
Tailoring Epoxy Network Architecture and Stiffness-Toughness Balance Using Competitive Short- and Long-Chain Curing Agents: A Multiscale Simulation Study


Abstract

1. Introduction
2. Materials and Methods
2.1. Multiscale Simulation Models for Polycondensation Process
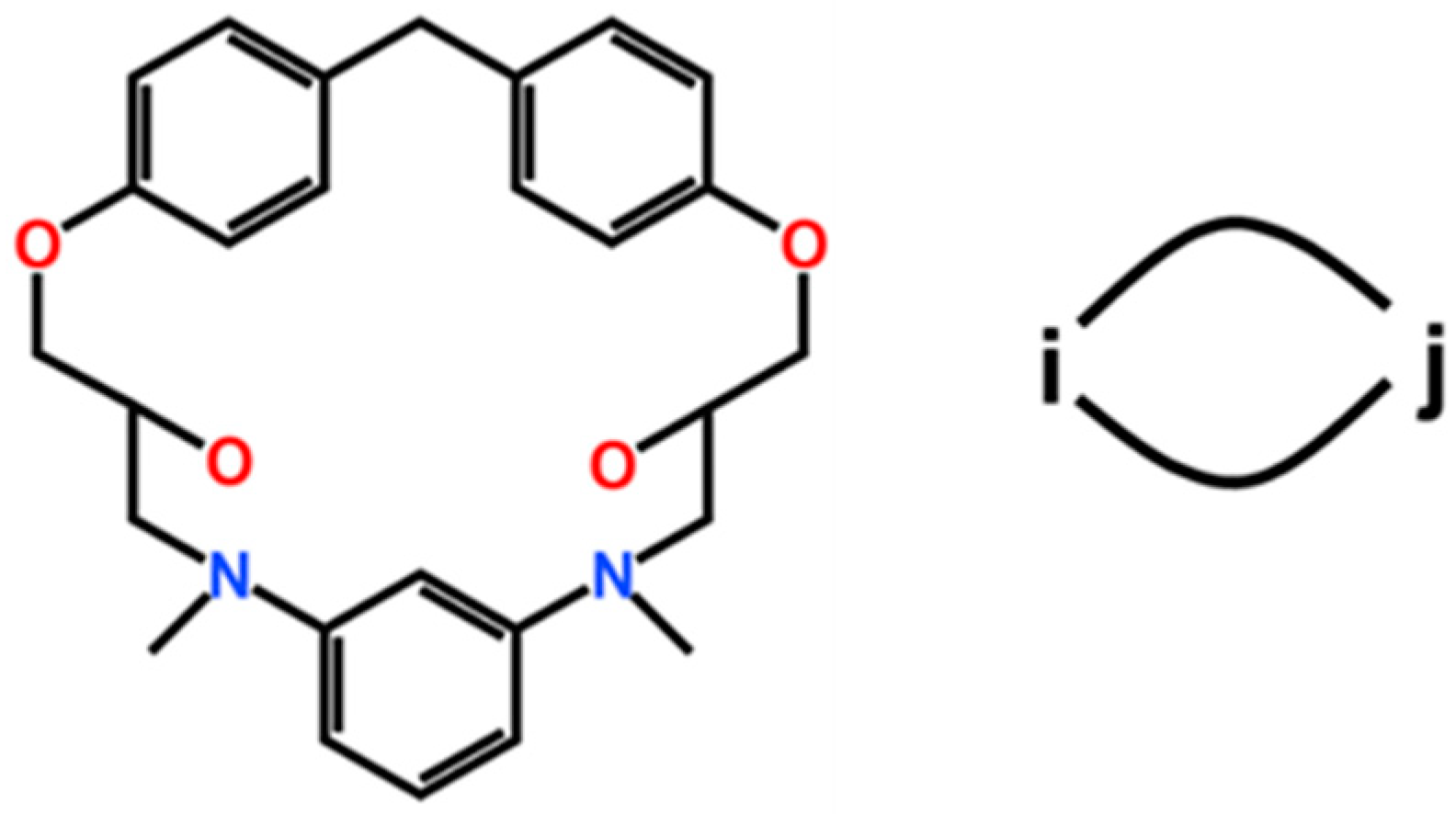
2.1.1. CG Model
2.1.2. Simulated Polycondensation Procedure
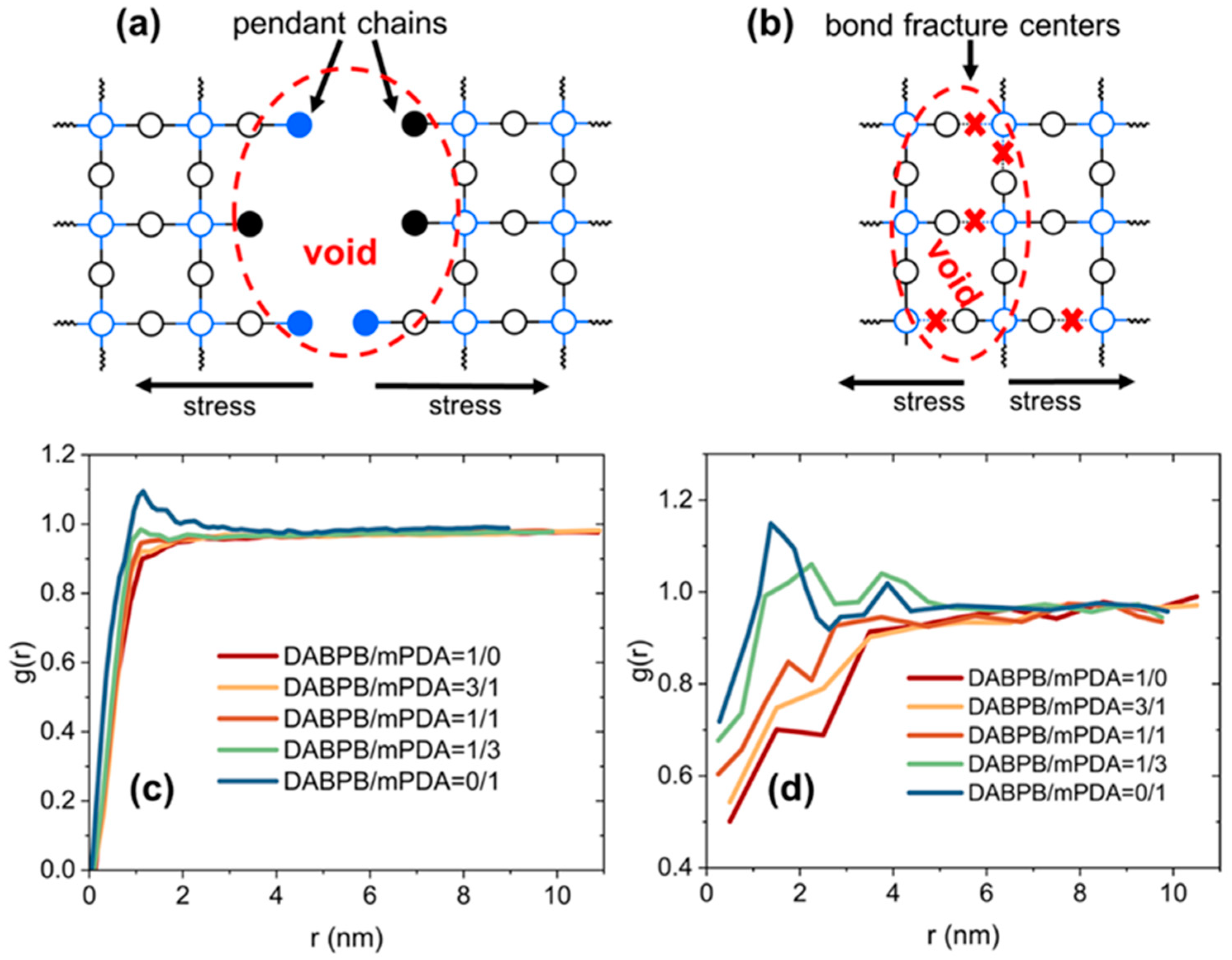
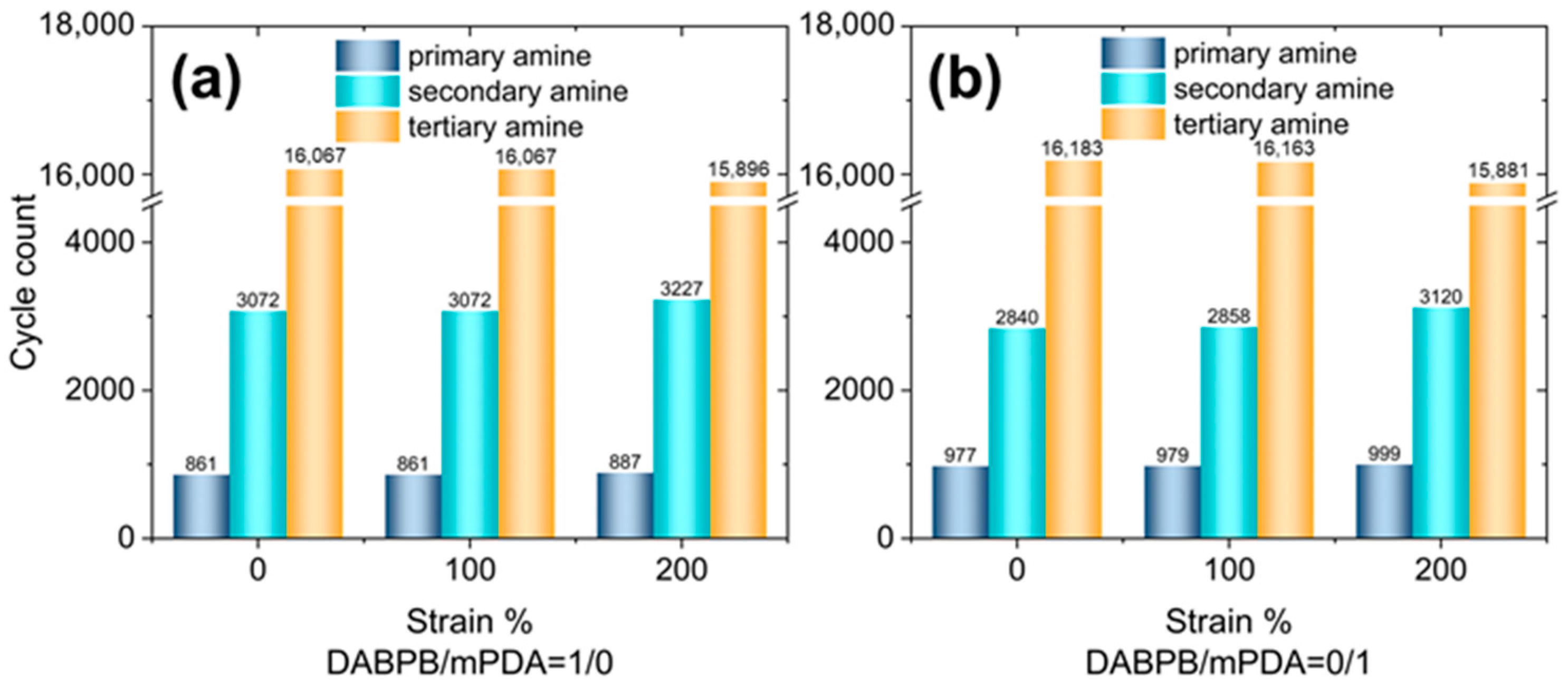
3. Results and Discussion
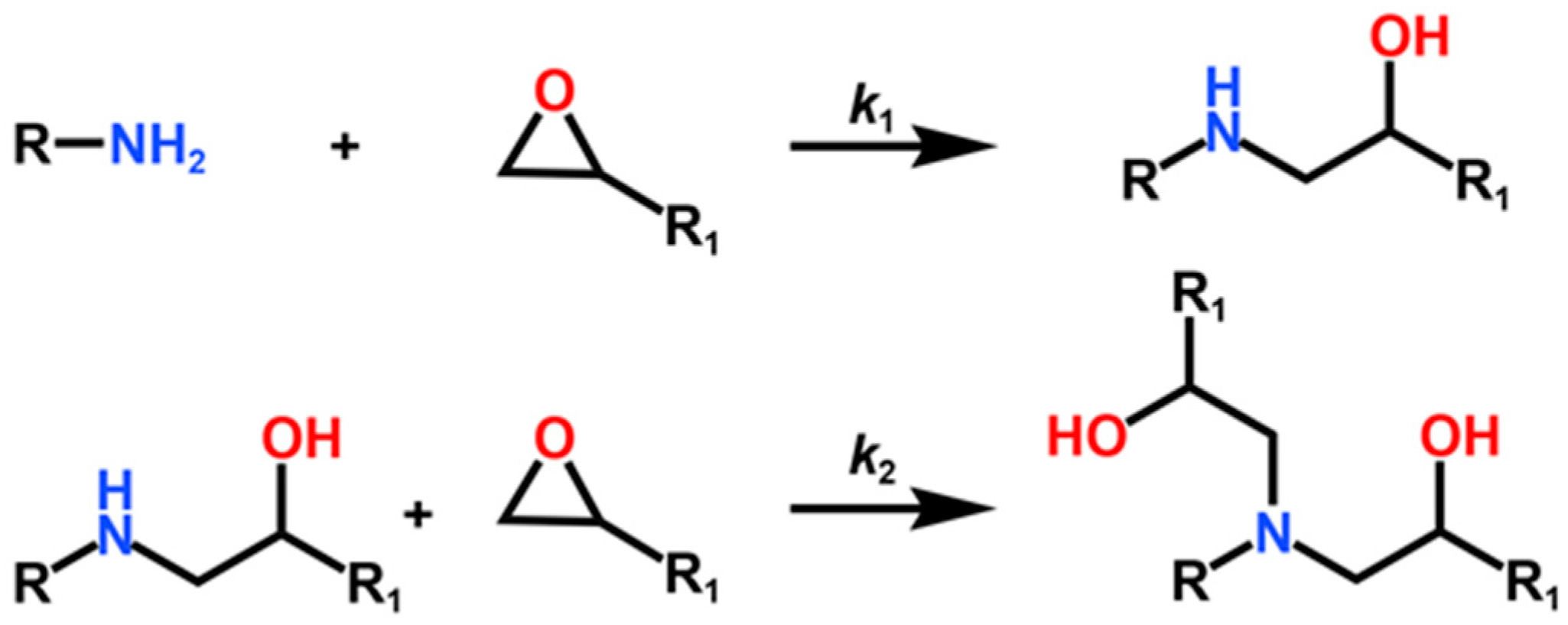
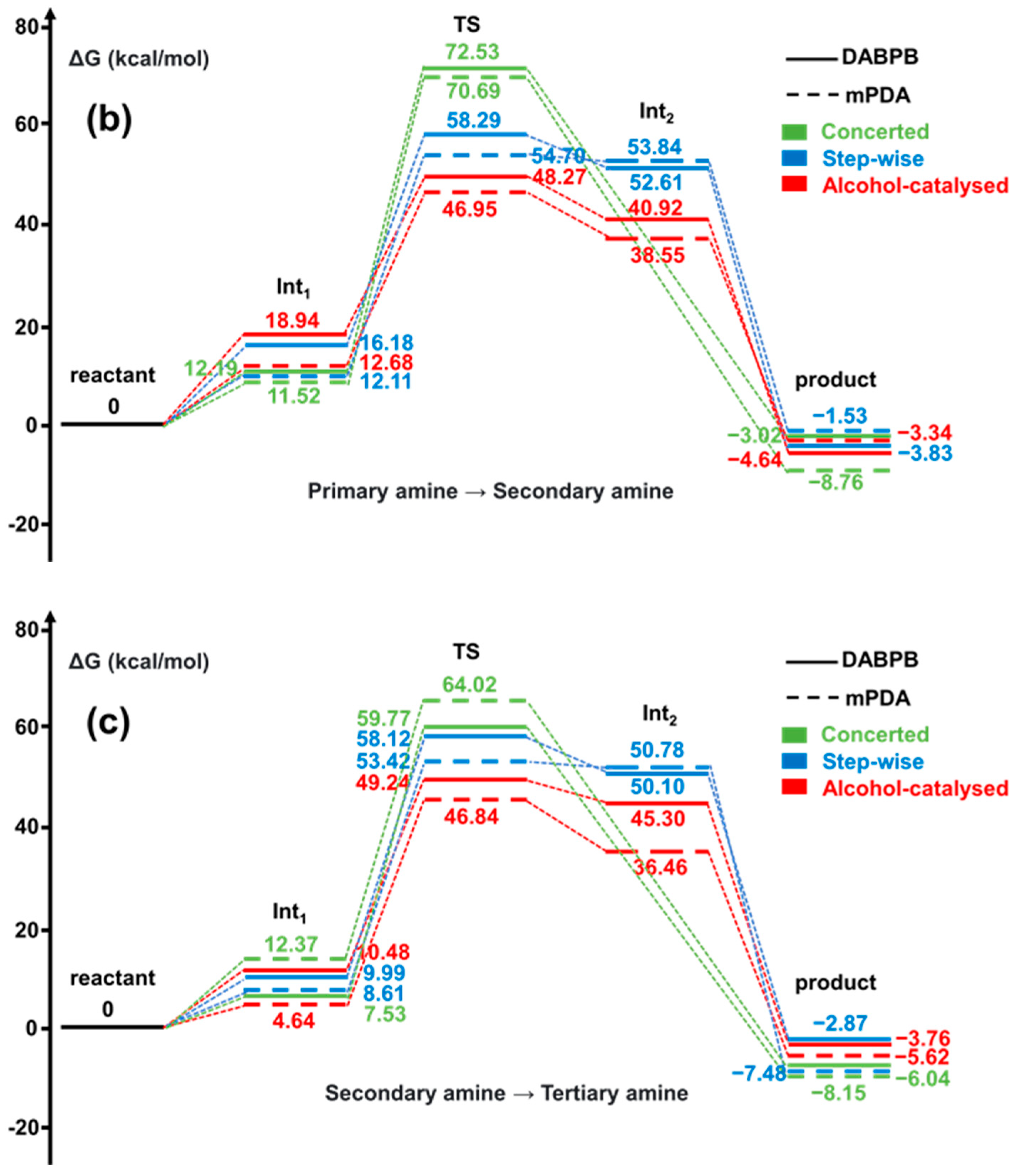
3.1. Competing Reactions in the Epoxy Resin Curing Process and Rate-Determining Steps
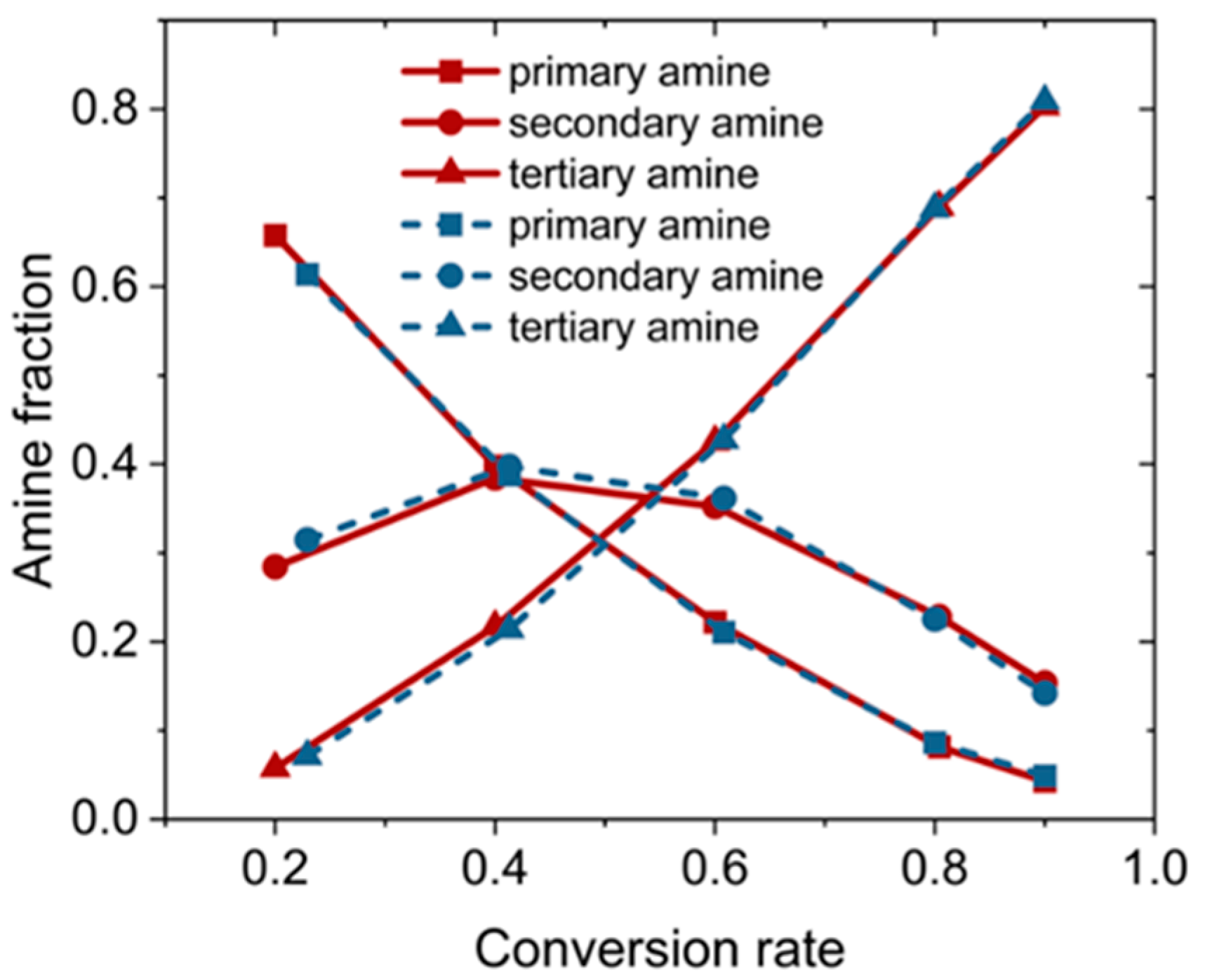
3.2. Simulated Curing of Epoxy Resin
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, B.; Wu, Q.; Li, J.; Lin, K.; Chen, D.; Zhou, C.; Wu, T.; Luo, X.; Liu, Y. A novel and green method to synthesize a epoxidized biomass eucommia gum as the nanofiller in the epoxy composite coating with excellent anticorrosive performance. Chem. Eng. J. 2020, 379, 122323. [Google Scholar] [CrossRef]
- Han, J.; Du, G.; Gao, W.; Bai, H. An anisotropically high thermal conductive boron nitride/epoxy composite based on nacre-mimetic 3D network. Adv. Funct. Mater. 2019, 29, 1900412. [Google Scholar] [CrossRef]
- Verma, C.; Olasunkanmi, L.O.; Akpan, E.D.; Quraishi, M.; Dagdag, O.; El Gouri, M.; Sherif, E.-S.M.; Ebenso, E.E. Epoxy resins as anticorrosive polymeric materials: A review. React. Funct. Polym. 2020, 156, 104741. [Google Scholar] [CrossRef]
