

A Novel In Situ Sol-Gel Synthesis Method for PDMS Composites Reinforced with Silica Nanoparticles

 ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. SiO2 Sol-Gel Synthesis
2.2. Synthesis of PDMS/NP-SiO2 Nanocomposites
3. Results
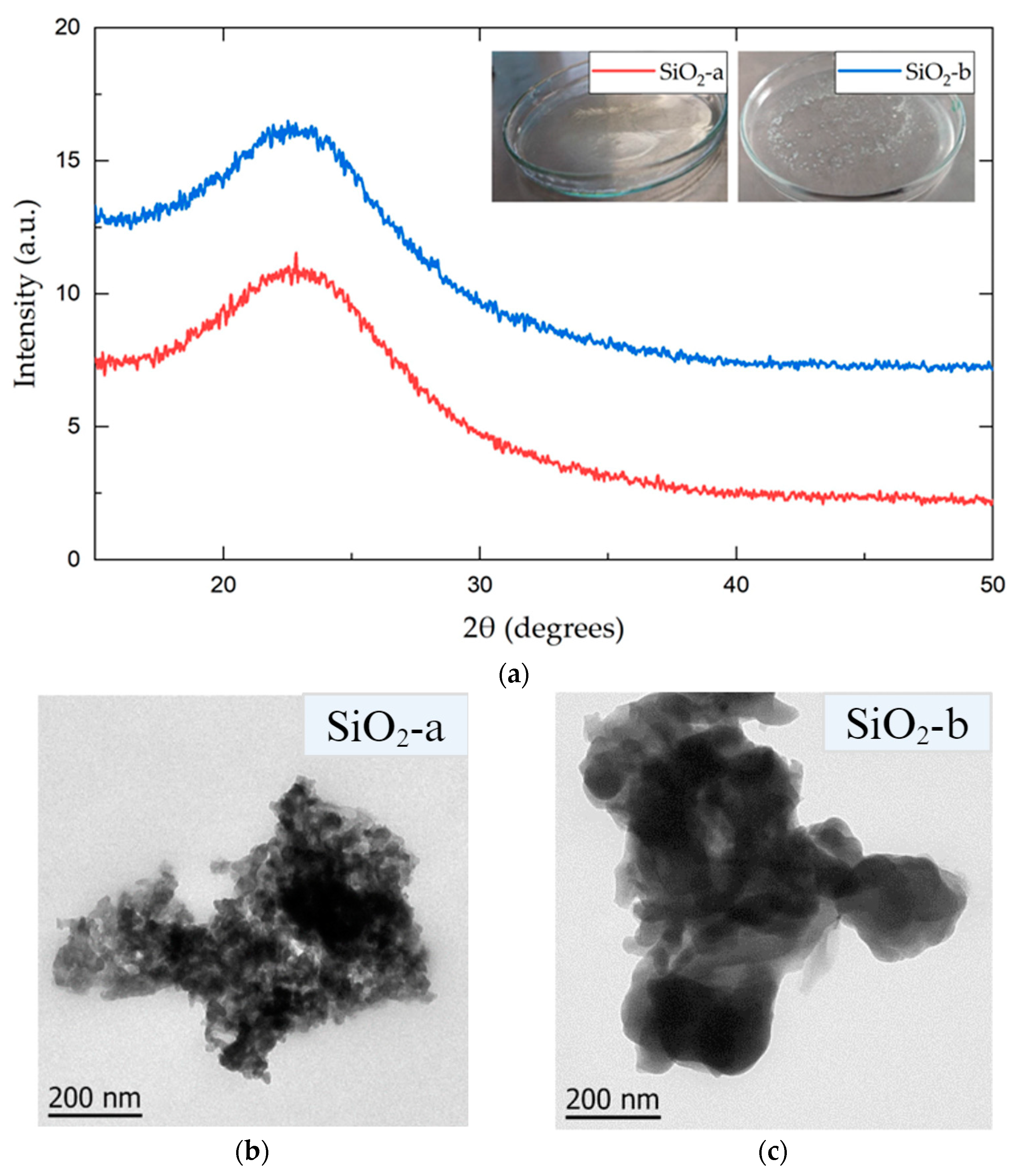
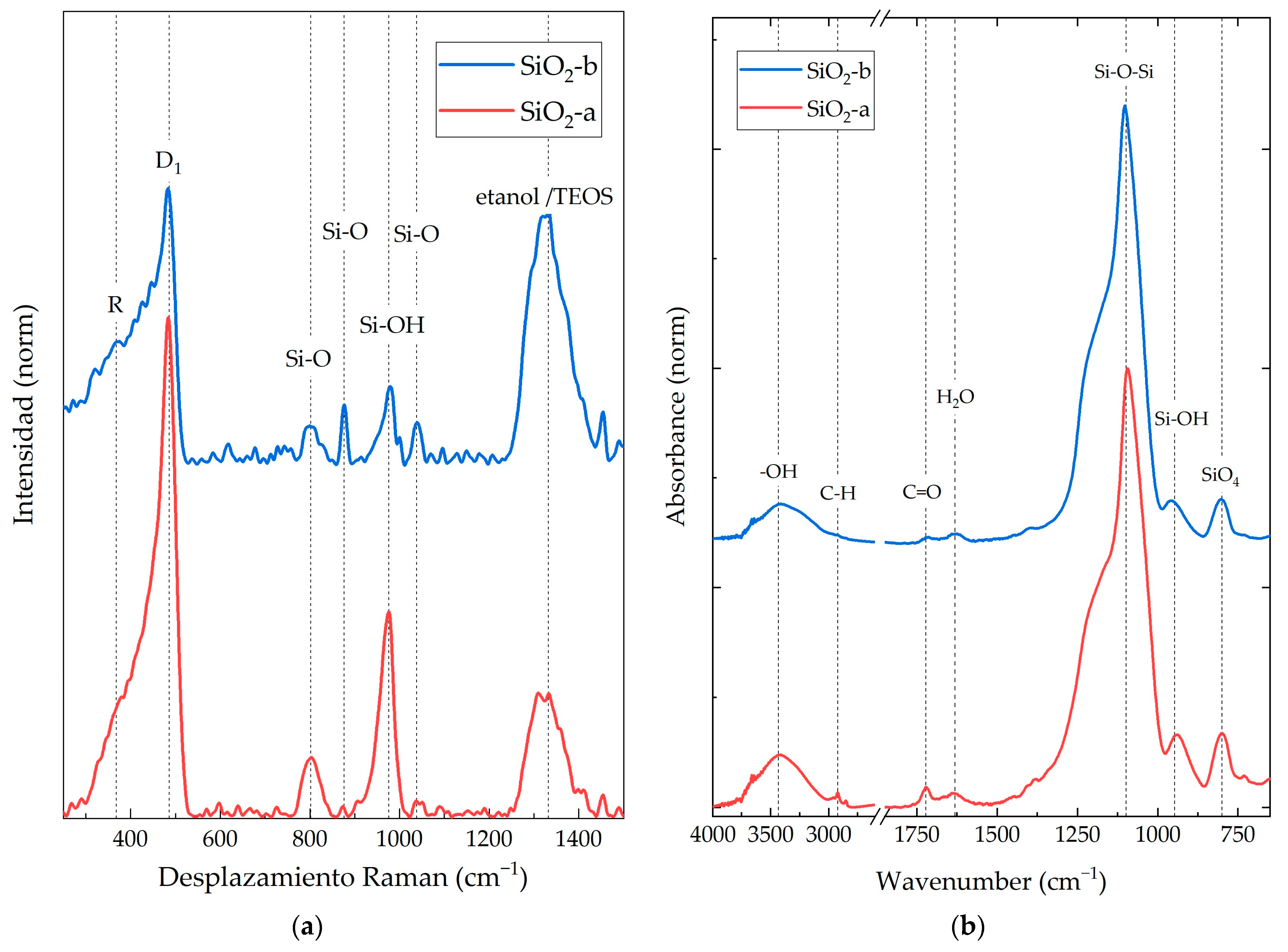
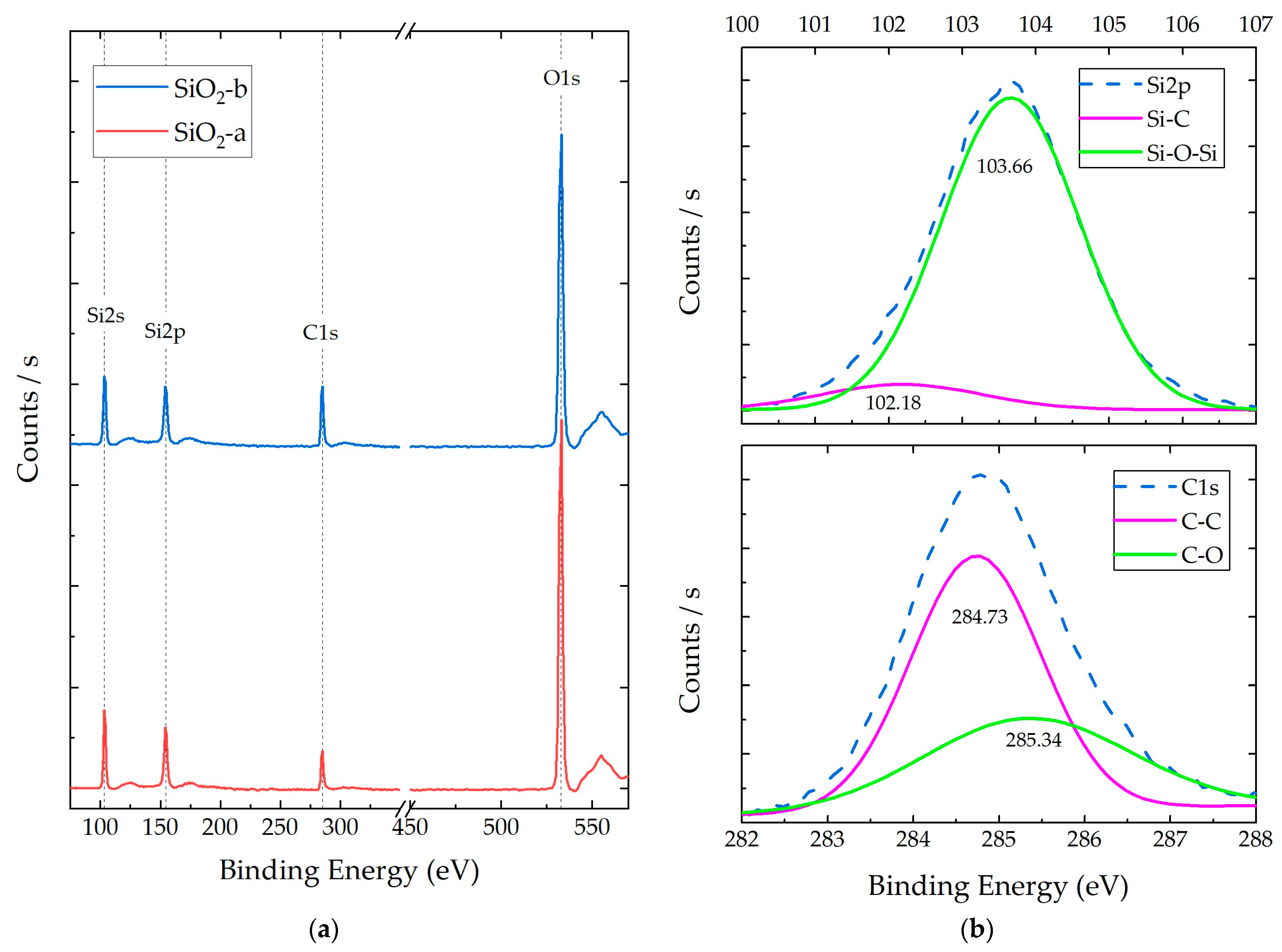
3.1. SiO2 NP Characterization
3.2. PDMS-%SiO2 Composite Chemical Characterization
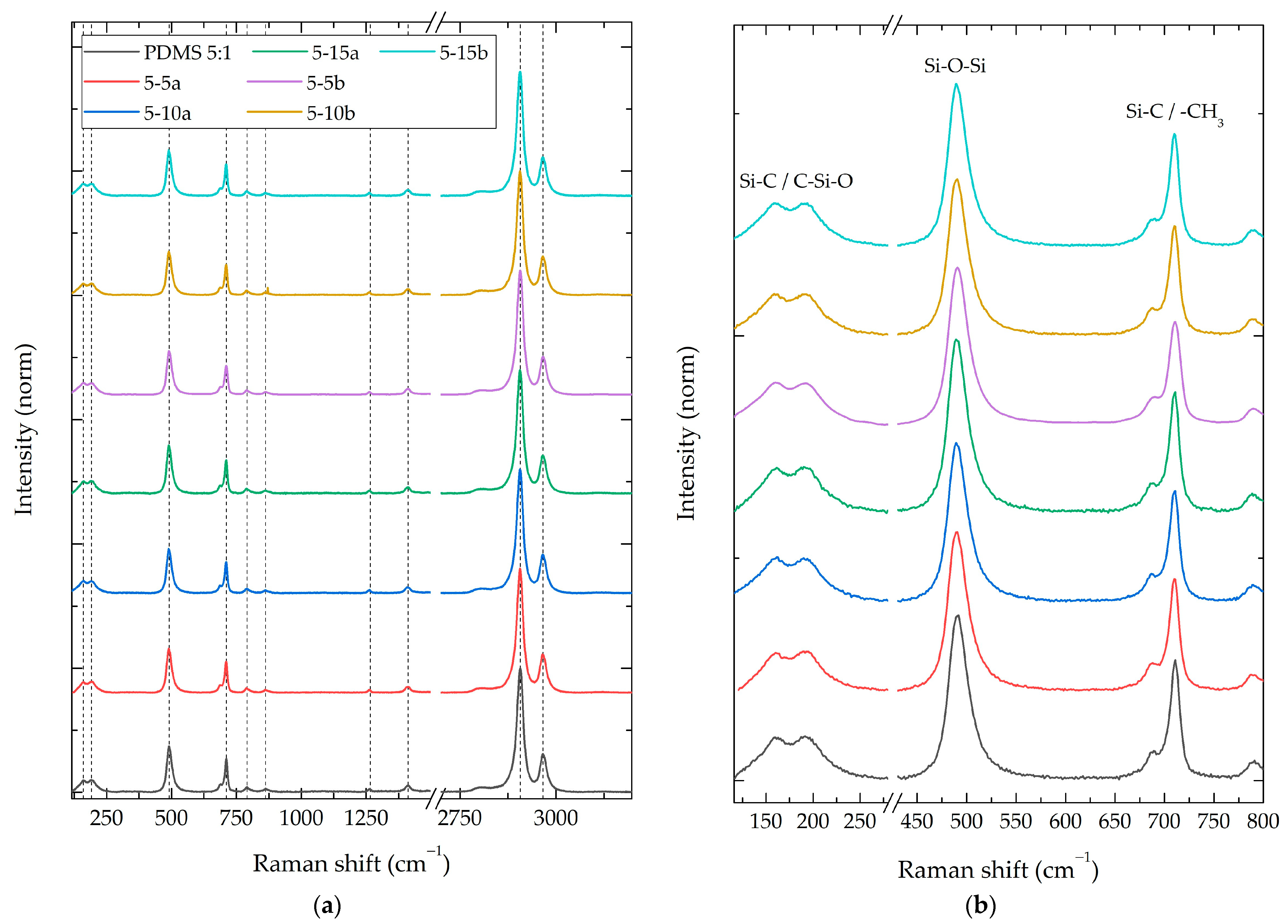
3.2.1. Composite Raman Spectroscopy
3.2.2. Composite Fourier-Transform Infrared Spectroscopy (FT-IR)
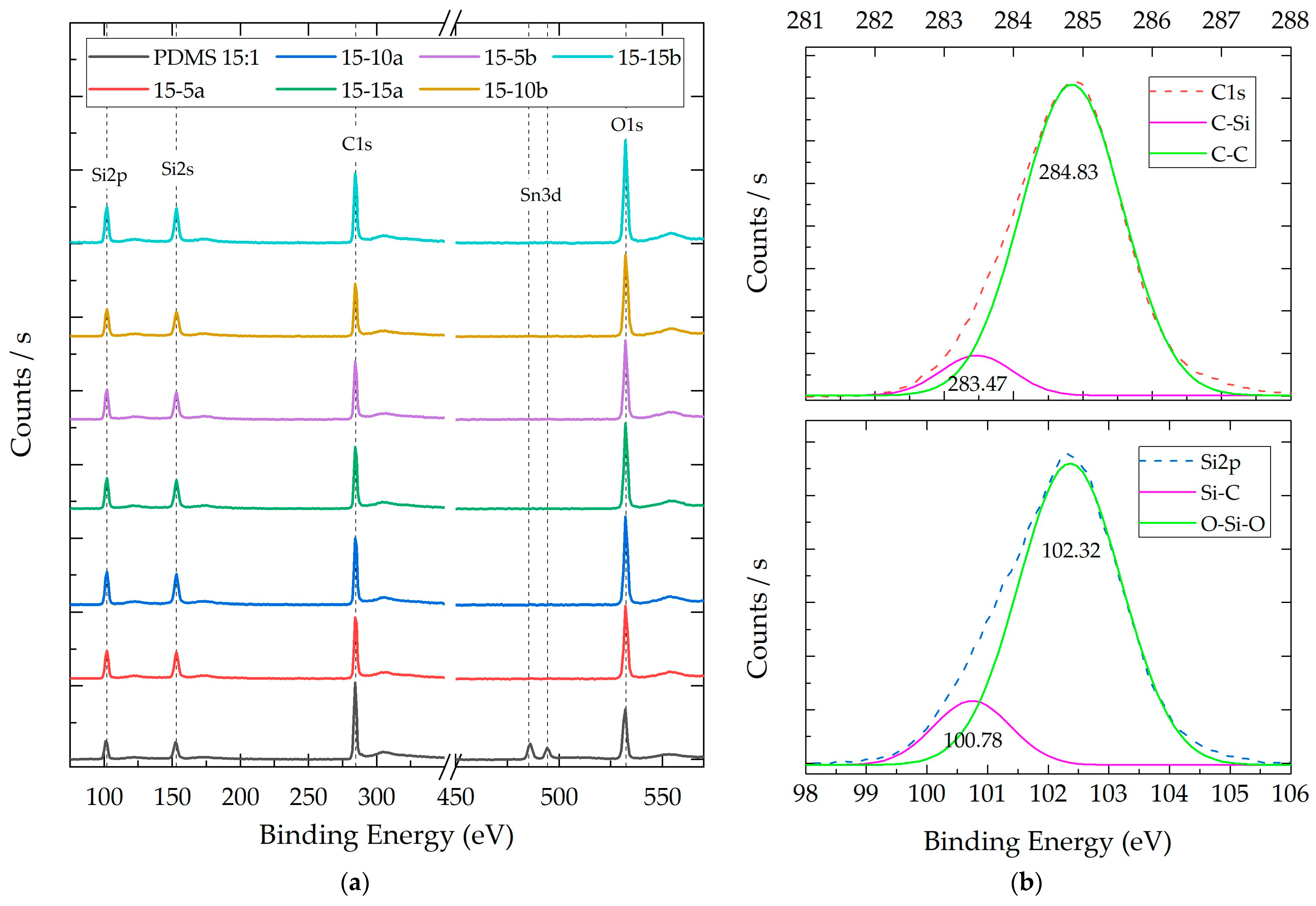
3.2.3. Composite X-ray Photoelectron Spectroscopy (XPS)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- PS Market Research. Polymer Market Research Report; Prescient & Strategic Intelligence: Delhi, India, 2022. [Google Scholar]
- Madidi, F.; Momen, G.; Farzaneh, M. Dielectric Properties of TiO2/Silicone Rubber Micro- and Nanocomposites. Adv. Mater. Sci. Eng. 2018, 2018, 4682076. [Google Scholar] [CrossRef]
- Fu, S.; Sun, Z.; Huang, P.; Li, Y.; Hu, N. Nano Materials Science Some Basic Aspects of Polymer Nanocomposites: A Critical Review. Nano Mater. Sci. 2019, 1, 2–30. [Google Scholar] [CrossRef]
- Ali AlMaadeed, M.; Carignano, D.P.; Carignano, M.A. Polymer Science and Innovative Applications. In Polymer Science and Innovative Applications; Elsevier: Amsterdam, The Netherlands, 2020; pp. i–iii. ISBN 9780128168080. [Google Scholar]
- Eduok, U.; Faye, O.; Szpunar, J. Recent Developments and Applications of Protective Silicone Coatings: A Review of PDMS Functional Materials. Prog. Org. Coat. 2017, 111, 124–163. [Google Scholar] [CrossRef]
- Aly, K.; Aboubakr, S.H.; Bradford, P.D. One-step Fabrication of Bulk Nanocomposites Reinforced by Carbon Nanotube Array Fragments. Polym. Compos. 2022, 43, 94–110. [Google Scholar] [CrossRef]
- Li, J.; Liu, H.; Guo, J.; Hu, Z.; Wang, Z.; Wang, B.; Liu, L.; Huang, Y.; Guo, Z. Flexible, Conductive, Porous, Fibrillar Polymer-Gold Nanocomposites with Enhanced Electromagnetic Interference Shielding and Mechanical Properties. J. Mater. Chem. C 2017, 5, 1095–1105. [Google Scholar] [CrossRef]
- Chungprempree, J.; Preechawong, J.; Nithitanakul, M. Developing an Effective and Durable Film for Marine Fouling Prevention from PDMS/SiO2 and PDMS/PU with SiO2 Composites. Polymers 2022, 14, 4252. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Zhu, G.; Pan, Y.; Shao, Q.; Zhao, C.; Dong, M.; Zhang, Y.; Guo, Z. Polydimethylsiloxane-Titania Nanocomposite Coating: Fabrication and Corrosion Resistance. Polymer 2018, 138, 203–210. [Google Scholar] [CrossRef]
- Krishna, D.N.G.; Philip, J. Review on Surface-Characterization Applications of X-Ray Photoelectron Spectroscopy (XPS): Recent Developments and Challenges. Appl. Surf. Sci. Adv. 2022, 12, 100332. [Google Scholar] [CrossRef]
