
New Insight into the Microstructure Changes and Molecular Mobility of Polyamides Exposed to H2S Scavengers
 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
3. Results
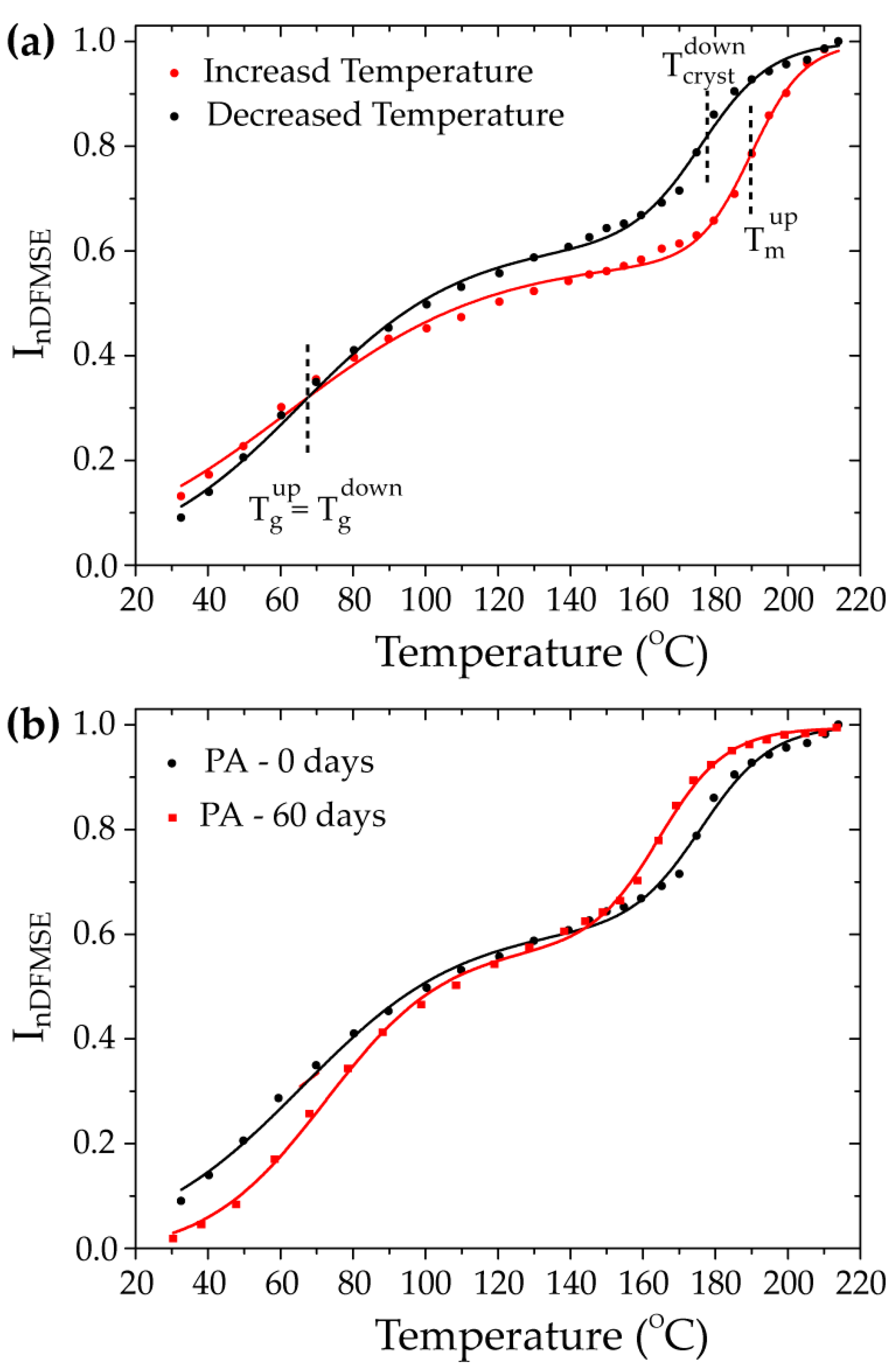
3.1. Effects of the H2S-SCVs on the Microstructure and Dynamics of PA
3.2. Changes in the PA Chemical Composition Due to Exposure to the H2S-SCVs
4. Discussions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Colucci, R.; Frulani, H.; Barbosa, P.; Günther, F.; Cavassin, P.; Couto Faria, G. Recent advances in modeling organic electrochemical transistors. Flex. Print. Electron. 2020, 5, 13001. [Google Scholar] [CrossRef]
- Kraft, A.; Grimsdale, A.C.; Holmes, A.B. Electroluminescent Conjugated Polymers—Seeing Polymers in a New Light. Angew. Chem.-Int. Ed. 1998, 37, 402–428. [Google Scholar] [CrossRef]
- Lukassen, T.V.; Gunnarsson, E.; Krenk, S.; Glejbøl, K.; Lyckegaard, A.; Berggreen, C. Tension-bending analysis of flexible pipe by a repeated unit cell finite element model. Mar. Struct. 2019, 64, 401–420. [Google Scholar] [CrossRef]
- Roseman, M.; Martin, R.; Morgan, G. Marine Applications of Advanced Fibre-Reinforced Composites; Elsevier Inc.: Amsterdam, The Netherlands, 2016; pp. 233–257. [Google Scholar]
- Romão, W.; Castro, E.V.R.; Filho, E.A.S.; Guimarães, R.C.L.; Silva, A.L.N.; Teixeira, S.C.S.; De Paoli, M.A.; De Sena, G.L. Ageing of polyamide 11 used in the manufacture of flexible piping. J. Appl. Polym. Sci. 2009, 114, 1777–1783. [Google Scholar] [CrossRef]
- Khalid, H.U.; Ismail, M.C.; Nosbi, N. Permeation Damage of Polymer Liner in Oil and Gas Pipelines: A Review. Polymers 2020, 12, 2307. [Google Scholar] [CrossRef]
- Perez-Pineiro, R.; Cruz-Perez, D.; Hoshowski, J.; Zhang, H.; Hendry, J. H2S Scavenger Tower Operational Efficiency Achieved Through Onsite Compositional Analysis. In Proceedings of the NACE—International Corrosion Conference Series 2018, Phoenix, AZ, USA, 15–19 April 2018. [Google Scholar]
- Benhabib, M.; Kleinman, S.L.; Peterman, M.C. Quantitative Analysis of Triazine-Based H2S Scavengers via Raman Spectroscopy. Ind. Eng. Chem. Res. 2021, 60, 15936–15941. [Google Scholar] [CrossRef]
- Portela, N.A.; Silva, S.R.C.; de Jesus, L.F.R.; Dalmaschio, G.P.; Sad, C.M.S.; Castro, E.V.R.; Morigaki, M.K.; Silva Filho, E.A.; Filgueiras, P.R. Spectroscopic evaluation of commercial H2S scavengers. Fuel 2018, 216, 681–685. [Google Scholar] [CrossRef]
