
Deep Eutectic Solvent Assisted Mechano-Enzymatic Preparation for Reprocessable Hot-Melting Starch: A Comprehensive Analysis of Molecular Structure and Thermal Properties

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. The Preparation of Deep Eutectic Solvent (DES)
2.3. The Preparation of Hot-Melting Starch (HMS)
2.4. The Preparation of Hot-Melting Starch/Sisal Fiber/PCL Composites
2.5. Characterization
2.6. Mechanical Property Tests
2.7. Statistical Analysis
3. Results and Discussions
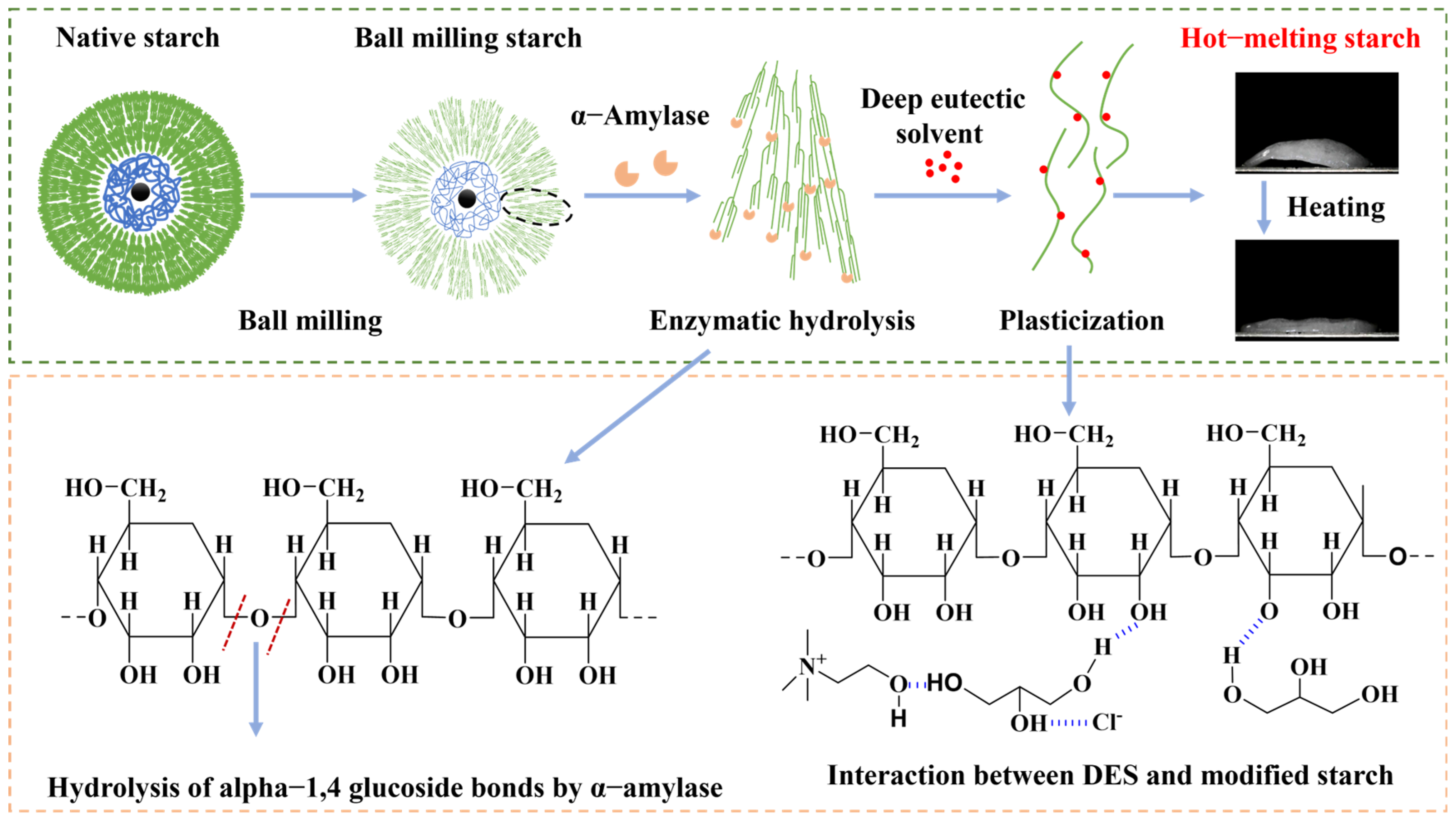
3.1. The Hot-Melting Phenomenon and Preparation Process of Hot-Melting Starch
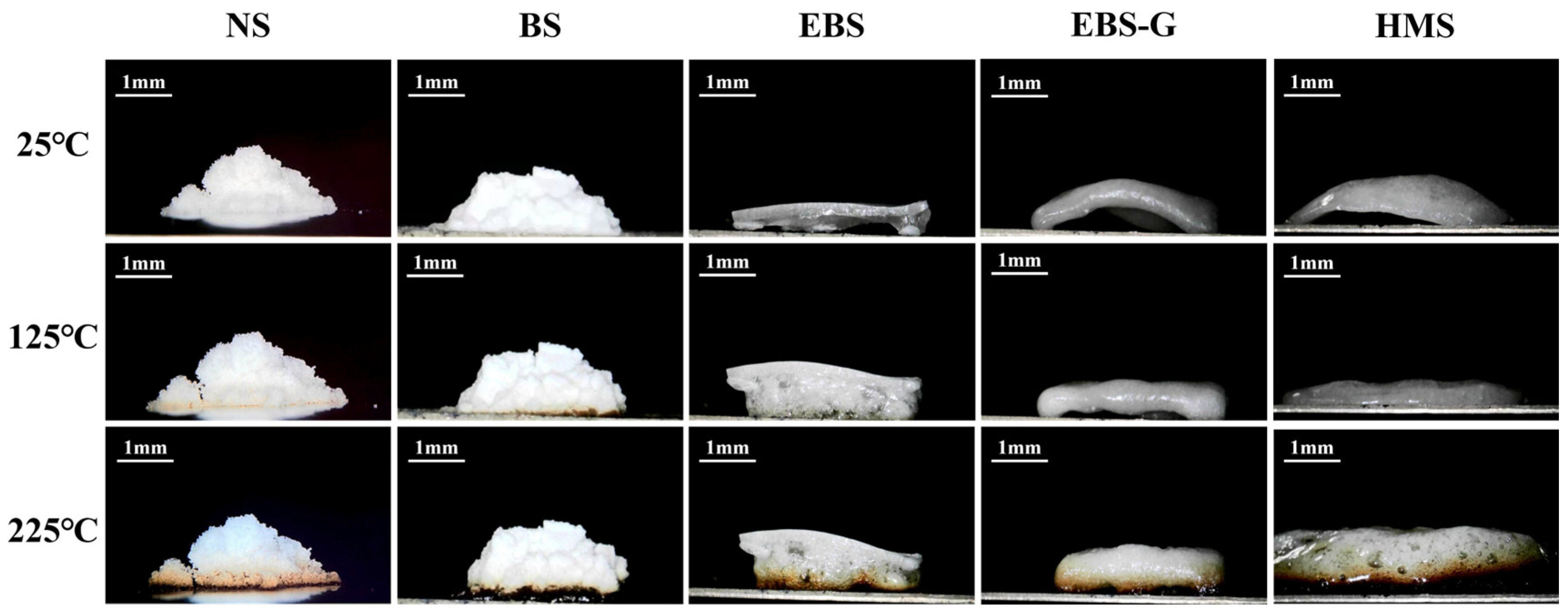
3.2. The Morphology of Hot-Melting Starch Products
3.3. Microscopic Performance Analysis
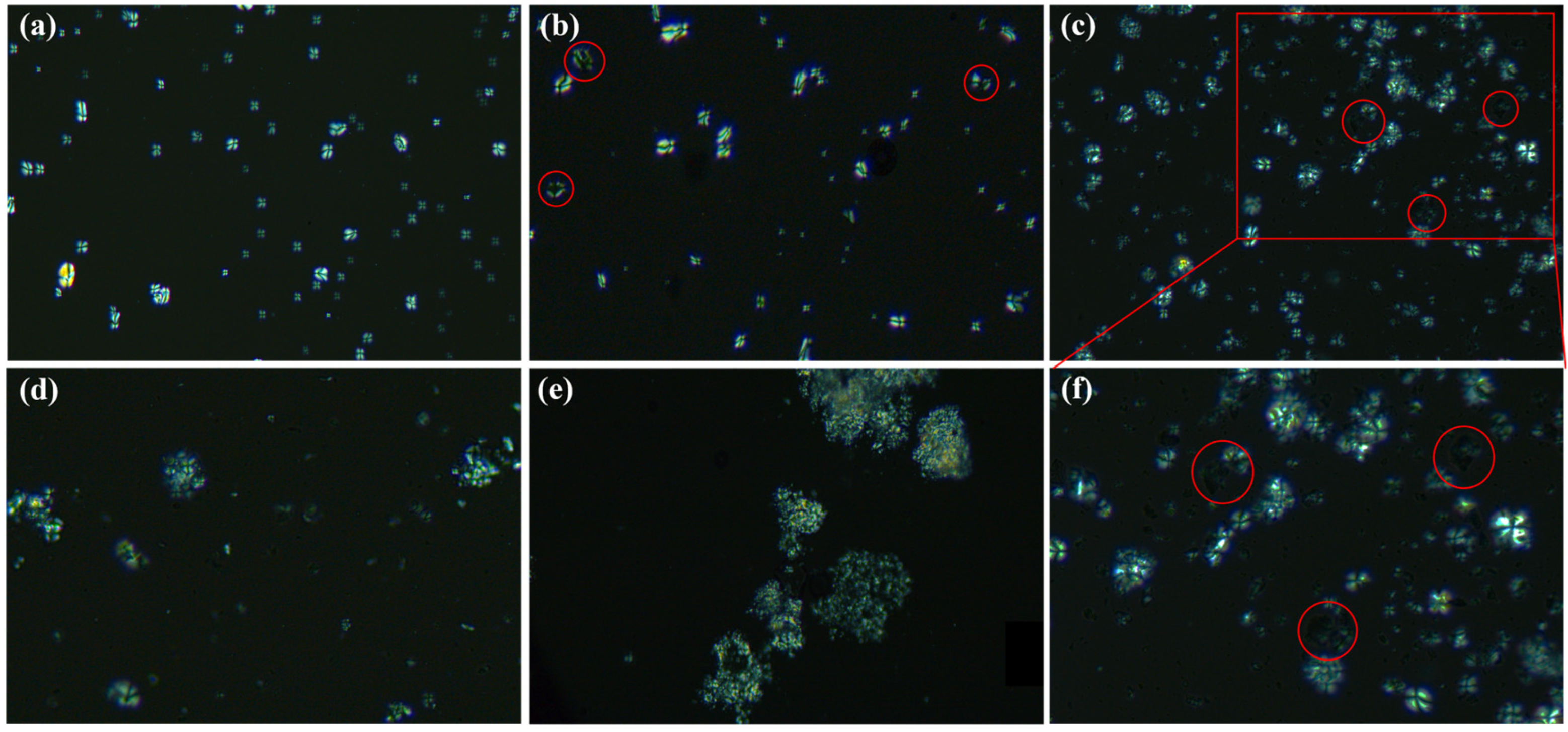
3.3.1. SEC
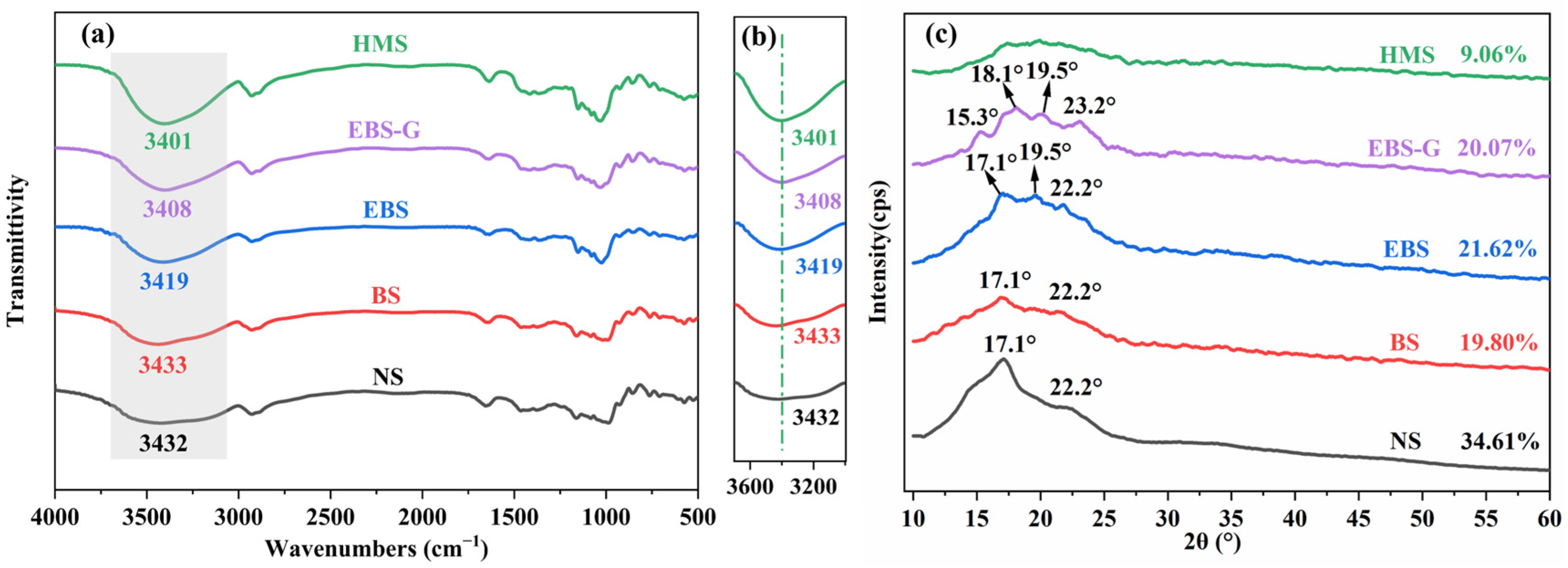
3.3.2. FTIR
3.3.3. XRD
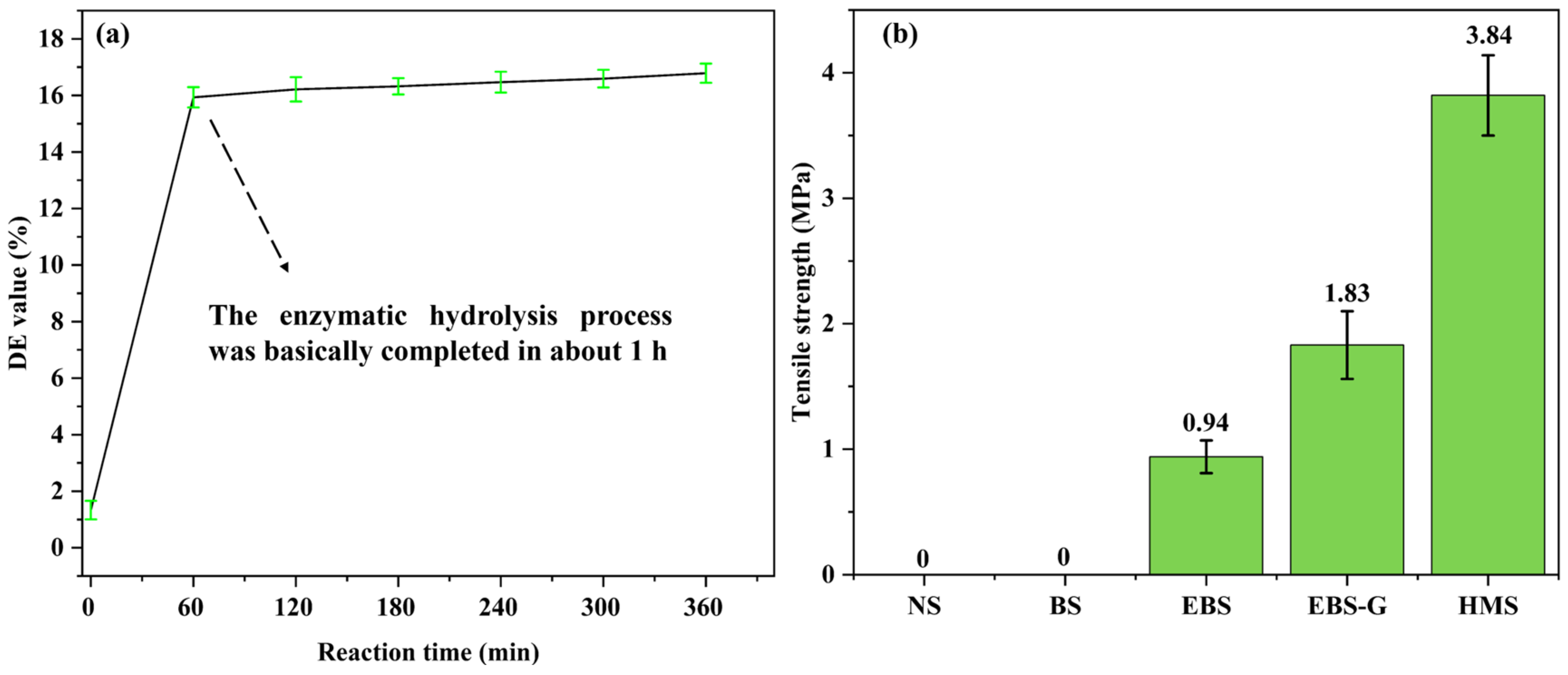
3.4. DE Value
3.5. Thermal Properties Analysis
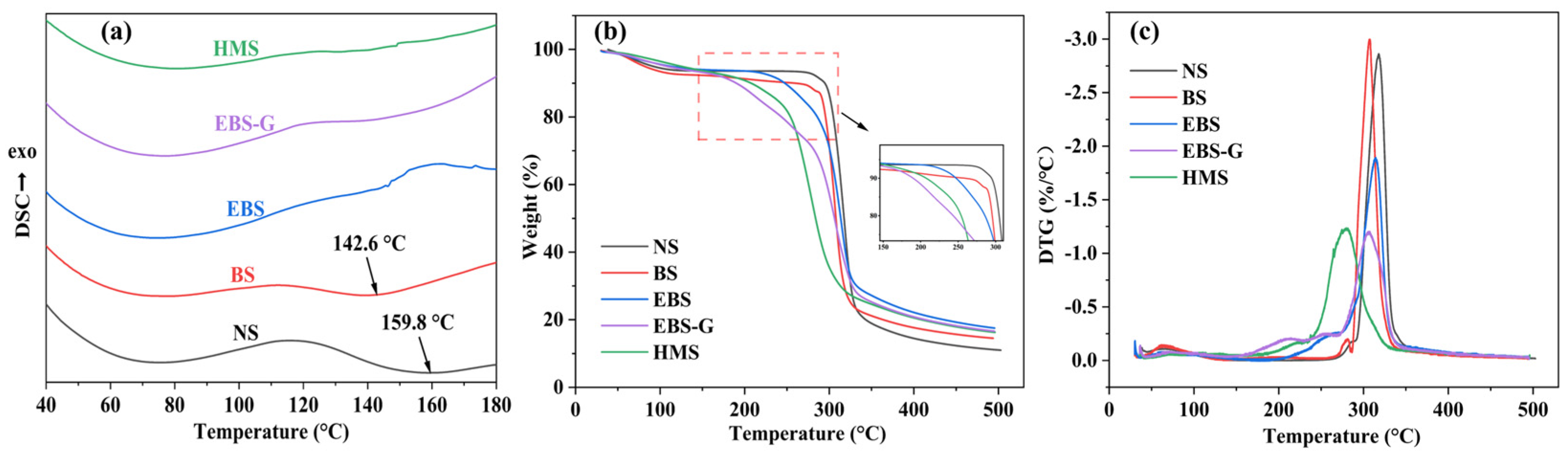
3.5.1. DSC
3.5.2. TGA
3.6. Mechanical Properties
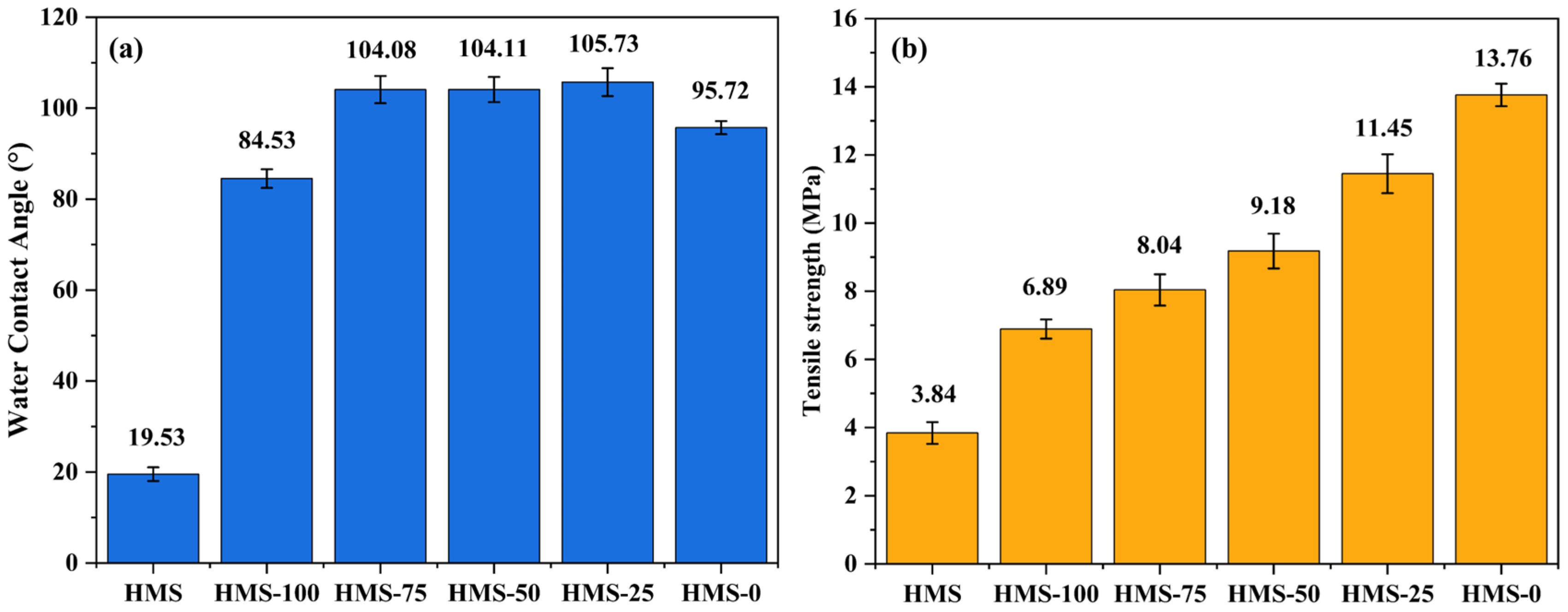
3.7. The Properties of Hot-Melting Starch/Sisal Fiber/PCL Composite Samples and Molded Products
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| NS | Natural starch |
| BS | Ball-milling starch |
| EBS | Enzymatically hydrolyzed ball-milling starch |
| DES | Deep eutectic solvent |
| EBS-G | Glycerol-plasticized hot-melting starch |
| HMS | Hot-melting starch |
| PCL | Polycaprolactone |
References
- Wang, S.; Zhang, P.; Li, Y.; Li, J.; Li, X.; Yang, J.; Ji, M.; Li, F.; Zhang, C. Recent advances and future challenges of the starch-based bio-composites for engineering applications. Carbohydr. Polym. 2023, 307, 120627. [Google Scholar] [CrossRef] [PubMed]