- Gupta, N.S.; Lee, K.; Labouriau, A. Tuning Thermal and Mechanical Properties of Polydimethylsiloxane with Carbon Fibers. Polymers 2021, 13, 1141. [Google Scholar] [CrossRef]
- Zhang, F.; Yang, R.; Lu, D. Investigation of Polymer Aging Mechanisms Using Molecular Simulations: A Review. Polymers 2023, 15, 1928. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, F.; Xu, L.; Xu, Z.; Shen, C.; Zhang, G.; Meng, Q.; Gao, C. Heterostructured ZIF-8/Lamellar Talc Composites Incorporated Polydimethylsiloxane Membrane with Enhanced Separation Performance for Butanol Recovery. J. Membr. Sci. 2022, 650, 120433. [Google Scholar] [CrossRef]
- Ashraf, M.A.; Peng, W.; Zare, Y.; Rhee, K.Y. Effects of Size and Aggregation/Agglomeration of Nanoparticles on the Interfacial/Interphase Properties and Tensile Strength of Polymer Nanocomposites. Nanoscale Res. Lett. 2018, 13, 214. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.; Zayat, M. The Sol-Gel Handbook: Synthesis, Characterization, and Applications; Wiley-VCH: Weinheim, Germany, 2015. [Google Scholar]
- Chen, G.; Dai, Z.; Ding, S.; Lei, M.; Lin, J.; Wang, S.; Zhou, Y.; Pan, H.; Zhou, B. Realization of Integrative Hierarchy by In-Situ Solidification of ‘Semi-Cured’ Microcilia Array in Candle Flame for Robust and Flexible Superhydrophobicity. Chem. Eng. J. 2022, 432, 134400. [Google Scholar] [CrossRef]
- Pandey, K.; Bindra, H.S.; Paul, D.; Nayak, R. Smart Multi-Tasking PDMS Nanocomposite Sponges for Microbial and Oil Contamination Removal from Water. J. Polym. Res. 2020, 27, 189. [Google Scholar] [CrossRef]
- Wang, Q.; Che, J.; Wu, W.; Hu, Z.; Liu, X.; Ren, T.; Chen, Y.; Zhang, J. Contributing Factors of Dielectric Properties for Polymer Matrix Composites. Polymers 2023, 15, 590. [Google Scholar] [CrossRef] [PubMed]
- Ríos, E.A.; Vega-Baudrit, J.R.; Villegas, J.G.; Sánchez, J.A. Silicon Nanostructures in Biomedicine and Biotechnology. Momento 2020, 60, 18–40. [Google Scholar] [CrossRef]
- Harito, C.; Bavykin, D.V.; Yuliarto, B.; Dipojono, H.K.; Walsh, F.C. Polymer Nanocomposites Having a High Filler Content: Synthesis, Structures, Properties, and Applications. Nanoscale 2019, 11, 4653–4682. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Xue, Y.; Li, Z.; Wen, Y.; Li, X.; Wu, F.; Li, X.; Shi, D.; Xue, Z.; Xie, X. Construction of 3D Boron Nitride Nanosheets/Silver Networks in Epoxy-Based Composites with High Thermal Conductivity via in-Situ Sintering of Silver Nanoparticles. Chem. Eng. J. 2019, 369, 1150–1160. [Google Scholar] [CrossRef]
- Yuan, T.; Cui, X.; Liu, X.; Qu, X.; Sun, J. Highly Tough, Stretchable, Self-Healing, and Recyclable Hydrogels Reinforced by in Situ-Formed Polyelectrolyte Complex Nanoparticles. Macromolecules 2019, 52, 3141–3149. [Google Scholar] [CrossRef]