- Perez, M.G.; Lima, A.P.; Moraes, T.B.; Chaves, E.G.; Ruiz, N.M.d.S.; dos Santos Teixeira, S.C.; Honorato, H.d.A.; de Menezes, S.M.C.; deAzevedo, E.R. 1H Time Domain NMR to probe microstructural and mobility changes in Polyamide 11 exposed to H2S scavengers. What type of information can be assessed? Polym. Degrad. Stab. 2022, 202, 110001. [Google Scholar] [CrossRef]
- Mordvinkin, A.; Saalwächter, K. Microscopic observation of the segmental orientation autocorrelation function for entangled and constrained polymer chains. J. Chem. Phys. 2017, 146, 094902. [Google Scholar] [CrossRef]
- Domingos, E.; Pereira, T.M.C.; De Castro, E.V.R.; Romão, W.; De Sena, G.L.; Guimarães, R.C.L. Monitorando a Degradação da Poliamida 11 (PA-11) via Espectroscopia na região do Infravermelho médio com Transformada de Fourier (FTIR). Polimeros 2013, 23, 37–41. [Google Scholar] [CrossRef]
- Deshoulles, Q.; Le Gall, M.; Dreanno, C.; Arhant, M.; Priour, D.; Le Gac, P.Y. Chemical coupling between oxidation and hydrolysis in polyamide 6—A key aspect in the understanding of microplastic formation. Polym. Degrad. Stab. 2022, 197, 109851. [Google Scholar] [CrossRef]
- Brette, M.M.; Holm, A.H.; Drozdov, A.D.; Christiansen, J.d.C. Pure Hydrolysis of Polyamides: A Comparative Study. Chemistry 2024, 6, 13–50. [Google Scholar] [CrossRef]
- Filgueiras, J.G.; Cobo, M.F.; Faria, G.C.; Moraes, T.B.; De Azevedo, E.R. NMR Methods for Characterization of Synthetic and Natural Polymers, 1st ed.; Zhang, R., Toshikazu, M., Sun, P., Eds.; Royal Society of Chemistry: Cambridge, UK, 2019; Volume 1, pp. 271–298. [Google Scholar] [CrossRef]
- Filgueiras, J.G.; da Silva, U.B.; Paro, G.; d’Eurydice, M.N.; Cobo, M.F.; deAzevedo, E.R. Dipolar filtered magic-sandwich-echoes as a tool for probing molecular motions using time domain NMR. J. Magn. Reson. 2017, 285, 47–54. [Google Scholar] [CrossRef]
- Saalwächter, K. Proton multiple-quantum NMR for the study of chain dynamics and structural constraints in polymeric soft materials. Prog. Nucl. Magn. Reson. Spectrosc. 2007, 51, 1–35. [Google Scholar] [CrossRef]
- Chassé, W.; Lang, M.; Sommer, J.U.; Saalwächter, K. Cross-link density estimation of PDMS networks with precise consideration of networks defects. Macromolecules 2012, 45, 899–912. [Google Scholar] [CrossRef]
- Saalwächter, K.; Ziegler, P.; Spyckerelle, O.; Haidar, B.; Vidal, A.; Sommer, J.U. H 1 multiple-quantum nuclear magnetic resonance investigations of molecular order distributions in poly (dimethylsiloxane) networks: Evidence for a linear mixing law in bimodal systems. J. Chem. Phys. 2003, 119, 3468–3482. [Google Scholar] [CrossRef]
