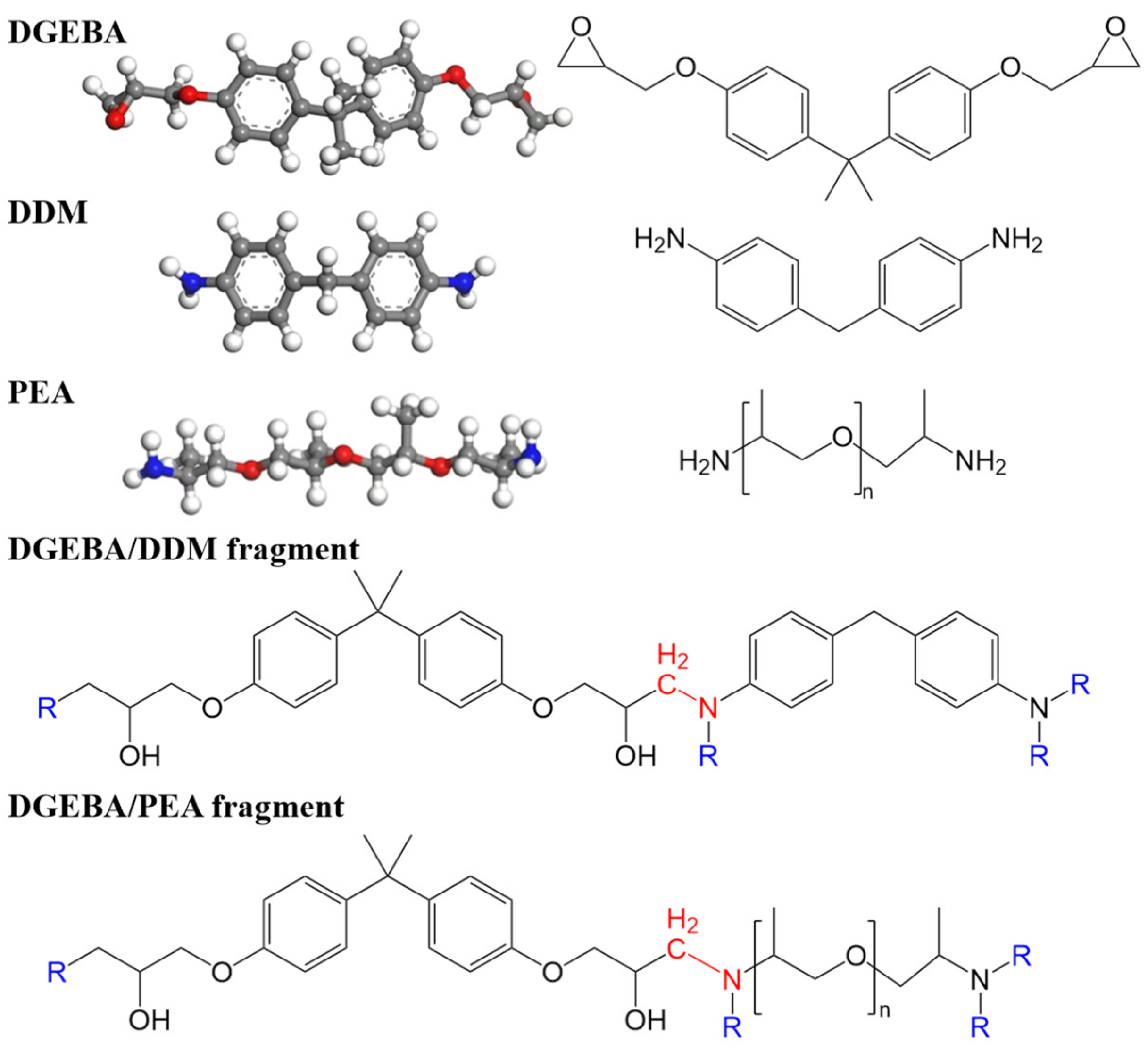
2.1.1. Model Preparation
2.1.2. Simulation of Permeation
2.1.3. Simulation of Free Volume
2.2. Experimental Procedure
2.2.1. Preparation of Epoxy Resins
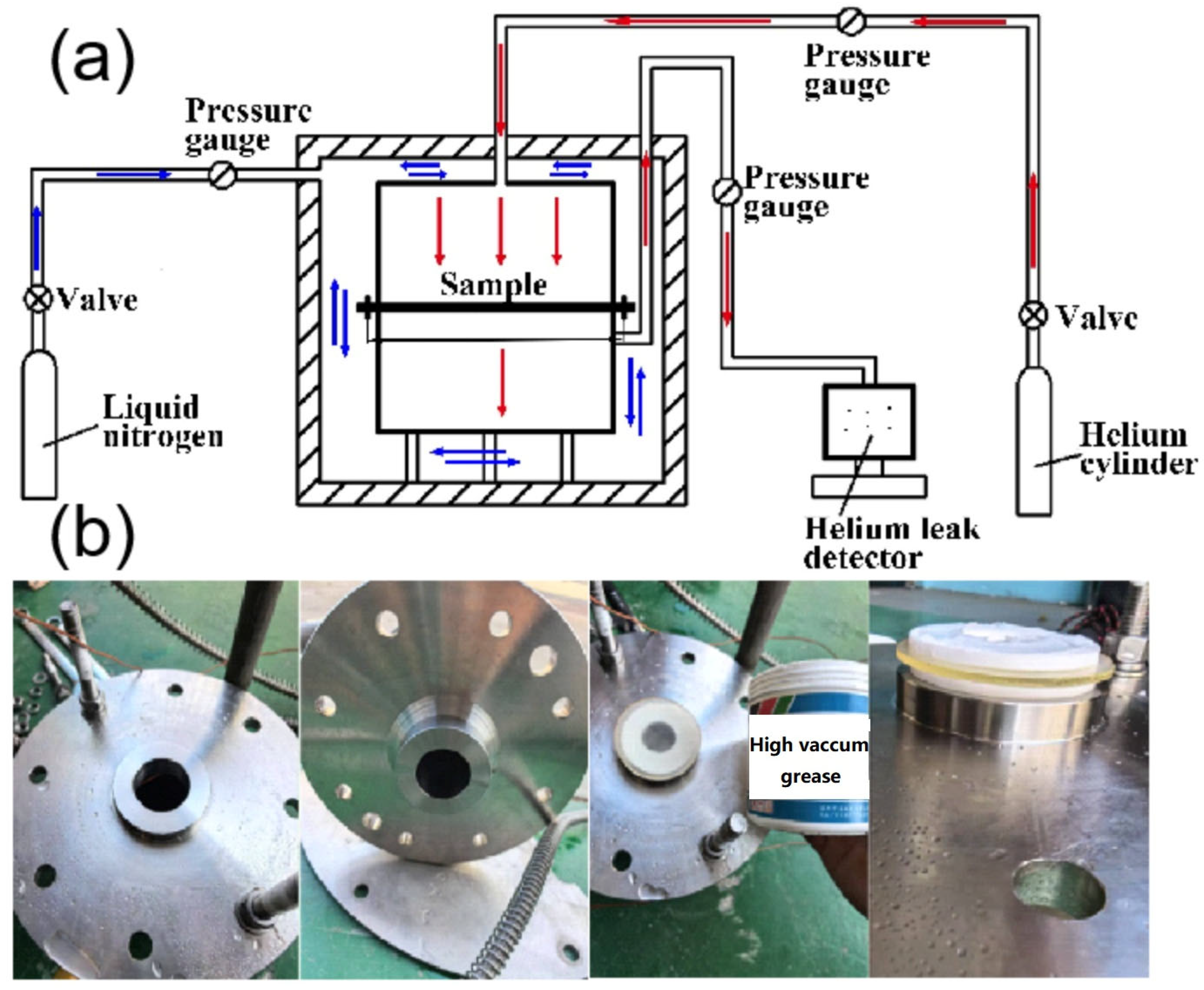
2.2.2. Helium Permeability Experiment
3. Results and Discussion
3.1. Effect of Cross-Linking Degree on Hydrogen Permeation in Epoxy Resins
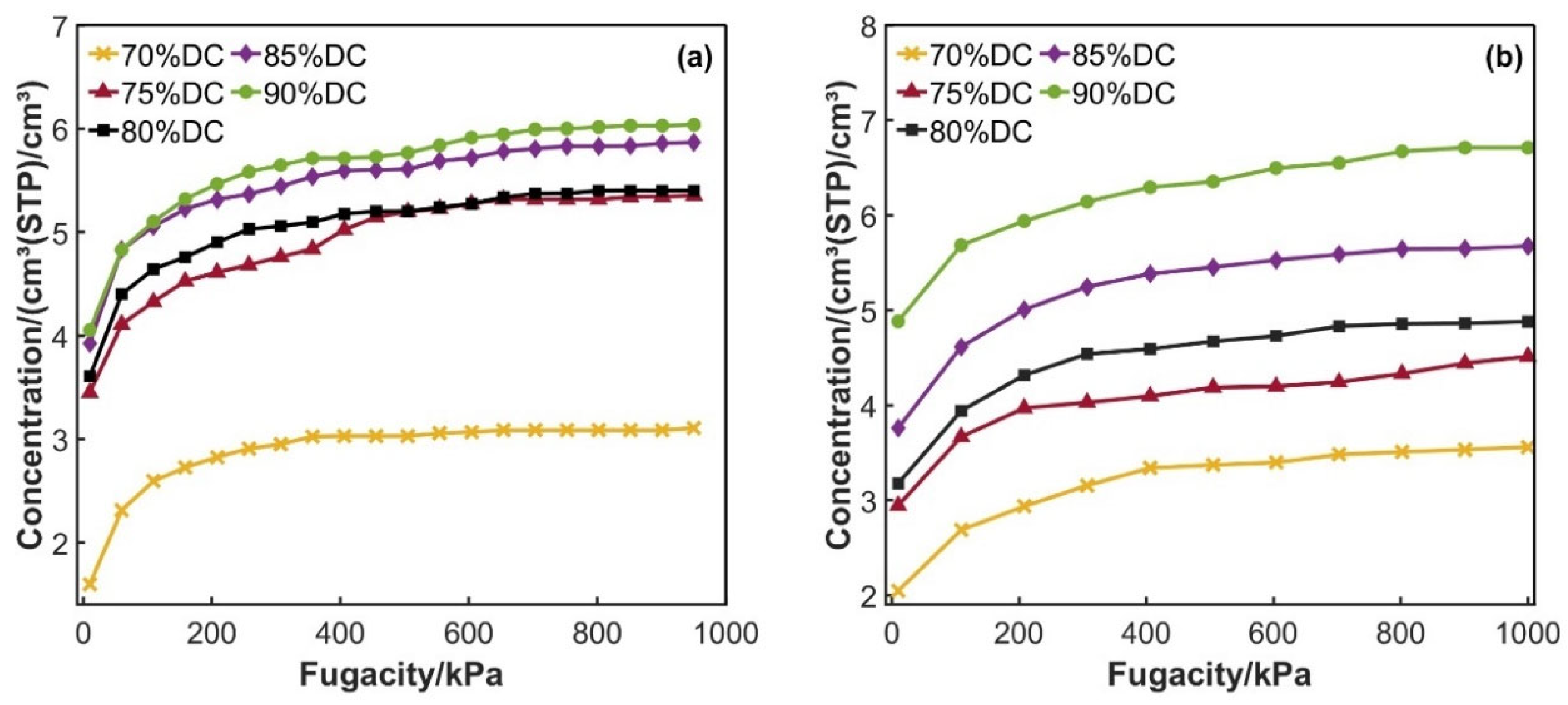
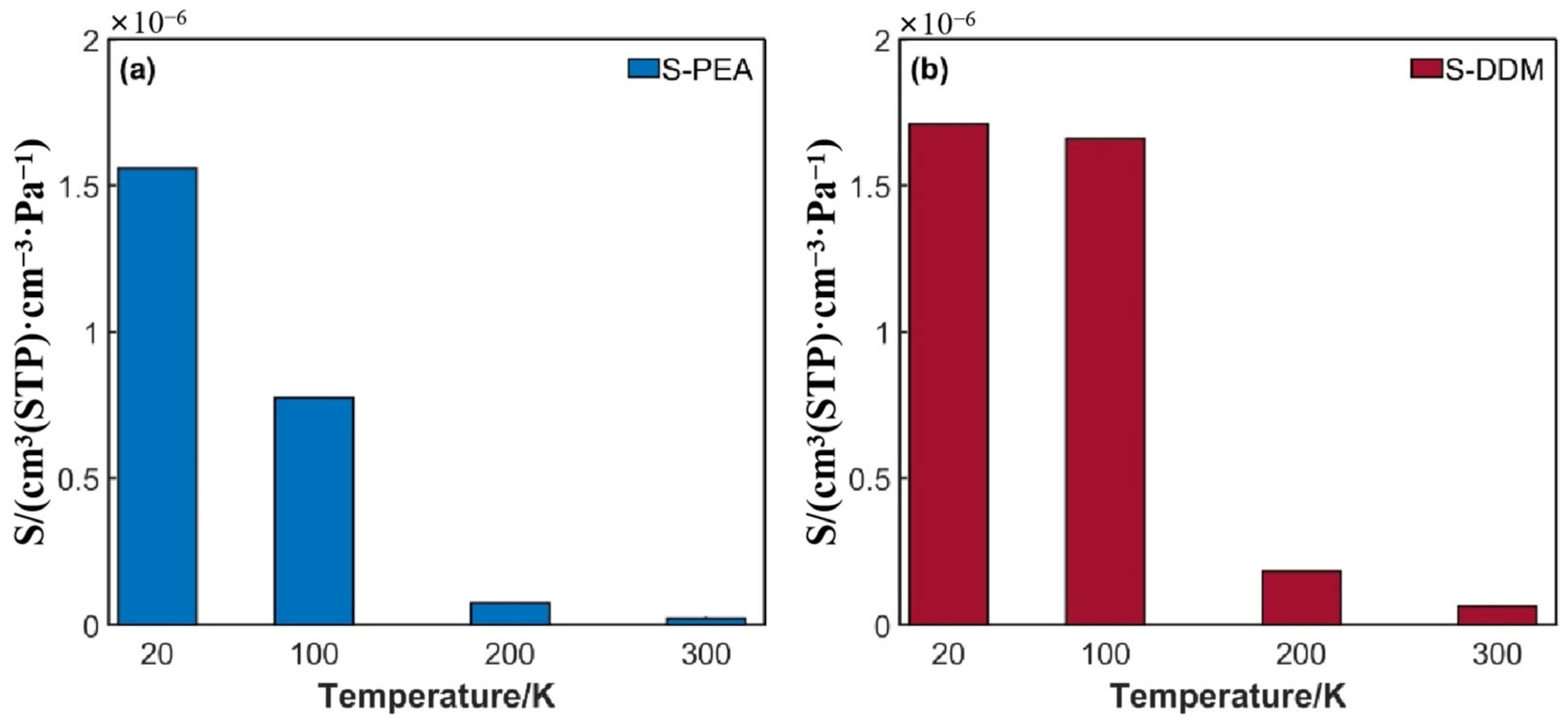
3.1.1. Solubility Coefficient
3.1.2. Diffusion Coefficient
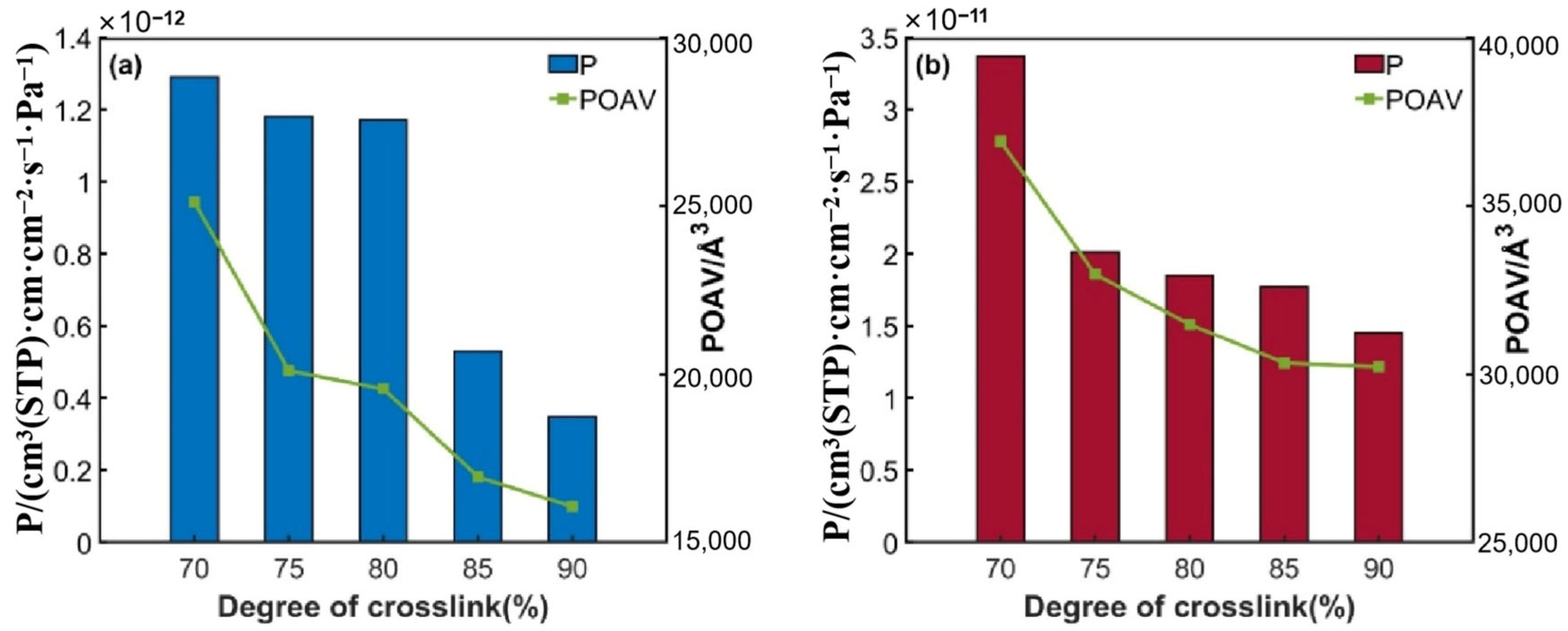
3.1.3. Permeability Coefficient
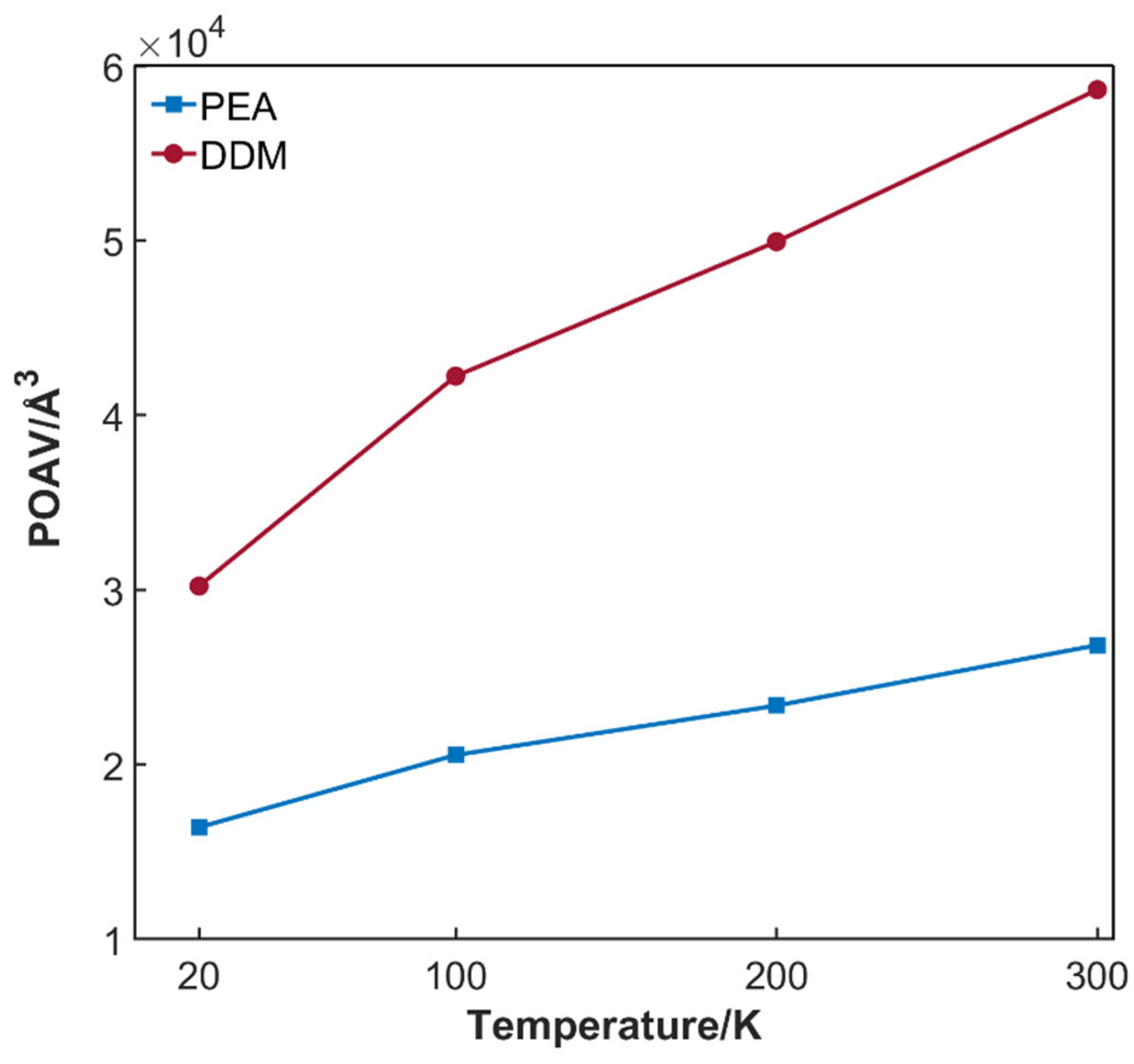
3.1.4. Free Volume
3.2. Parametric Discussion
3.2.1. Effect of Temperature on Hydrogen Permeation
3.2.2. Effect of Curing Agents on Hydrogen Permeation
3.3. Experimental Results
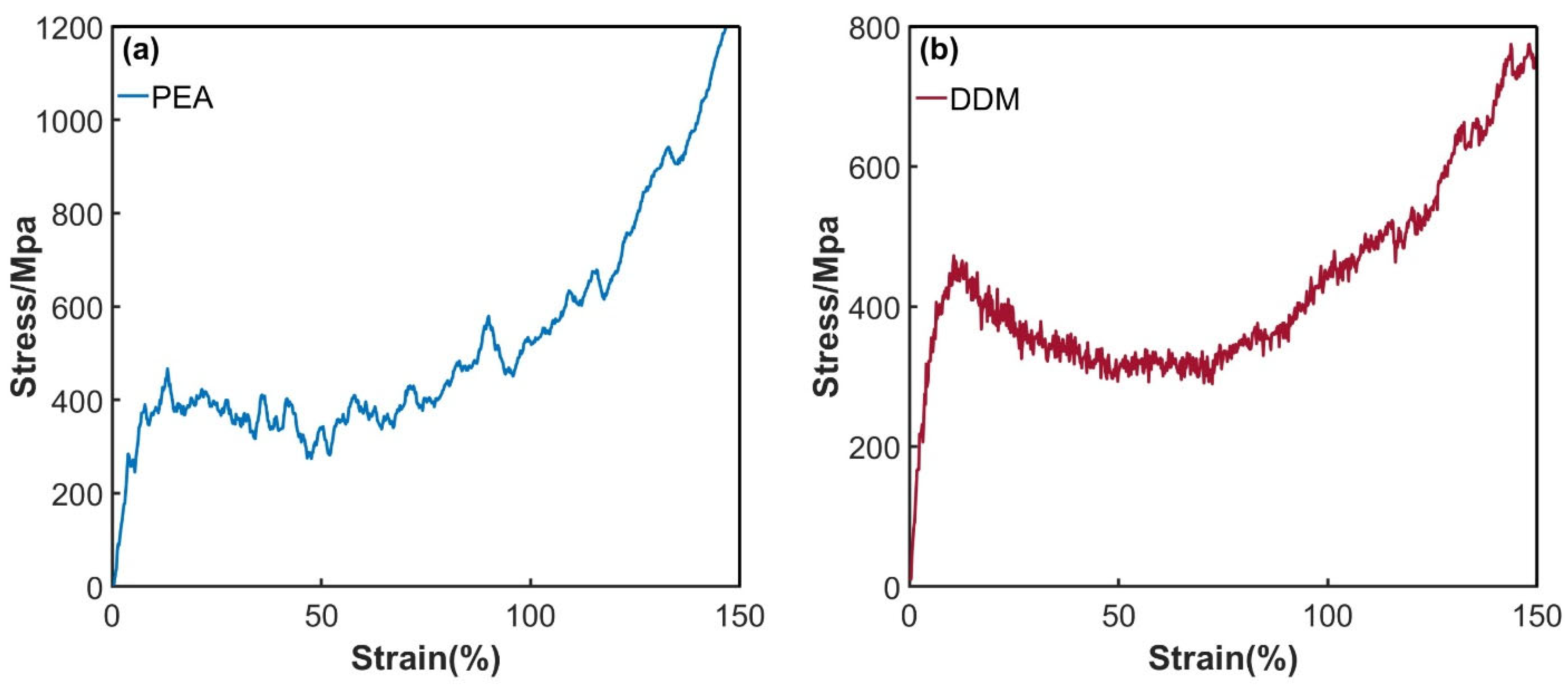