- Santiago-Castillo, K.; Del Angel-López, D.; Torres-Huerta, A.M.; Domínguez-Crespo, M.A.; Palma-Ramírez, D.; Willcock, H.; Brachetti-Sibaja, S.B. Effect on the Processability, Structure and Mechanical Properties of Highly Dispersed in Situ ZnO:CS Nanoparticles into PVA Electrospun Fibers. J. Mater. Res. Technol. 2021, 11, 929–945. [Google Scholar] [CrossRef]
- Bathini, S.; Raju, D.; Badilescu, S.; Packirisamy, M. Effect of Cross-Linking and Thermal Budget on Plasmonic Sensing and Sub-Surface Segregation of in Situ Synthesized Gold in Polymer Nanocomposite. J. Nanoparticle Res. 2021, 23, 21. [Google Scholar] [CrossRef]
- Yorseng, K.; Siengchin, S.; Ashok, B.; Rajulu, A.V. Nanocomposite Egg Shell Powder with in Situ Generated Silver Nanoparticles Using Inherent Collagen as Reducing Agent. J. Bioresour. Bioprod. 2020, 5, 101–107. [Google Scholar] [CrossRef]
- Lazareva, S.V.; Shikina, N.V.; Tatarova, L.E.; Ismagilov, Z.R. Synthesis of High-Purity Silica Nanoparticles by Sol-Gel Method. Eurasian Chem. J. 2017, 19, 295–302. [Google Scholar] [CrossRef]
- Bhatt, N.; Mishra, A.; Goswami, R.; Prasad, B. Preparation of Silica Nano-Particles by Sol-Gel Method and Its Characterization. J. Graph. Era Univ. 2021, 9, 215–230. [Google Scholar] [CrossRef]
- Rosales, A.; Maury-Ramírez, A.; Mejía-De Gutiérrez, R.; Guzmán, C.; Esquivel, K. SiO2@TiO2 Coating: Synthesis, Physical Characterization and Photocatalytic Evaluation. Coatings 2018, 8, 120. [Google Scholar] [CrossRef]
- Cordoba, A.; Rivera-Muñoz, E.M.; Velázquez-Castillo, R.; Esquivel, K. PDMS/TiO2 and PDMS/SiO2 Nanocomposites: Mechanical Properties’ Evaluation for Improved Insulating Coatings. Nanomaterials 2023, 13, 1699. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, H.; Serra, J.; González, P.; León, B. Structural Study of Sol–Gel Silicate Glasses by IR and Raman Spectroscopies. J. Non-Cryst. Solids 2009, 355, 475–480. [Google Scholar] [CrossRef]
- Hoffmann, G.G. Infrared and Raman Spectroscopy; De Gruyter: Berlin, Germany, 2023; ISBN 9783110717556/9783110717549. [Google Scholar]
- Alessi, A.; Agnello, S.; Buscarino, G.; Gelardi, F.M. Raman and IR Investigation of Silica Nanoparticles Structure. J. Non-Cryst. Solids 2013, 362, 20–24. [Google Scholar] [CrossRef]
- Rubio, F.; Rubio, J.; Oteo, J.L. A FT-IR Study of the Hydrolysis of Tetraethylorthosilicate (TEOS). Spectrosc. Lett. 1998, 31, 199–219. [Google Scholar] [CrossRef]
- Lu, Y.-K.; Yan, X.-P. An Imprinted Organic−Inorganic Hybrid Sorbent for Selective Separation of Cadmium from Aqueous Solution. Anal. Chem. 2004, 76, 453–457. [Google Scholar] [CrossRef]
- Hudson, R.L.; Gerakines, P.A.; Ferrante, R.F. IR Spectra and Properties of Solid Acetone, an Interstellar and Cometary Molecule. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 193, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Roy, T.; Wanchoo, S.K.; Pal, K. Synergetic Proton-Conducting Effect of Sulfonated PEEK-MO2-CNT Membranes for PEMFC Applications. Ionics 2021, 27, 4859–4873. [Google Scholar] [CrossRef]