- Zhong, M.; Wang, R.; Kawamoto, K.; Olsen, B.D.; Johnson, J.A. Quantifying the impact of molecular defects on polymer network elasticity. Science 2016, 353, 1264–1268. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Chen, X.; Yuk, H.; Lin, S.; Liu, X.; Parada, G. Soft materials by design: Unconventional polymer networks give extreme properties. Chem. Rev. 2021, 121, 4309–4372. [Google Scholar] [CrossRef] [PubMed]
- Hodgkin, J.; Simon, G.P.; Varley, R.J. Thermoplastic toughening of epoxy resins: A critical review. Polym. Adv. Technol. 1998, 9, 3–10. [Google Scholar] [CrossRef]
- Radue, M.S.; Jensen, B.D.; Gowtham, S.; Klimek-McDonald, D.R.; King, J.A.; Odegard, G.M. Comparing the mechanical response of di-, tri-, and tetra-functional resin epoxies with reactive molecular dynamics. J. Polym. Sci. Part B Polym. Phys. 2018, 56, 255–264. [Google Scholar] [CrossRef]
- Lee, A.; McKenna, G.B. Effect of crosslink density on physical ageing of epoxy networks. Polymer 1988, 29, 1812–1817. [Google Scholar] [CrossRef]
- Grillet, A.C.; Galy, J.; Gérard, J.-F.; Pascault, J.-P. Mechanical and viscoelastic properties of epoxy networks cured with aromatic diamines. Polymer 1991, 32, 1885–1891. [Google Scholar] [CrossRef]
- Gavrilov, A.A.; Komarov, P.V.; Khalatur, P.G. Thermal properties and topology of epoxy networks: A multiscale simulation methodology. Macromolecules 2015, 48, 206–212. [Google Scholar] [CrossRef]
- Kallivokas, S.V.; Sgouros, A.P.; Theodorou, D.N. Molecular dynamics simulations of EPON-862/DETDA epoxy networks: Structure, topology, elastic constants, and local dynamics. Soft Matter 2019, 15, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, M.; Lu, Z.-Y.; Zhang, Z.-G.; Sun, C.-C.; Cui, T. Multiscale simulation study on the curing reaction and the network structure in a typical epoxy system. Macromolecules 2011, 44, 8650–8660. [Google Scholar] [CrossRef]