- Papon, A.; Montes, H.; Hanafi, M.; Lequeux, F.; Guy, L.; Saalwächter, K. Glass-transition temperature gradient in nanocomposites: Evidence from nuclear magnetic resonance and differential scanning calorimetry. Phys. Rev. Lett. 2012, 108, 065702. [Google Scholar] [CrossRef] [PubMed]
- Baum, J.; Munowitz, M.; Garroway, A.N.; Pines, A. Multiple-quantum dynamics in solid state NMR. J. Chem. Phys. 1985, 83, 2015–2025. [Google Scholar] [CrossRef]
- Wu, P.; Siesler, H.W. The assignment of overtone and combination bands in the near infrared spectrum of polyamide 11. J. Near Infrared Spectrosc. 1999, 7, 65–76. [Google Scholar] [CrossRef]
- Wypych, G. Handbook of Plasticizers, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2023. [Google Scholar]
- Serpe, G.; Chaupart, N. Relaxation–structure relationship in bulk and plasticized polyamide 11. J. Polym. Sci. B Polym. Phys. 1996, 34, 2351–2365. [Google Scholar] [CrossRef]
- Behler, K.; Havel, M.; Gogotsi, Y. New solvent for polyamides and its application to the electrospinning of polyamides 11 and 12. Polymer 2007, 48, 6617–6621. [Google Scholar] [CrossRef]
- Hsin, H.Y. Crystal phase transformations in nylon 11. Mater. Chem. Phys. 1998, 56, 289–293. [Google Scholar]
- Johnson, C.G.; Mathias, L.J. Solid-state NMR characterization of copolymers of nylon 11 and nylon 12. Solid. State Nucl. Magn. Reson. 1997, 8, 161–171. [Google Scholar] [CrossRef]
- Mota, G.P.; da Silva, A.H.M.; da, F.T.; da Rocha, E.B.D.; da Silva, A.L.N. Evaluation of aging process of PA11 from offshore flexible pipe pressure barrier. Appl. Ocean Res. 2024, 144, 103917. [Google Scholar] [CrossRef]
- Mazan, T.; Jørgensen, J.K.; Echtermeyer, A. Aging of polyamide 11. Part 3: Multiscale model predicting the mechanical properties after hydrolytic degradation. J. Appl. Polym. Sci. 2015, 132, 42792. [Google Scholar] [CrossRef]
- Celina, M.C. Polymer Degradation and Stability 2013; Elsevier: Amsterdam, The Netherlands, 2013; Volume 98, pp. 2419–2429. [Google Scholar]
- Dias, F.G.A.; Veiga, A.G.; Gomes, A.P.A.C.P.; Rocco, M.L.M.; da Costa, M.F. The chemical impact of oil fluids in the morphology and thermal properties of the polyamide 11 used in flexible lines. Discov. Mech. Eng. 2024, 3, 3–13. [Google Scholar] [CrossRef]