- Schubert, U. Part One Sol—Gel Chemistry and Methods. Sol-Gel Handb. Synth. Charact. Appl. 2015, 1–28. [Google Scholar] [CrossRef]
- Ferrara, M.C.; Mirenghi, L.; Mevoli, A.; Tapfer, L. Synthesis and Characterization of Sol–Gel Silica Films Doped with Size-Selected Gold Nanoparticles. Nanotechnology 2008, 19, 365706. [Google Scholar] [CrossRef] [PubMed]
- Moulder, J.F.; Strickle, W.F.; Sobol, P.E. Handbook of X-ray Photoelectron Spectrocopy, 1st ed.; Perkin-Elmer Corporation: Eden Prairie, MN, USA, 1992. [Google Scholar]
- Bistričić, L.; Borjanović, V.; Mikac, L.; Dananić, V. Vibrational Spectroscopic Study of Poly(Dimethylsiloxane)-ZnO Nanocomposites. Vib. Spectrosc. 2013, 68, 1–10. [Google Scholar] [CrossRef]
- Mark, J.E. Polymer Data Book, 1st ed.; University of Cincinnati: Cincinnati, OH, USA, 1999. [Google Scholar]
- Hong, H.; Zhang, J.; Zhu, Y.; Tse, S.D.; Guo, H.; Lai, Y.; Xi, Y.; He, L.; Zhu, Z.; Yin, K.; et al. In Situ Polymer-Solution-Processed Graphene–PDMS Nanocomposites for Application in Intracranial Pressure Sensors. Nanomaterials 2024, 14, 399. [Google Scholar] [CrossRef] [PubMed]
- Dhahir, M.; Khyoon, H. Study the Effect of PH Variation on the Particle Size of SiO2 Thin Films. Iraqi J. Laser 2016, 15, 1–8. [Google Scholar]
- Atta, A.; Abdeltwab, E. Influence of Ion Irradiation on the Surface Properties of Silver-Coated Flexible PDMS Polymeric Films. Braz. J. Phys. 2022, 52, 3. [Google Scholar] [CrossRef]
- Melvin, G.J.H.; Ni, Q.-Q.; Suzuki, Y.; Natsuki, T. Microwave-Absorbing Properties of Silver Nanoparticle/Carbon Nanotube Hybrid Nanocomposites. J. Mater. Sci. 2014, 49, 5199–5207. [Google Scholar] [CrossRef]
- Hoflund, G.B.; Gonzalez, R.I.; Phillips, S.H. In Situ Oxygen Atom Erosion Study of a Polyhedral Oligomeric Silsesquioxane-Polyurethane Copolymer. J. Adhes. Sci. Technol. 2001, 15, 1199–1211. [Google Scholar] [CrossRef][Green Version]
- Post, P.; Wurlitzer, L.; Maus-Friedrichs, W.; Weber, A. Characterization and Applications of Nanoparticles Modified In-Flight with Silica or Silica-Organic Coatings. Nanomaterials 2018, 8, 530. [Google Scholar] [CrossRef] [PubMed]
- Seghir, R.; Arscott, S. Extended PDMS Stiffness Range for Flexible Systems. Sens. Actuators A Phys. 2015, 230, 33–39. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Binding Energy (eV) | Intensity (CPS) | FWHM (eV) | Area (CPS•eV) | Area (Norm) | Atomic Conc. (%) | Atomic Ratio * | |
|---|---|---|---|---|---|---|---|---|
| SiO2-a | Si2p | 101.57 | 1359.45 | 1.06 | 1507.74 | 0.027 | 31.82% | 1 |
| 103.52 | 28,287.24 | 1.86 | 54,919.59 | 0.973 | ||||
| C1s | 284.66 | 14,530.61 | 1.48 | 22,874.40 | 0.619 | 19.21% | 0.6 | |
| 285.37 | 4922.10 | 2.68 | 14,052.02 | 0.381 | ||||
| O1s | 532.85 | 135,607.75 | 1.71 | 259,798.74 | 1 | 48.97% | 1.54 | |