- Lange, F.; Schwenke, K.; Kurakazu, M.; Akagi, Y.; Chung, U.-I.; Lang, M.; Sommer, J.-U.; Sakai, T.; Saalwächter, K. Connectivity and structural defects in model hydrogels: A combined proton NMR and Monte Carlo simulation study. Macromolecules 2011, 44, 9666–9674. [Google Scholar] [CrossRef]
- Saalwächter, K.; Seiffert, S. Dynamics-based assessment of nanoscopic polymer-network mesh structures and their defects. Soft Matter 2018, 14, 1976–1991. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, K.; Zhong, M.; Wang, R.; Olsen, B.D.; Johnson, J.A. Loops versus branch functionality in model click hydrogels. Macromolecules 2015, 48, 8980–8988. [Google Scholar] [CrossRef]
- Wang, J.; Lin, T.-S.; Gu, Y.; Wang, R.; Olsen, B.D.; Johnson, J.A. Counting secondary loops is required for accurate prediction of end-linked polymer network elasticity. ACS Macro Lett. 2018, 7, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Woo, J.; Cok, A.M.; Wang, M.; Olsen, B.D.; Johnson, J.A. Counting primary loops in polymer gels. Proc. Natl. Acad. Sci. USA 2012, 109, 19119–19124. [Google Scholar] [CrossRef]
- Shundo, A.; Yamamoto, S.; Tanaka, K. Network formation and physical properties of epoxy resins for future practical applications. JACS Au 2022, 2, 1522–1542. [Google Scholar] [CrossRef]
- Zhang, X.-Z.; Lu, Z.-Y.; Qian, H.-J. A perspective on the dynamics properties in polymer nanocomposites. Chin. J. Polym. Sci. 2023, 41, 1355–1360. [Google Scholar] [CrossRef]