- Fonseca-García, A.; Jiménez-Regalado, E.J.; Aguirre-Loredo, R.Y. Preparation of a novel biodegradable packaging film based on corn starch-chitosan and poloxamers. Carbohydr. Polym. 2021, 251, 117009. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Y.; Zhang, J.; Man, J.; Nie, Y.; Ji, M.; Chen, H.; Li, F.; Zhang, C. Treatment and mechanism for hot melting starch by reducing the molecular chain winding and crystallinity. Carbohydr. Polym. 2024, 325, 121574. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Cui, G.; Li, J.; Li, F.; Ji, M.; Zhang, C.; Meng, T.; Li, J.; Man, J. Combined role of stearic acid and maleic anhydride in the development of thermoplastic starch-based materials with ultrahigh ductility and durability. Carbohydr. Polym. 2024, 339, 122296. [Google Scholar] [CrossRef]
- Wu, H.; Hou, A.; Hu, X.; Lu, X.; Qu, J.P. Effect of elongational rheology on plasticization and properties of thermoplastic starch prepared by biaxial eccentric rotor extruder. Ind. Crops Prod. 2022, 176, 114323. [Google Scholar] [CrossRef]
- Sun, Y.; Hu, Q.; Qian, J.; Li, T.; Ma, P.; Shi, D.; Dong, W.; Chen, M. Preparation and properties of thermoplastic poly(caprolactone) composites containing high amount of esterified starch without plasticizer. Carbohydr. Polym. 2016, 139, 28–34. [Google Scholar] [CrossRef]
- Chen, H.; Fu, M.; Zhang, Y.; Ma, C.; Kan, J. Effect of temperature during acetylation and heat moisture treatment on the structural and physicochemical properties and application of wheat starch. Food Hydrocoll. 2023, 144, 109036. [Google Scholar] [CrossRef]