| SiO2-b | Si2p | 102.18 | 1919.39 | 2.54 | 5097.19 | 0.086 | 31.15% | 1 |
| 103.66 | 23,610.95 | 2.21 | 54,497.88 | 0.914 | ||||
| C1s | 284.73 | 18,567.63 | 1.78 | 35,174.67 | 0.635 | 23.98% | 0.76 | |
| 285.34 | 6675.72 | 2.84 | 20,190.43 | 0.365 | ||||
| O1s | 532.93 | 111,300.88 | 2.08 | 256,032.04 | 1 | 44.87% | 1.44 |
| PDMS 15:1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Vibrational Mode [30] | Raman Shift (cm−1) | 0% (Norm) | a | b | |||||
| 5% | 10% | 15% | 5% | 10% | 15% | ||||
| 1 | Si-C torsion | 157.40 | 0.075 | 1.66% | 15.05% | 22.79% | 9.89% | 11.56% | 10.66% |
| 2 | C-Si-C deflection/C-Si-O deflection | 192.63 | 0.078 | 5.22% | 14.96% | 24.07% | 9.44% | 11.54% | 12.32% |
| 3 | Si-O-Si stretch | 489.29 | 0.315 | 8.57% | 12.73% | 20.52% | 10.85% | 10.87% | 10.18% |
| 4 | Si-C stretch | 689.19 | 0.062 | −6.23% | −0.55% | 15.36% | −0.52% | −5.67% | −5.42% |
| 5 | Si–C symmetric stretch/CH3 rock | 709.69 | 0.232 | −2.45% | 0.52% | 11.44% | −0.35% | −0.87% | −1.28% |
| 6 | Si–C stretch/CH3 rock | 788.66 | 0.038 | −11.45% | −5.78% | 16.30% | −8.77% | −14.81% | −7.58% |
| 7 | CH3 asymmetric stretch | 861.97 | 0.029 | −19.45% | −24.79% | −3.16% | −26.19% | −30.09% | −33.75% |
| 8 | Si-CH3 symmetric bending | 1262.66 | 0.027 | −18.41% | −18.05% | 5.35% | −20.48% | −17.07% | −22.68% |
| 9 | CH3 asymmetric bending | 1410.73 | 0.05 | −11.46% | −8.39% | 2.10% | −6.95% | −4.50% | −4.90% |
| 10 | CH3 symmetric stretch | 2905.73 | 1 | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% |
| 11 | CH3 asymmetric stretch | 2965.55 | 0.309 | 1.02% | −0.27% | 1.29% | 0.86% | 0.70% | 2.21% |
| PDMS 10:1 | |||||||||
| Vibrational Mode [30] | Raman Shift (cm−1) | 0% (Norm) | a | b | |||||
| 5% | 10% | 15% | 5% | 10% | 15% | ||||
| 1 | Si-C torsion | 158.70 | 0.089 | 10.49% | 3.61% | 6.68% | −7.15% | −5.83% | −4.21% |
| 2 | C-Si-C deflection/C-Si-O deflection | 192.63 | 0.092 | 4.82% | 7.44% | 2.78% | −5.11% | −3.93% | −3.82% |
| 3 | Si-O-Si stretch | 490.54 | 0.36 | 0.85% | 1.74% | 2.15% | −5.51% | −2.98% | −4.54% |
| 4 | Si-C stretch | 687.98 | 0.063 | 2.67% | 1.84% | 2.82% | −7.86% | −6.05% | −6.14% |
| 5 | Si–C symmetric stretch/CH3 rock | 710.89 | 0.252 | 0.12% | 2.64% | −2.48% | −11.94% | −9.53% | −12.48% |
| 6 | Si–C stretch/CH3 rock | 791.04 | 0.035 | 7.32% | 18.21% | 10.76% | −7.61% | −0.43% | 2.20% |
| 7 | CH3 asymmetric stretch | 864.32 | 0.025 | 0.81% | 13.67% | 3.35% | −8.93% | −9.13% | −4.39% |
| 8 | Si-CH3 symmetric bending | 1262.66 | 0.027 | −5.66% | 4.56% | −21.92% | −15.65% | −24.98% | −15.94% |
| 9 | CH3 asymmetric bending | 1412.88 | 0.05 | −1.01% | 5.61% | −5.93% | −5.47% | −7.43% | −3.61% |
| 10 | CH3 symmetric stretch | 2906.33 | 1 | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% |
| 11 | CH3 asymmetric stretch | 2965.55 | 0.311 | −1.82% | −0.29% | −0.33% | −0.36% | 0.49% | 0.08% |