- Mazan, T.; Berggren, R.; Jørgensen, J.K.; Echtermeyer, A.J. Aging of polyamide 11. Part 1: Evaluating degradation by thermal, mechanical, and viscometric analysis. Appl. Polym. Sci. 2015, 132, 6249–6260. [Google Scholar] [CrossRef]
- Okamba-Diogo, O.; Richaud, E.; Verdu, J.; Fernagut, F.; Guilment, J.; Fayolle, B. Investigation of polyamide 11 embrittlement during oxidative degradation. Polymer 2016, 82, 49–56. [Google Scholar] [CrossRef]
- Okamba-Diogo, O.; Richaud, E.; Verdu, J.; Fernagut, F.; Guilment, J.; Fayolle, B. Molecular and macromolecular structure changes in polyamide 11 during thermal oxidation–Kinetic modeling. Polym. Degrad. Stab. 2014, 108, 123–132. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | ||||||
|---|---|---|---|---|---|---|
| PA—0 days | 66 ± 2 | 176 ± 1 | 22 ± 1 | 10 ± 1 | 0.62 ± 0.01 | 0.38 ± 0.01 |
| PA—60 days | 74 ± 1 | 165 ± 1 | 17 ± 1 | 9 ± 1 | 0.59 ± 0.01 | 0.41 ± 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazari, M.P.d.; Silva, A.H.M.d.F.T.d.; Garcia, R.H.d.S.; Teixeira, S.C.d.S.; Oliveira, J.E.d.; Chaves, É.G.; Nunes, L.A.d.O.; Honorato, H.d.A.; Menezes, S.M.C.d.; Lima, A.P.; et al. New Insight into the Microstructure Changes and Molecular Mobility of Polyamides Exposed to H2S Scavengers. Polymers 2025, 17, 1634. https://doi.org/10.3390/polym17121634
Lazari MPd, Silva AHMdFTd, Garcia RHdS, Teixeira SCdS, Oliveira JEd, Chaves ÉG, Nunes LAdO, Honorato HdA, Menezes SMCd, Lima AP, et al. New Insight into the Microstructure Changes and Molecular Mobility of Polyamides Exposed to H2S Scavengers. Polymers. 2025; 17(12):1634. https://doi.org/10.3390/polym17121634
Chicago/Turabian StyleLazari, Marina Perassoli de, Antonio Henrique Monteiro da Fonseca Thomé da Silva, Rodrigo Henrique dos Santos Garcia, Sylvia Correa dos Santos Teixeira, João Eduardo de Oliveira, Érica Gervasoni Chaves, Luiz Antônio de Oliveira Nunes, Hercílio de Angeli Honorato, Sonia Maria Cabral de Menezes, Aline Pinde Lima, and et al. 2025. "New Insight into the Microstructure Changes and Molecular Mobility of Polyamides Exposed to H2S Scavengers" Polymers 17, no. 12: 1634. https://doi.org/10.3390/polym17121634
APA StyleLazari, M. P. d., Silva, A. H. M. d. F. T. d., Garcia, R. H. d. S., Teixeira, S. C. d. S., Oliveira, J. E. d., Chaves, É. G., Nunes, L. A. d. O., Honorato, H. d. A., Menezes, S. M. C. d., Lima, A. P., Chinelatto Junior, L. S., & de Azevedo, E. R. (2025). New Insight into the Microstructure Changes and Molecular Mobility of Polyamides Exposed to H2S Scavengers. Polymers, 17(12), 1634. https://doi.org/10.3390/polym17121634