- Gao, H.; Shi, R.; Zhu, Y.; Qian, H.; Lu, Z. Coarse-grained dynamics simulation in polymer systems: From structures to material properties. Chem. Res. Chin. Univ. 2022, 38, 653–670. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Liu, H.; Zhu, Y.L.; Lu, Z.Y.; Müller-Plathe, F. A kinetic chain growth algorithm in coarse-grained simulations. J. Comput. Chem. 2016, 37, 2634–2646. [Google Scholar] [CrossRef]
- Karimi-Varzaneh, H.A.; Qian, H.J.; Chen, X.; Carbone, P.; Müller-Plathe, F. IBIsCO: A molecular dynamics simulation package for coarse-grained simulation. J. Comput. Chem. 2011, 32, 1475–1487. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.-J.; Carbone, P.; Chen, X.; Karimi-Varzaneh, H.A.; Liew, C.C.; Müller-Plathe, F. Temperature-transferable coarse-grained potentials for ethylbenzene, polystyrene, and their mixtures. Macromolecules 2008, 41, 9919–9929. [Google Scholar] [CrossRef]
- Xie, Z.; Chai, D.; Wang, Y.; Tan, H. Directly modifying the nonbonded potential based on the standard iterative boltzmann inversion method for coarse-grained force fields. J. Phys. Chem. B 2016, 120, 11834–11844. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Aramoon, A.; Breitzman, T.D.; Woodward, C.; El-Awady, J.A. Coarse-grained molecular dynamics study of the curing and properties of highly cross-linked epoxy polymers. J. Phys. Chem. B 2016, 120, 9495–9505. [Google Scholar] [CrossRef]