| PDMS 5:1 | |||||||||
| Vibrational Mode [30] | Raman Shift (cm−1) | 0% (Norm) | a | b | |||||
| 5% | 10% | 15% | 5% | 10% | 15% | ||||
| 1 | Si-C torsion | 160.01 | 0.097 | −12.43% | 4.69% | 6.42% | −2.03% | −2.11% | 1.69% |
| 2 | C-Si-C deflection/C-Si-O deflection | 191.33 | 0.1 | −10.55% | 0.28% | 4.83% | −6.90% | −5.89% | −0.85% |
| 3 | Si-O-Si stretch | 491.78 | 0.373 | −3.89% | −3.55% | 4.92% | −5.31% | −5.55% | −1.36% |
| 4 | Si-C stretch | 689.19 | 0.067 | −4.59% | −4.90% | 0.98% | −8.15% | −6.55% | −5.46% |
| 5 | Si–C symmetric stretch/CH3 rock | 710.89 | 0.271 | −6.82% | −7.87% | 1.02% | −16.78% | −9.40% | −6.32% |
| 6 | Si–C stretch/CH3 rock | 791.04 | 0.045 | −18.12% | −13.81% | −0.22% | −24.81% | −18.59% | −16.84% |
| 7 | CH3 asymmetric stretch | 861.97 | 0.03 | −19.22% | −18.80% | 0.49% | −34.65% | −23.14% | −15.60% |
| 8 | Si-CH3 symmetric bending | 1265.97 | 0.03 | −26.69% | −7.55% | −6.38% | −32.01% | −20.28% | −18.35% |
| 9 | CH3 asymmetric bending | 1411.80 | 0.058 | −16.67% | −16.06% | −3.87% | −23.19% | −15.18% | −16.39% |
| 10 | CH3 symmetric stretch | 2907.15 | 1 | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% |
| 11 | CH3 asymmetric stretch | 2966.36 | 0.311 | −0.43% | 0.67% | 0.54% | −1.04% | 0.80% | −0.06% |
| PDMS 15:1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Wavenumber (cm−1) | Vibration Mode | 0% | a | b | |||||
| 5% | 10% | 15% | 5% | 10% | 15% | ||||
| f | 660.5 | Si-CH3 | 0.14 | −5.97% | −13.31% | −15.51% | −6.13% | −7.39% | −3.58% |
| c | 684.61 | Si-(CH3)3 | 0.14 | −9.92% | −15.95% | −16.54% | −13.98% | −12.65% | −5.63% |
| c | 699.55 | Si-(CH3)3 | 0.14 | −9.39% | −14.22% | −14.77% | −14.40% | −13.11% | −4.44% |
| b-d | 784.4 | Si(CH3)2-O-Si(CH3)2-/Si-(CH3)3 | 1 | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% |
| c-e | 864.44 | Si-(CH3)3/Si-OH | 0.1 | −2.91% | −9.36% | −10.64% | −8.84% | −8.16% | −2.86% |
| a | 1008.11 | Si-(CH3)n | 0.76 | 2.33% | 2.86% | 4.52% | 3.36% | 5.09% | 6.64% |
| a | 1077.53 | Si-(CH3)n | 0.35 | 5.84% | 6.04% | 9.21% | 8.91% | 8.90% | 14.60% |
| b-d | 1257.36 | Si(CH3)2-O-Si(CH3)2-/Si-(CH3)3 | 0.41 | 2.64% | 1.42% | 0.21% | 5.77% | 4.60% | 0.03% |
| f | 1412.12 | Si-CH3 | 0.03 | −11.68% | 18.42% | −0.16% | −7.53% | −0.13% | −7.23% |
| g | 2905.72 | C-H | 0.02 | 19.77% | 19.94% | 15.16% | 21.14% | 38.46% | 26.67% |
| a,d | 2962.61 | SI(CH3)n/Si-(CH3)3 | 0.08 | 3.52% | 2.91% | 3.15% | 7.21% | 10.57% | 6.78% |
| PDMS 10:1 | |||||||||
| Wavenumber (cm−1) | Vibration Mode | 0% | a | b | |||||
| 5% | 10% | 15% | 5% | 10% | 15% | ||||
| f | 660.5 | Si-CH3 | 0.13 | 3.30% | −1.93% | −2.04% | 14.20% | 9.34% | 0.18% |
| c | 684.61 | Si-(CH3)3 | 0.13 | −3.72% | −5.46% | −2.15% | 4.32% | −0.76% | −3.27% |
| c | 699.55 | Si-(CH3)3 | 0.13 | −4.19% | −4.39% | −1.32% | 2.21% | −1.33% | −3.40% |