- Qi, T.; Lü, S.; Zhang, S.F.; Bai, X.; Chen, J.; Huang, M.; Liu, M. Zein coated porous carboxymethyl starch fertilizer for iron promoting and phosphate sustainable release. J. Clean. Prod. 2020, 258, 120778. [Google Scholar] [CrossRef]
- Zavareze, E.D.R.; Dias, A.R.G. Impact of heat-moisture treatment and annealing in starches: A review. Carbohydr. Polym. 2011, 83, 317–328. [Google Scholar] [CrossRef]
- Dai, L.; Li, C.; Zhang, J.; Cheng, F. Preparation and characterization of starch nanocrystals combining ball milling with acid hydrolysis. Carbohydr. Polym. 2018, 180, 122–127. [Google Scholar] [CrossRef]
- Amini, A.M.; Razavi, S.M.A.; Mortazavi, S.A. Morphological, physicochemical, and viscoelastic properties of sonicated corn starch. Carbohydr. Polym. 2015, 122, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Dome, K.; Podgorbunskikh, E.; Bychkov, A.; Lomovsky, O. Changes in the crystallinity degree of starch having different types of crystal structure after mechanical pretreatment. Polymers 2020, 12, 641. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.; Yu, L.; Gu, Z.; Li, Z.; Ban, X.; Cheng, L.; Hong, Y.; Li, C. Fine structure impacts highly concentrated starch liquefaction process and product performance. Ind. Crops Prod. 2021, 164, 113347. [Google Scholar] [CrossRef]
- Li, Z.; Liu, W.; Gu, Z.; Li, C.; Hong, Y.; Cheng, L. The effect of starch concentration on the gelatinization and liquefaction of corn starch. Food Hydrocoll. 2015, 48, 189–196. [Google Scholar] [CrossRef]
- Liu, L.; An, X.; Zhang, H.; Lu, Z.; Nie, S.; Cao, H.; Xu, Q.; Liu, H. Ball milling pretreatment facilitating α-amylase hydrolysis for production of starch-based bio-latex with high performance. Carbohydr. Polym. 2020, 242, 116384. [Google Scholar] [CrossRef]