- Yamamoto, S.; Ida, R.; Aoki, M.; Kuwahara, R.; Shundo, A.; Tanaka, K. Formation mechanism of a heterogeneous network in epoxy resins. Macromolecules 2023, 56, 3913–3921. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695. [Google Scholar] [CrossRef]
- Ma, S.; Chen, P.; Xu, J.; Xiong, X. Molecular dynamics simulations of key physical properties and microstructure of epoxy resin cured with different curing agents. J. Mater. Sci. 2022, 57, 1123–1133. [Google Scholar] [CrossRef]
- Yang, S.; Cui, Z.; Qu, J. A coarse-grained model for epoxy molding compound. J. Phys. Chem. B 2014, 118, 1660–1669. [Google Scholar] [CrossRef]
- Zhu, Y.L.; Liu, H.; Li, Z.W.; Qian, H.J.; Milano, G.; Lu, Z.Y. GALAMOST: GPU-accelerated large-scale molecular simulation toolkit. J. Comput. Chem. 2013, 34, 2197–2211. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, J.-E.; Rondan, N.G.; Huynh, L.K.; Pham, H.; Marks, M.; Truong, T.N. Theoretical study on mechanisms of the epoxy-amine curing reaction. Macromolecules 2007, 40, 4370–4377. [Google Scholar] [CrossRef]
- Zhao, Y.; Kikugawa, G.; Kawagoe, Y.; Shirasu, K.; Kishimoto, N.; Xi, Y.; Okabe, T. Uncovering the mechanism of size effect on the thermomechanical properties of highly cross-linked epoxy resins. J. Phys. Chem. B 2022, 126, 2593–2607. [Google Scholar] [CrossRef]
- Zihan, W.; Peibin, K.; Tianyu, W.; Dongli, C.; Xiaoping, Y.; Gang, S. Atomistic understanding of cross-linking network in different epoxy resin: Effect of loop structure. Polymer 2022, 243, 124629. [Google Scholar] [CrossRef]
- Shi, R.; Qian, H.-J.; Lu, Z.-Y. Interfacial tuning of the cavitation and strain-softening behavior of polymer/nanoparticle composites in the glassy state. Macromolecules 2019, 52, 7353–7360. [Google Scholar] [CrossRef]