3.4. Effect of Material Defects on Hydrogen Permeation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Yu, H.; Gao, Q.; Pu, L.; Dai, M.; Sun, R. Numerical investigation on the characteristics of leakage and dispersion of cryogenic liquid oxygen in open environment. Cryogenics 2022, 125, 103514. [Google Scholar] [CrossRef]
- Zhang, J.; Yan, Y.; Zhang, C.; Xu, Z.; Li, X.; Zhao, G.; Ni, Z. Properties improvement of composite layer of cryo-compressed hydrogen storage vessel by polyethylene glycol modified epoxy resin. Int. J. Hydrogen Energy 2023, 48, 5576–5594. [Google Scholar] [CrossRef]
- Li, G.; Zhang, J.; Chai, J.; Ni, Z.; Yan, Y. Cryogenic mechanical performance and gas-barrier property of epoxy resins modified with multi-walled carbon nanotubes. Int. J. Hydrogen Energy 2024, 89, 738–745. [Google Scholar] [CrossRef]
- Lei, L.; Zhang, J.; Li, G.; Ni, Z.; Yan, Y. Improve the gas-barrier and mechanical performance of epoxy resin by Co-modification from montmorillonite and polyethylene glycol. Int. J. Hydrogen Energy 2024, 65, 751–758. [Google Scholar] [CrossRef]