- Zdanowicz, M.; Wilpiszewska, K.; Spychaj, T. Deep eutectic solvents for polysaccharides processing. A review. Carbohydr. Polym. 2018, 200, 361–380. [Google Scholar] [CrossRef]
- Abbott, A.P.; Ballantyne, A.D.; Conde, J.P.; Ryder, K.S.; Wise, W.R. Salt modified starch: Sustainable, recyclable plastics. Green Chem. 2012, 14, 1302–1307. [Google Scholar] [CrossRef]
- Ramesh, S.; Shanti, R.; Morris, E. Studies on the plasticization efficiency of deep eutectic solvent in suppressing the crystallinity of corn starch based polymer electrolytes. Carbohydr. Polym. 2012, 87, 701–706. [Google Scholar] [CrossRef]
- Zdanowicz, M. Starch treatment with deep eutectic solvents, ionic liquids and glycerol. A comparative study. Carbohydr. Polym. 2020, 229, 115574. [Google Scholar] [CrossRef]
- Hassan, M.M.; Le Guen, M.J.; Tucker, N.; Parker, K. Thermo-mechanical, morphological and water absorption properties of thermoplastic starch/cellulose composite foams reinforced with PLA. Cellulose 2019, 26, 4463–4478. [Google Scholar] [CrossRef]
- Zhai, X.; Wang, W.; Zhang, H.; Dai, Y.; Dong, H.; Hou, H. Effects of high starch content on the physicochemical properties of starch/PBAT nanocomposite films prepared by extrusion blowing. Carbohydr. Polym. 2020, 239, 116231. [Google Scholar] [CrossRef] [PubMed]
- Peng, S.; Li, F.; Man, J.; Li, J.; Zhang, C.; Ji, M.; Li, J.; Wang, S. Enhancing the properties of starch-fiber foaming material by adjusting fiber length: The synergistic effect of macro-micro stress conduction. Mater. Today Commun. 2022, 33, 104408. [Google Scholar] [CrossRef]
- Goudarzi, V.; Shahabi-Ghahfarrokhi, I. Photo-producible and photo-degradable starch/TiO2 bionanocomposite as a food packaging material: Development and characterization. Int. J. Biol. Macromol. 2018, 106, 661–669. [Google Scholar] [CrossRef]