- Shi, R.; Qian, H.-J.; Lu, Z.-Y. Tuning cavitation and crazing in polymer nanocomposite glasses containing bimodal grafted nanoparticles at the nanoparticle/polymer interface. Phys. Chem. Chem. Phys. 2019, 21, 7115–7126. [Google Scholar] [CrossRef]
- Frisch, M. Gaussian 09, Revision D. 01; Gaussian. Inc.: Wallingford, UK, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J. Chem. Phys. 1992, 96, 2155–2160. [Google Scholar] [CrossRef]
- McCormick, T.M.; Bridges, C.R.; Carrera, E.I.; DiCarmine, P.M.; Gibson, G.L.; Hollinger, J.; Kozycz, L.M.; Seferos, D.S. Conjugated polymers: Evaluating DFT methods for more accurate orbital energy modeling. Macromolecules 2013, 46, 3879–3886. [Google Scholar] [CrossRef]
- Fukui, K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Eyring, H. The activated complex in chemical reactions. J. Chem. Phys. 1935, 3, 107–115. [Google Scholar] [CrossRef]
- Pechukas, P. Transition state theory. Annu. Rev. Phys. Chem. 1981, 32, 159–177. [Google Scholar] [CrossRef]
- Truhlar, D.G.; Garrett, B.C.; Klippenstein, S.J. Current status of transition-state theory. J. Phys. Chem. 1996, 100, 12771–12800. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling exascale software challenges in molecular dynamics simulations with GROMACS. In Proceedings of the Solving Software Challenges for Exascale: International Conference on Exascale Applications and Software, EASC 2014, Stockholm, Sweden, 2–3 April 2014; Revised Selected Papers 2, 2015. Springer: Berlin/Heidelberg, Germany, 2014; pp. 3–27. [Google Scholar]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Träskelin, P.; Kuhl, T.; Faller, R. Molecular dynamics simulations of polystyrene brushes in dry conditions and in toluene solution. Phys. Chem. Chem. Phys. 2009, 11, 11324–11332. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Martoňák, R.; Laio, A.; Parrinello, M. Predicting crystal structures: The Parrinello-Rahman method revisited. Phys. Rev. Lett. 2003, 90, 075503. [Google Scholar] [CrossRef] [PubMed]


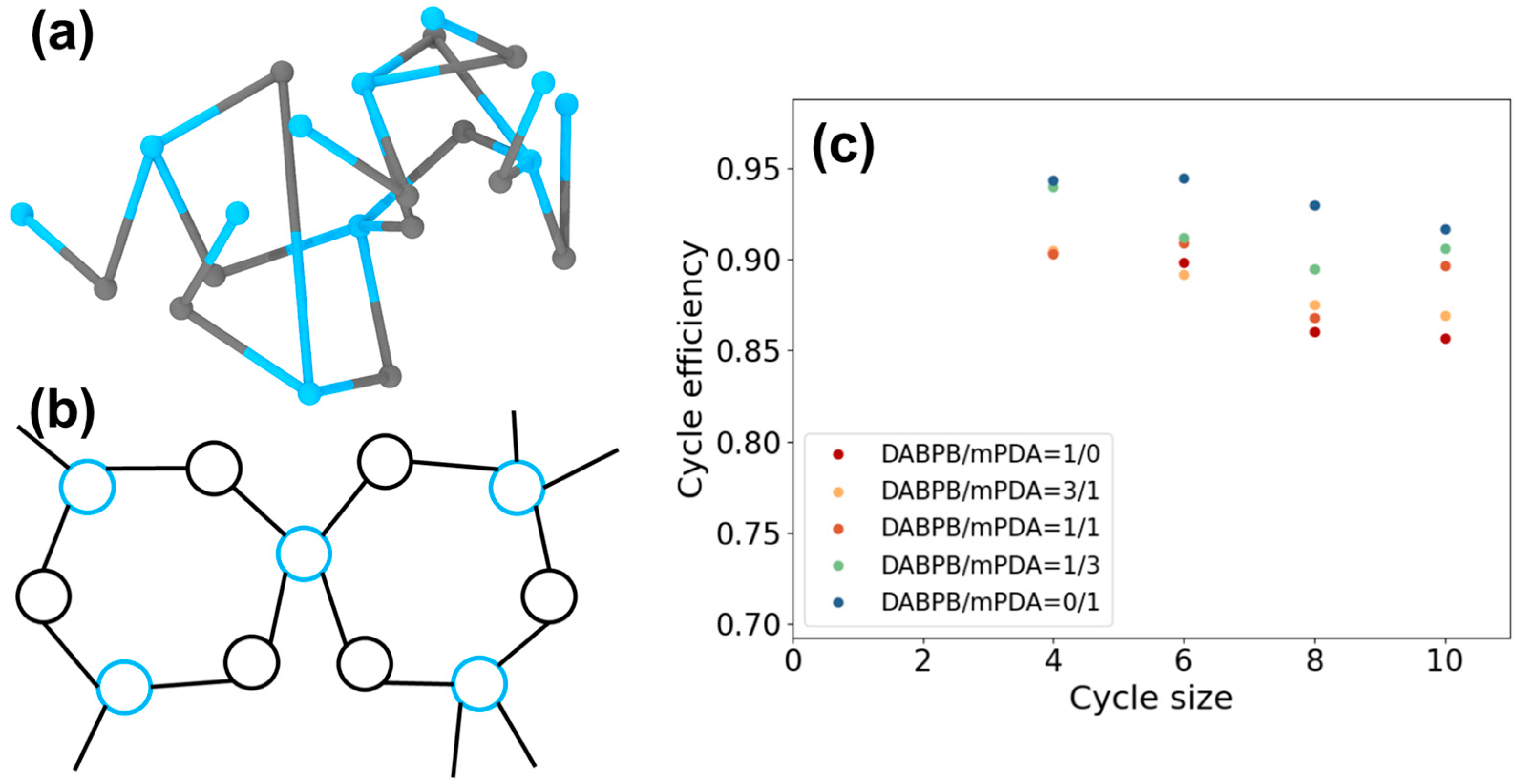
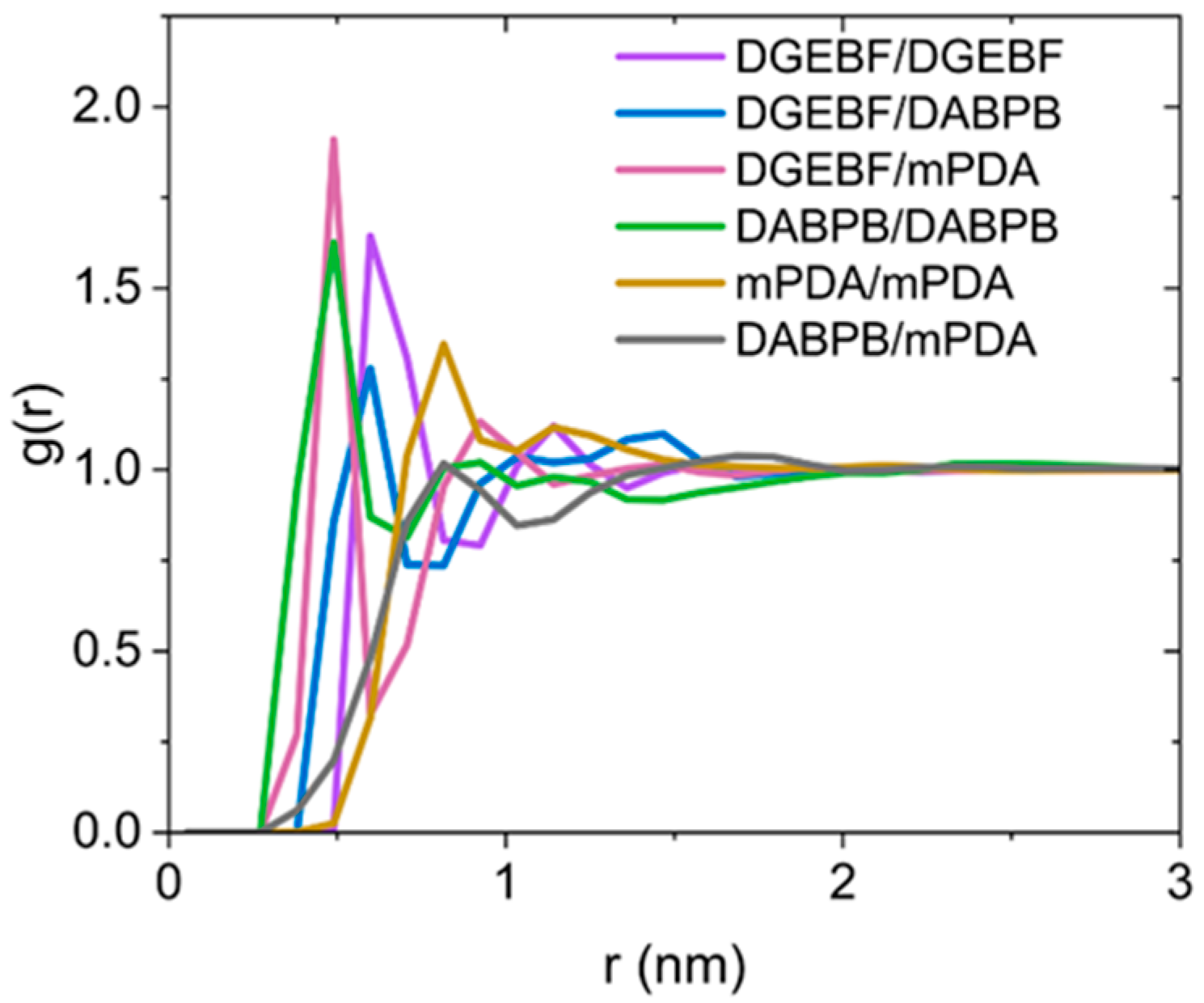
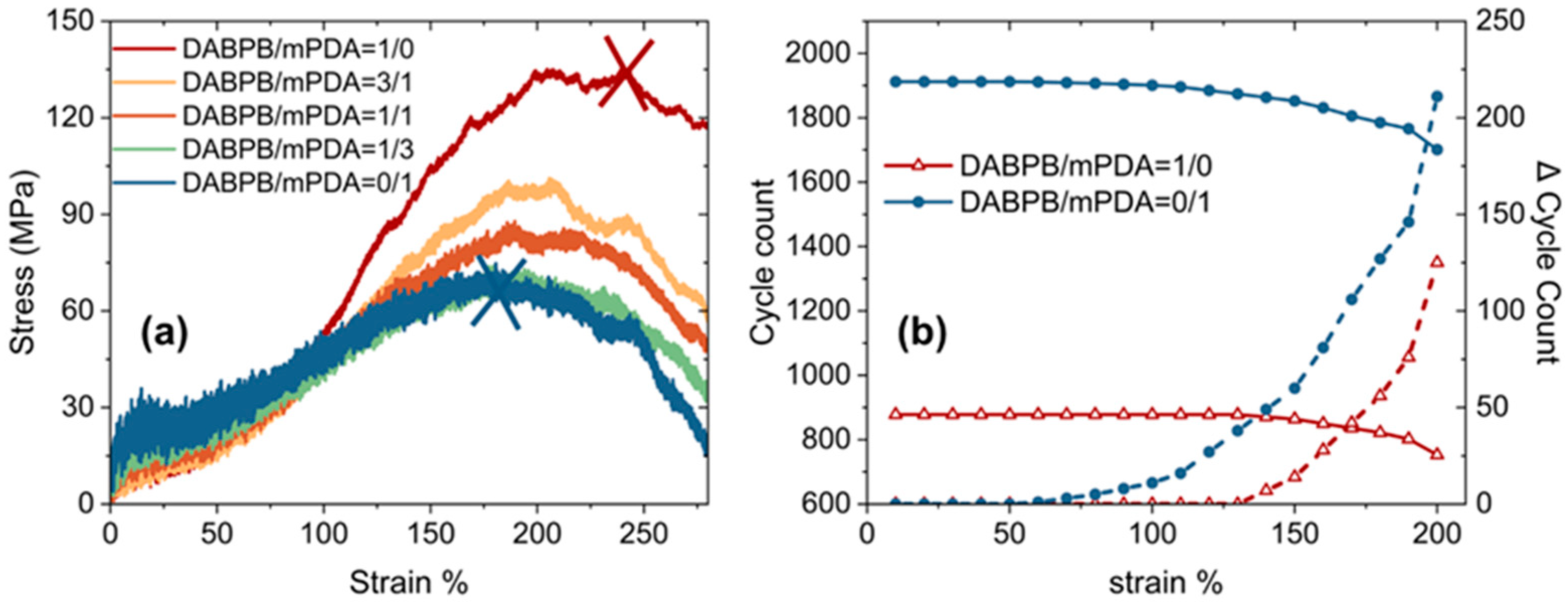
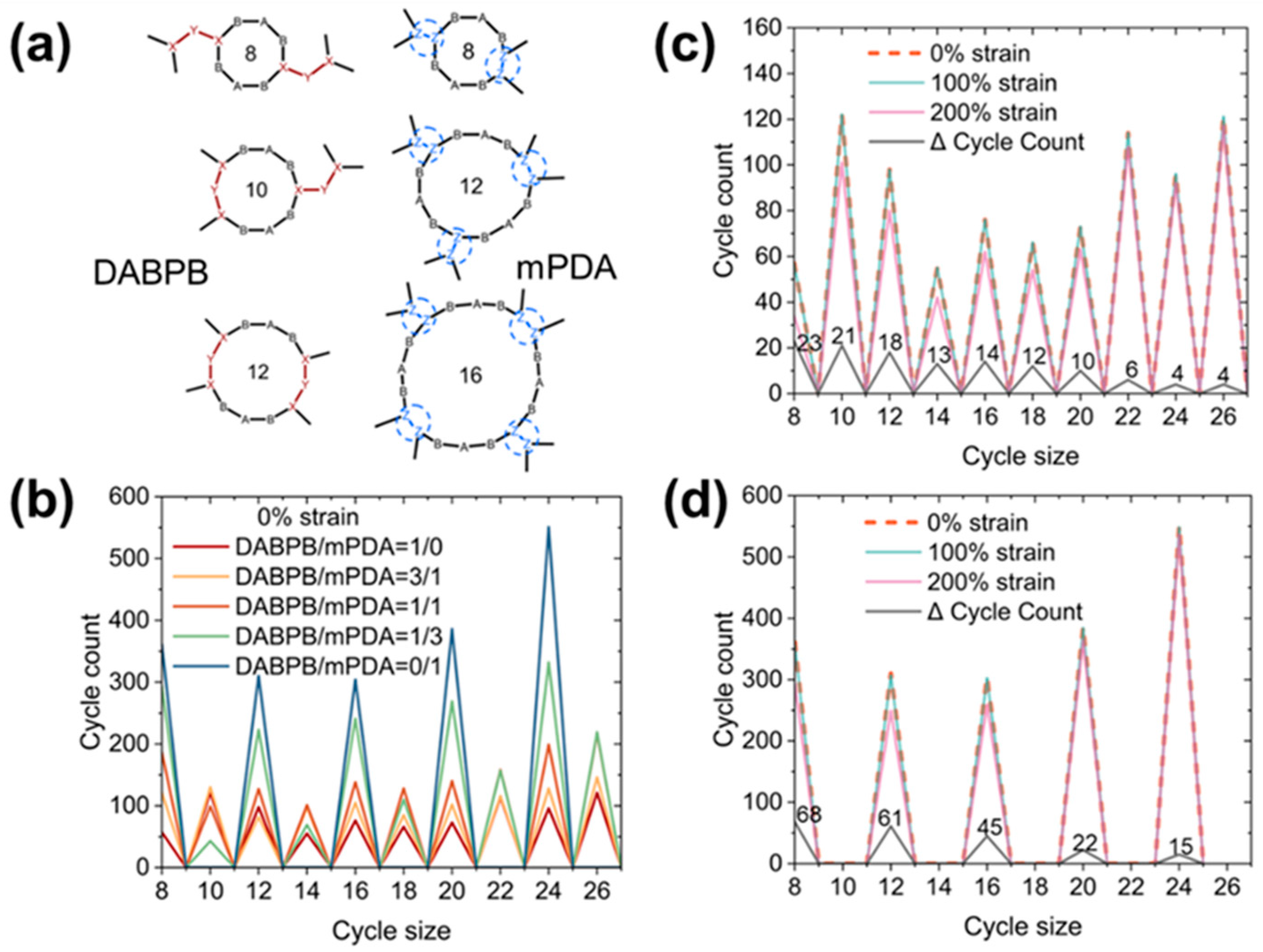


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ReactionType | Gibbs Free Energy Barrier (kcal/mol) | Rate Constant (s−1Lmol−1) |
|---|---|---|
| DABPB/Primary amine → Secondary amine | 48.27 | 1.90 × 10−7 |
| DABPB/Secondary amine → Tertiary amine | 49.24 | 7.05 × 10−8 |
| mPDA/Primary amine → Secondary amine | 46.95 | 7.33 × 10−7 |
| mPDA/Secondary amine → Tertiary amine | 46.84 | 8.09 × 10−7 |
| DABPB/mPDA | 1/0 | 3/1 | 1/1 | 1/3 | 0/1 |
|---|---|---|---|---|---|
| residual unreacted monomers | 1.83% | 1.48% | 1.43% | 1.27% | 1.17% |
| pendant chains (single-reactive molecules) | 14.97% | 15.27% | 14.92% | 14.57% | 10.82% |
| double bonds | 0.97% | 1.12% | 1.28% | 1.59% | 2.72% |
| Molar Ratio of Curing Agent Compositions (DABPB/mPDA) | Young’s Modulus (MPa) | Yield Stress (MPa) | Tensile Strength (MPa) | Toughness (J/m3) |
|---|---|---|---|---|
| 1/0 | 40.71 (±0.58) | 8.69 (±0.26) | 134.40 (±0.95) | 120.74 (±0.50) |