- Shokrani Havigh, R.; Mahmoudi Chenari, H. A comprehensive study on the effect of carbonization temperature on the physical and chemical properties of carbon fibers. Sci. Rep. 2022, 12, 10704. [Google Scholar] [CrossRef]
- Dagdag, O.; Kim, H. Cryogenic Impact on Carbon Fiber-Reinforced Epoxy Composites for Hydrogen Storage Vessels. J. Compos. Sci. 2024, 8, 459. [Google Scholar] [CrossRef]
- Fan, H.B.; Yuen, M.M.F. Material properties of the cross-linked epoxy resin compound predicted by molecular dynamics simulation. Polymer 2007, 48, 2174–2178. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, L.; Cui, L.; Zhang, Y.; Du, X. Molecular dynamics simulation of cross-linked epoxy resin and its interaction energy with graphene under two typical force fields. Comput. Mater. Sci. 2018, 143, 2400000. [Google Scholar] [CrossRef]
- Odegard, G.M.; Patil, S.U.; Deshpande, P.P.; Kanhaiya, K.; Winetrout, J.J.; Heinz, H.; Shah, S.P.; Maiaru, M. Molecular dynamics modeling of epoxy resins using the reactive interface force field. Macromolecules 2021, 54, 9815–9824. [Google Scholar] [CrossRef]