| b-d | 784.89 | Si(CH3)2-O-Si(CH3)2-/Si-(CH3)3 | 1 | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% |
| c-e | 864.44 | Si-(CH3)3/Si-OH | 0.1 | −3.36% | −0.27% | −6.30% | 3.40% | 3.23% | −1.29% |
| a | 1008.11 | Si-(CH3)n | 0.81 | −8.03% | −4.26% | −2.26% | −3.50% | 0.60% | 0.62% |
| a | 1077.05 | Si-(CH3)n | 0.37 | −5.24% | −1.53% | −1.61% | 5.56% | 10.05% | 10.24% |
| b-d | 1257.36 | Si(CH3)2-O-Si(CH3)2-/Si-(CH3)3 | 0.43 | −9.49% | −6.03% | −5.15% | 1.88% | 1.91% | 0.67% |
| f | 1412.6 | Si-CH3 | 0.03 | −12.53% | 4.92% | 1.33% | 34.26% | 11.54% | 25.17% |
| g | 2905.72 | C-H | 0.02 | −10.22% | −5.54% | −1.88% | 26.84% | 49.80% | 19.59% |
| a,d | 2962.125 | SI(CH3)n/Si-(CH3)3 | 0.08 | −9.16% | −5.96% | −3.00% | 5.50% | 12.44% | 5.35% |
| PDMS 5:1 | |||||||||
| Wavenumber (cm−1) | Vibration Mode | 0% | a | b | |||||
| 5% | 10% | 15% | 5% | 10% | 15% | ||||
| f | 660.5 | Si-CH3 | 0.13 | −8.61% | −10.32% | −14.05% | −1.20% | −8.64% | −7.90% |
| c | 685.09 | Si-(CH3)3 | 0.13 | −9.93% | −6.73% | −7.98% | −5.34% | −5.77% | −5.15% |
| c | 699.55 | Si-(CH3)3 | 0.13 | −9.22% | −4.16% | −5.11% | −5.03% | −4.01% | −4.00% |
| b-d | 784.4 | Si(CH3)2-O-Si(CH3)2-/Si-(CH3)3 | 1 | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% |
| c-e | 864.44 | Si-(CH3)3/Si-OH | 0.1 | −6.99% | −3.89% | 0.87% | −4.18% | −4.98% | −1.71% |
| a | 1008.11 | Si-(CH3)n | 0.74 | 1.52% | 2.59% | 5.93% | 4.57% | 7.73% | 8.11% |
| a | 1077.05 | Si-(CH3)n | 0.35 | 1.38% | 0.99% | 7.03% | 4.53% | 7.50% | 10.79% |
| b-d | 1257.36 | Si(CH3)2-O-Si(CH3)2-/Si-(CH3)3 | 0.4 | 0.16% | −1.37% | −1.94% | 4.09% | 2.58% | 1.95% |
| f | 1412.12 | Si-CH3 | 0.03 | −8.22% | 1.21% | −9.22% | −13.18% | −10.33% | 17.51% |
| g | 2905.24 | C-H | 0.02 | 5.83% | 8.11% | 7.29% | 59.76% | 24.65% | 17.05% |
| a,d | 2962.61 | SI(CH3)n/Si-(CH3)3 | 0.08 | 3.15% | 2.38% | 4.47% | 14.40% | 8.26% | 5.90% |
| PDMS 15:1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Species | Binding Energy (eV) | Intensity (CPS) | FWHM (eV) | Area (CPS•eV) | Area (Norm) | Atomic Conc. (%) | Atomic Ratio * | Relation Shift | |
| 0% | Si2p | 100.66 | 5009.84 | 1.54 | 8229.91 | 0.19 | 20.21% | 1.000 | - |
| 102.12 | 16,946.03 | 1.99 | 35,922.96 | 0.81 | |||||
| C1s | 283.78 | 27,009.70 | 1.46 | 42,091.72 | 0.27 | 61.82% | 3.059 | - | |
| 284.98 | 71,324.21 | 1.48 | 112,418.83 | 0.73 | |||||
| O1s | 529.08 | 57,659.77 | 1.59 | 106,054.23 | 1.00 | 17.06% | 0.844 | - | |
| Sn3d | 482.38 | 13,153.65 | 2.07 | 50,075.61 | 1.00 | 0.91% | 0.045 | - | |
| 5% a | Si2p | 100.84 | 7870.52 | 1.60 | 13,418.41 | 0.22 | 25.54% | 1.000 | 0.000 |
| 102.30 | 25,513.39 | 1.80 | 48,861.13 | 0.78 | |||||
| C1s | 283.39 | 10,737.10 | 1.21 | 13,828.33 | 0.09 | 53.62% | 2.099 | −0.959 | |
| 284.78 | 73,470.19 | 1.69 | 131,790.71 | 0.91 | |||||
| O1s | 526.00 | 82,874.91 | 1.62 | 152,715.54 | 1.00 | 20.84% | 0.816 | −0.028 | |