| 3/1 | 44.52 (±0.44) | 14.90 (±0.19) | 98.11 (±1.09) | 97.13 (±1.07) |
| 1/1 | 76.49 (±0.47) | 17.71 (±0.43) | 85.87 (±1.51) | 92.40 (±0.20) |
| 1/3 | 87.20 (±0.38) | 23.75 (±0.29) | 71.37 (±1.14) | 86.44 (±1.01) |
| 0/1 | 172.54 (±0.55) | 31.58 (0.47) | 69.15 (±0.29) | 83.15 (±0.89) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, Z.; Li, Y.; Huang, R.; Zhang, X.; Li, M.; Liu, D.; Shi, R.; Zhu, X.; Mu, J.; Qian, H. Tailoring Epoxy Network Architecture and Stiffness-Toughness Balance Using Competitive Short- and Long-Chain Curing Agents: A Multiscale Simulation Study. Polymers 2025, 17, 1297. https://doi.org/10.3390/polym17101297
Dong Z, Li Y, Huang R, Zhang X, Li M, Liu D, Shi R, Zhu X, Mu J, Qian H. Tailoring Epoxy Network Architecture and Stiffness-Toughness Balance Using Competitive Short- and Long-Chain Curing Agents: A Multiscale Simulation Study. Polymers. 2025; 17(10):1297. https://doi.org/10.3390/polym17101297
Chicago/Turabian StyleDong, Zhiyong, Yuqing Li, Renhai Huang, Xuze Zhang, Mingyang Li, Duo Liu, Rui Shi, Xuanbo Zhu, Jianxin Mu, and Hujun Qian. 2025. "Tailoring Epoxy Network Architecture and Stiffness-Toughness Balance Using Competitive Short- and Long-Chain Curing Agents: A Multiscale Simulation Study" Polymers 17, no. 10: 1297. https://doi.org/10.3390/polym17101297
APA StyleDong, Z., Li, Y., Huang, R., Zhang, X., Li, M., Liu, D., Shi, R., Zhu, X., Mu, J., & Qian, H. (2025). Tailoring Epoxy Network Architecture and Stiffness-Toughness Balance Using Competitive Short- and Long-Chain Curing Agents: A Multiscale Simulation Study. Polymers, 17(10), 1297. https://doi.org/10.3390/polym17101297