- Marx, D.; Hutter, J. Ab initio molecular dynamics: Basic theory and advanced methods. Phys. Today 2010, 63, 54–56. [Google Scholar]
- Allen, M.P. Introduction to molecular dynamics simulation. Comput. Soft Matter Synth. Polym. Proteins 2004, 23, 1–28. [Google Scholar]
- Tuckerman, M.E.; Martyna, G.J. Understanding modern molecular dynamics: Techniques and applications. J. Phys. Chem. B 2000, 104, 159–178. [Google Scholar] [CrossRef]
- Zheng, D.; Li, J.; Liu, B.; Yu, B.; Yang, Y.; Han, D.; Li, J.; Huang, Z. Molecular dynamics investigations into the hydrogen permeation mechanism of polyethylene pipeline material. J. Mol. Liq. 2022, 368, 120773. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, T.; Sun, S. Molecular mechanisms of hydrogen leakage through caprock in moisture and residual gas conditions: A molecular dynamics–Monte Carlo study. Phys. Fluids 2024, 36, 022024. [Google Scholar] [CrossRef]
- Hassanzadeh, A.; Sabzi, F. Prediction of CO2 and H2 solubility, diffusion, and permeability in MFI zeolite by molecular dynamics simulation. Struct. Chem. 2021, 32, 1641–1650. [Google Scholar] [CrossRef]
- Neyertz, S.; Brown, D. Influence of system size in molecular dynamics simulations of gas permeation in glassy polymers. Macromolecules 2004, 37, 10109–10122. [Google Scholar] [CrossRef]
- Cozmuta, I.; Blanco, M.; Goddard, W.A. Gas sorption and barrier properties of polymeric membranes from molecular dynamics and Monte Carlo simulations. J. Phys. Chem. B 2007, 111, 3151–3166. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Jagirdar, P.; Naserifar, S.; Sahimi, M. Molecular simulation study of gas solubility and diffusion in a polymer-boron nitride nanotube composite. J. Phys. Chem. B 2016, 120, 1273–1284. [Google Scholar] [CrossRef]