- Zolelmein, A.; Movahhed, S.; Azizi, M.H.; Chenarbon, H.A. Assessment of simultaneous addition of sucrose and xanthan effects on the thermal, pasting, and rheological behavior of corn starch. J. Texture Stud. 2020, 51, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Ji, M.; Li, J.; Li, F.; Wang, X.; Man, J.; Li, J.; Zhang, C.; Peng, S. A biodegradable chitosan-based composite film reinforced by ramie fibre and lignin for food packaging. Carbohydr. Polym. 2022, 281, 119078. [Google Scholar] [CrossRef] [PubMed]
- Noorbakhsh-Soltani, S.M.; Zerafat, M.M.; Sabbaghi, S. A comparative study of gelatin and starch-based nano-composite films modified by nano-cellulose and chitosan for food packaging applications. Carbohydr. Polym. 2018, 189, 48–55. [Google Scholar] [CrossRef]
- Miller, G.L. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal. Chem. 1959, 31, 426–428. [Google Scholar] [CrossRef]
- ASTM D638-14; Standard Test Method for Tensile Properties of Plastics. ASTM: West Conshohocken, PA, USA, 2022.
- Atrous, H.; Benbettaieb, N.; Hosni, F.; Danthine, S.; Blecker, C.; Attia, H.; Ghorbel, D. Effect of γ-radiation on free radicals formation, structural changes and functional properties of wheat starch. Int. J. Biol. Macromol. 2015, 80, 64–76. [Google Scholar] [CrossRef]
- Chen, L.; Dai, Y.; Hou, H.; Wang, W.; Ding, X.; Zhang, H.; Li, X.; Dong, H. Effect of high pressure microfluidization on the morphology, structure and rheology of sweet potato starch. Food Hydrocoll. 2021, 115, 106606. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, P.; Li, Y.; Sandeep, S.N.; Li, J.; Ji, M.; Peng, S.; Yan, N.; Li, F. Fully biodegradable, hydrophobic, enhanced barrier starch bio-composites with sandwich structure by simulating wood. Ind. Crops Prod. 2023, 197, 116603. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, P.; Cheng, L.; Li, J.; Jian, R.; Ji, M.; Li, F. A strong, hydrophobic, transparent and biodegradable nano-lignocellulosic membrane from wheat straw by novel strategy. J. Clean. Prod. 2022, 356, 131879. [Google Scholar] [CrossRef]
- López Terán, J.L.; Cabrera Maldonado, E.V.; Araque Rangel, J.d.C.; Poveda Otazo, J.; Beltrán Rico, M.I. Development of Antibacterial Thermoplastic Starch with Natural Oils and Extracts: Structural, Mechanical and Thermal Properties. Polymers 2024, 16, 180. [Google Scholar] [CrossRef] [PubMed]
- Bangar, S.P.; Singh, A.; Ashogbon, A.O.; Bobade, H. Ball-milling: A sustainable and green approach for starch modification. Int. J. Biol. Macromol. 2023, 237, 124069. [Google Scholar] [CrossRef] [PubMed]
- Domene-López, D.; García-Quesada, J.C.; Martin-Gullon, I.; Montalbán, M.G. Influence of starch composition and molecular weight on physicochemical properties of biodegradable films. Polymers 2019, 11, 1084. [Google Scholar] [CrossRef]
- Chatpapamon, C.; Uttapap, D.; Wandee, Y.; Puttanlek, C.; Rungsardthong, V. Glycerol-enhancing heat-moisture treatment of A-type rice and cassava starches and B-type potato and canna starches. Int. J. Food Sci. Technol. 2021, 56, 4038–4049. [Google Scholar] [CrossRef]
- He, X.; Zhang, Z.; Yan, T.; He, Y.; Zeng, C.; Guo, S.; Li, Q.; Xia, H. Rapid preparation of anti-retrogradation starch by choline chloride based deep eutectic solvents: A comparative study. Int. J. Biol. Macromol. 2024, 281, 136527. [Google Scholar] [CrossRef]