- Abbott, L.J.; Hart, K.E.; Colina, C.M. Polymatic: A generalized simulated polymerization algorithm for amorphous polymers. Ther. Chem. Acc. 2013, 132, 1–19. [Google Scholar] [CrossRef]
- Mozaffari, F.; Eslami, H.; Moghadasi, J. Molecular dynamics simulation of diffusion and penetration of gases in polystyrene. Polymer 2010, 51, 300–307. [Google Scholar] [CrossRef]
- Kamiya, Y.; Naito, Y.; Terada, A.K.; Mizoguchi, K.; Tsuboi, A. Volumetric Properties and Interaction Parameters of Dissolved Gases in Poly(dimethylsiloxane) and Polyethylene. Macromolecules 2000, 33, 3111–3119. [Google Scholar] [CrossRef]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of State by Fast Computing Machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]
- Boyd, R.H.; Pant, P.V.K. Molecular packing and diffusion in polyisobutylene. Macromolecules 2002, 24, 6325–6331. [Google Scholar] [CrossRef]
- Sun, J.; Li, S.; Stirner, T.; Chen, J.; Wang, D. Molecular dynamics simulation of energy exchanges during hydrogen collision with graphite sheets. J. Appl. Phys. 2010, 107, 207. [Google Scholar] [CrossRef]
- Willems, T.F.; Rycroft, C.H.; Kazi, M.; Meza, J.C.; Haranczyk, M. Algorithms and tools for high-throughput geometry-based analysis of crystalline porous materials. Microporous Mesoporous Mater. Off. J. Int. Zeolite Assoc. 2012, 149, 134–141. [Google Scholar] [CrossRef]
- Martin, R.L.; Smit, B.; Haranczyk, M. Addressing challenges of identifying geometrically diverse sets of crystalline porous materials. J. Chem. Inf. Model. 2011, 52, 308–318. [Google Scholar] [CrossRef]
- Pinheiro, M.; Martin, R.L.; Rycroft, C.H.; Jones, A.; Iglesia, E.; Haranczyk, M. Characterization and comparison of pore landscapes in crystalline porous materials. J. Mol. Graph. Model. 2013, 44, 208–219. [Google Scholar] [CrossRef] [PubMed]
- GB/T 1038.1-2022; Plastics—Film and Sheeting—Determination of Gas-Transmission Rate—Part 1: Differential-Pressure Methods. Standardization Administration of the People’s Republic of China: Beijing, China, 2022.