- Donovan, J.W. Phase transitions of the starch–water system. Biopolymers 1979, 18, 263–275. [Google Scholar] [CrossRef]
- Homer, S.; Kelly, M.; Day, L. Determination of the thermo-mechanical properties in starch and starch/gluten systems at low moisture content-A comparison of DSC and TMA. Carbohydr. Polym. 2014, 108, 1–9. [Google Scholar] [CrossRef]
- Chancelier, L.; Diallo, A.O.; Santini, C.C.; Marlair, G.; Gutel, T.; Mailley, S.; Len, C. Targeting adequate thermal stability and fire safety in selecting ionic liquid-based electrolytes for energy storage. Phys. Chem. Chem. Phys. 2014, 16, 1967–1976. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Materials | Mn (×103 g/mol) | Mw (×103 g/mol) | Dispersity (×10−2) |
|---|---|---|---|
| NS | 932.3 ± 1.1 | 5265.4 ± 1.3 | 564.7 ± 0.1 |
| BS | 778.7 ± 1.3 | 4683.4 ± 1.5 | 601.4 ± 0.1 |
| EBS | 242.0 ± 2.0 | 1279.4 ± 2.0 | 528.8 ± 0.1 |
| EBS-G | 242.9 ± 0.8 | 1290.0 ± 1.0 | 531.2 ± 0.1 |
| HMS | 261.2 ± 1.0 | 1419.5 ± 1.1 | 543.5 ± 0.1 |
| Materials | To (°C) | Tp (°C) | Tc (°C) | ΔH (J/g) |
|---|---|---|---|---|
| NS | 33.6 ± 0.3 | 75.8 ± 0.3 | 108.3 ± 1.3 | 47.7 ± 2.4 |
| BS | 33.9 ± 0.1 | 77.9 ± 0.5 | 107.4 ± 1.3 | 39.9 ± 1.4 |
| EBS | 34.4 ± 0.2 | 74.8 ± 0.2 | 115.8 ± 0.9 | 41.5 ± 1.4 |
| EBS-G | 35.7 ± 0.3 | 77.0 ± 0.1 | 116.8 ± 1.5 | 36.5 ± 1.7 |
| HMS | 35.3 ± 0.1 | 80.4 ± 0.2 | 110.3 ± 2.4 | 27.2 ± 1.1 |
| Materials | Tonset (°C) | Tmax (°C) | Td5 (°C) | Td30 (°C) | Residue (%) |
|---|---|---|---|---|---|
| NS | 287.0 ± 0.6 | 317.6 ± 0.7 | 300.0 ± 3.3 | 313.2 ± 1.5 | 11.0 ± 1.6 |
| BS | 281.4 ± 2.6 | 307.2 ± 2.3 | 293.5 ± 1.9 | 305.1 ± 1.8 | 14.5 ± 3.3 |
| EBS | 261.4 ± 4.7 | 314.1 ± 0.4 | 280.3 ± 0.5 | 309.7 ± 1.0 | 17.5 ± 0.7 |
| EBS-G | 259.9 ± 1.7 | 306.5 ± 1.0 | 279.4 ± 2.0 | 309.4 ± 0.4 | 16.6 ± 2.2 |