- Pan, F.; Peng, F.; Lu, L.; Wang, J.; Jiang, Z. Molecular simulation on penetrants diffusion at the interface region of organic–inorganic hybrid membranes. Chem. Eng. Sci. 2008, 63, 1072–1080. [Google Scholar] [CrossRef]
- Shan, Y.P.; Zeng, P.; Song, Y.H.; Gu, Y.T.; Galarneau, A.; Manero, M.H. Amino-modified hydrogen-bonding resin and its adsorption on berberine. Appl. Mech. Mater. 2014, 448, 145–149. [Google Scholar] [CrossRef]
- Liu, R. Investigation of adsorption and young’s modulus of epoxy resin–sand interfaces using molecular dynamics simulation. Appl. Sci. 2024, 14, 10383. [Google Scholar]
- Agapov, A.L.; Wang, Y.; Kunal, K.; Robertson, C.G.; Sokolov, A.P. Effect of polar interactions on polymer dynamics. Macromolecules 2012, 45, 8430–8437. [Google Scholar] [CrossRef]
- Van Rooyen, L.J.; Karger-Kocsis, J.; Vorster, O.C.; Kock, L.D. Helium gas permeability reduction of epoxy composite coatings by incorporation of glass flakes. J. Membr. Sci. 2013, 430, 203–210. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Y.C.; Bailey, C.G.; Istrate, O.M.; Li, Z.; Kinloch, I.A.; Budd, P.M. Quantification of gas permeability of epoxy resin composites with graphene nanoplatelets. Compos. Sci. Technol. 2019, 184, 107875. [Google Scholar] [CrossRef]
- Nielsen, L.E. Cross-linking–effect on physical properties of polymers. Polym. Rev. 1969, 3, 69–103. [Google Scholar] [CrossRef]
- Wan, X.; Demir, B.; An, M.; Walsh, T.R.; Yang, N. Thermal conductivities and mechanical properties of epoxy resin as a function of the degree of cross-linking. Int. J. Heat Mass Transf. 2021, 180, 121821. [Google Scholar] [CrossRef]
- Liu, Z.K.; Ågren, J.; Hillert, M. Application of the Le Chatelier principle on gas reactions. Fluid Phase Equilibria 1996, 121, 167–177. [Google Scholar] [CrossRef]
- Li, B.; Li, X.; Gao, W.; Jiang, Q. An effective scheme to determine surface energy and its relation with adsorption energy. Acta Mater. 2021, 212, 116895. [Google Scholar] [CrossRef]
- Podkościelna, B.; Wawrzkiewicz, M.; Klapiszewski, Ł. Synthesis, characterization and sorption ability of epoxy resin-based sorbents with amine groups. Polymers 2021, 13, 4139. [Google Scholar] [CrossRef]
- Valenti, G. Hydrogen liquefaction and liquid hydrogen storage. In Compendium of Hydrogen Energy; Woodhead Publishing: Sawston, UK, 2016; pp. 27–51. [Google Scholar]
- Ashcroft, N.W. The hydrogen liquids. J. Phys. Condens. Matter 2000, 12, A129. [Google Scholar] [CrossRef]
- Zhai, Y.; Zhu, X.; Chen, H.; Teng, G.; Feng, Z.; Qian, P.; Wang, J. Bubble defects and control of thermal curing of waterborne resin coatings. High Temp. Mater. Process. Int. Q. High-Technol. Plasma Process. 2024, 28, 55–67. [Google Scholar] [CrossRef]
- Sun, G.; Zuo, W.; Chen, D.; Luo, Q.; Pang, T.; Li, Q. On the effects of temperature on tensile behavior of carbon fiber reinforced epoxy laminates. Thin-Walled Struct. 2021, 164, 107769. [Google Scholar] [CrossRef]
- Yan, M.; Liu, Y.; Jiang, W.; Qin, W.; Yan, Y.; Wan, L.; Jiao, W.; Wang, R. Mechanism of matrix influencing the cryogenic mechanical property of carbon fibre reinforced epoxy resin composite. Compos. Commun. 2022, 33, 101220. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Degree of Crosslinking /% | D | S | P |
|---|---|---|---|
| 70 | |||
| 75 | |||
| 80 | |||
| 85 | |||
| 90 |
| Degree of Crosslinking /% | D | S | P |
|---|---|---|---|
| 70 | |||
| 75 | |||
| 80 | |||
| 85 | |||
| 90 |
| Sample | L | P | |
|---|---|---|---|
| A-1 | |||
| A-2 | |||
| A-3 | |||
| B-1 | |||
| B-2 | |||
| B-3 |
| System | Strain /% | P |
|---|---|---|
| 0 | ||
| 50 | ||
| PEA system | 100 | |
| 150 | ||
| 200 |
| System | Strain /% | P |
|---|---|---|
| 0 | ||
| 50 | ||
| DDM system | 100 | |
| 150 | ||
| 200 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, C.; Chen, H.; Xu, H.; Wu, Z.; Dong, X. Molecular Dynamics Simulation of Hydrogen Permeation Behavior in Epoxy Resin Systems. Polymers 2025, 17, 1755. https://doi.org/10.3390/polym17131755
Gao C, Chen H, Xu H, Wu Z, Dong X. Molecular Dynamics Simulation of Hydrogen Permeation Behavior in Epoxy Resin Systems. Polymers. 2025; 17(13):1755. https://doi.org/10.3390/polym17131755
Chicago/Turabian StyleGao, Chang, Hongzhi Chen, Hao Xu, Zhanjun Wu, and Xufeng Dong. 2025. "Molecular Dynamics Simulation of Hydrogen Permeation Behavior in Epoxy Resin Systems" Polymers 17, no. 13: 1755. https://doi.org/10.3390/polym17131755
APA StyleGao, C., Chen, H., Xu, H., Wu, Z., & Dong, X. (2025). Molecular Dynamics Simulation of Hydrogen Permeation Behavior in Epoxy Resin Systems. Polymers, 17(13), 1755. https://doi.org/10.3390/polym17131755