| HMS | 245.4 ± 1.5 | 280.1 ± 3.6 | 257.2 ± 2.3 | 281.0 ± 1.2 | 16.3 ± 1.2 |
| Samples | HMS (%) | PCL (%) | Sisal Fibers (%) |
|---|---|---|---|
| HMS-0 | 0 | 90 | 10 |
| HMS-25 | 22.5 | 67.5 | 10 |
| HMS-50 | 45 | 45 | 10 |
| HMS-75 | 67.5 | 22.5 | 10 |
| HMS-100 | 90 | 0 | 10 |
| HMS | 100 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Man, J.; Li, Y.; Wang, L.; Ji, M.; Peng, S.; Li, J.; Wang, S.; Li, F.; Zhang, C. Deep Eutectic Solvent Assisted Mechano-Enzymatic Preparation for Reprocessable Hot-Melting Starch: A Comprehensive Analysis of Molecular Structure and Thermal Properties. Polymers 2025, 17, 1296. https://doi.org/10.3390/polym17101296
Liu X, Man J, Li Y, Wang L, Ji M, Peng S, Li J, Wang S, Li F, Zhang C. Deep Eutectic Solvent Assisted Mechano-Enzymatic Preparation for Reprocessable Hot-Melting Starch: A Comprehensive Analysis of Molecular Structure and Thermal Properties. Polymers. 2025; 17(10):1296. https://doi.org/10.3390/polym17101296
Chicago/Turabian StyleLiu, Xuan, Jia Man, Yanhui Li, Liming Wang, Maocheng Ji, Sixian Peng, Junru Li, Shen Wang, Fangyi Li, and Chuanwei Zhang. 2025. "Deep Eutectic Solvent Assisted Mechano-Enzymatic Preparation for Reprocessable Hot-Melting Starch: A Comprehensive Analysis of Molecular Structure and Thermal Properties" Polymers 17, no. 10: 1296. https://doi.org/10.3390/polym17101296
APA StyleLiu, X., Man, J., Li, Y., Wang, L., Ji, M., Peng, S., Li, J., Wang, S., Li, F., & Zhang, C. (2025). Deep Eutectic Solvent Assisted Mechano-Enzymatic Preparation for Reprocessable Hot-Melting Starch: A Comprehensive Analysis of Molecular Structure and Thermal Properties. Polymers, 17(10), 1296. https://doi.org/10.3390/polym17101296