| 10% a | Si2p | 100.78 | 7243.57 | 1.46 | 11,229.16 | 0.16 | 26.34% | 1.000 | 0.000 |
| 102.32 | 29,902.90 | 1.92 | 60,969.90 | 0.84 | |||||
| C1s | 283.47 | 11,458.63 | 1.21 | 14,780.85 | 0.09 | 52.11% | 1.978 | −1.081 | |
| 284.83 | 78,640.31 | 1.71 | 143,052.16 | 0.91 | |||||
| O1s | 526.00 | 94,731.26 | 1.63 | 175,305.35 | 1.00 | 21.55% | 0.818 | −0.026 | |
| 15% a | Si2p | 100.75 | 5996.81 | 1.49 | 9536.94 | 0.14 | 26.40% | 1.000 | 0.000 |
| 102.36 | 28,171.11 | 1.99 | 59,791.79 | 0.86 | |||||
| C1s | 283.47 | 9441.53 | 1.23 | 12,359.82 | 0.08 | 51.30% | 1.943 | −1.116 | |
| 284.84 | 73,199.86 | 1.72 | 133,768.13 | 0.92 | |||||
| O1s | 526.00 | 91,951.90 | 1.63 | 170,697.23 | 1.00 | 22.30% | 0.845 | 0.001 | |
| 5% b | Si2p | 100.92 | 8986.55 | 1.54 | 14,696.63 | 0.22 | 27.37% | 1.000 | 0.000 |
| 102.40 | 26,703.67 | 1.81 | 51,393.55 | 0.78 | |||||
| C1s | 283.45 | 11,868.31 | 1.36 | 17,127.24 | 0.13 | 50.46% | 1.844 | −1.215 | |
| 284.81 | 65,734.69 | 1.71 | 119,319.86 | 0.87 | |||||
| O1s | 526.08 | 86,589.05 | 1.60 | 158,313.86 | 1.00 | 22.17% | 0.810 | −0.034 | |
| 10% b | Si2p | 100.54 | 3492.26 | 1.24 | 4616.74 | 0.08 | 26.08% | 1.000 | 0.000 |
| 102.31 | 24,503.38 | 2.17 | 56,610.91 | 0.92 | |||||
| C1s | 283.48 | 10,530.95 | 1.17 | 13,088.25 | 0.10 | 50.40% | 1.933 | −1.126 | |
| 284.83 | 67,031.50 | 1.62 | 115,775.55 | 0.90 | |||||
| O1s | 526.00 | 85,752.56 | 1.66 | 163,780.62 | 1.00 | 23.52% | 0.902 | 0.058 | |
| 15% b | Si2p | 100.80 | 8372.29 | 1.53 | 13,591.69 | 0.17 | 27.19% | 1.000 | 0.000 |
| 102.38 | 33,761.94 | 1.88 | 67,485.20 | 0.83 | |||||
| C1s | 283.48 | 15,322.67 | 1.20 | 19,562.28 | 0.12 | 49.84% | 1.833 | −1.226 | |
| 284.86 | 83,141.20 | 1.63 | 144,223.20 | 0.88 | |||||
| O1s | 526.08 | 109,943.27 | 1.61 | 203,118.13 | 1.00 | 22.98% | 0.845 | 0.001 | |
| PDMS 10:1 | |||||||||
| Species | Binding Energy (eV) | Intensity (CPS) | FWHM (eV) | Area (CPS•eV) | Area (Norm) | Atomic conc. (%) | Atomic Ratio * | Relation Shift | |
| 0% | Si2p | 100.39 | 233.10 | 1.33 | 329.17 | 0.07 | 25.95% | 1.000 | - |
| 102.25 | 2147.49 | 1.93 | 4408.81 | 0.93 | |||||
| C1s | 283.15 | 438.10 | 1.41 | 657.33 | 0.06 | 52.68% | 2.030 | - | |
| 284.79 | 5821.64 | 1.67 | 10,319.33 | 0.94 | |||||
| O1s | 532.37 | 6454.43 | 1.52 | 11,236.48 | 0.82 | 21.37% | 0.824 | - | |
| 5% a | Si2p | 100.68 | 5679.92 | 1.49 | 9038.24 | 0.15 | 25.62% | 1.000 | 0.000 |
| 102.30 | 25,206.02 | 1.84 | 49,283.79 | 0.85 | |||||
| C1s | 283.35 | 9915.91 | 1.14 | 12,046.90 | 0.09 | 52.19% | 2.037 | 0.007 | |
| 284.77 | 68,133.75 | 1.63 | 118,566.48 | 0.91 | |||||
| O1s | 532.40 | 80,913.78 | 1.60 | 148,934.83 | 0.87 | 22.19% | 0.866 | 0.043 | |
| 10% a | Si2p | 100.73 | 4663.48 | 1.43 | 7079.97 | 0.12 | 26.46% | 1.000 | 0.000 |
| 102.28 | 24,304.36 | 2.03 | 52,505.68 | 0.88 | |||||
| C1s | 283.42 | 7865.35 | 1.19 | 9949.09 | 0.08 | 51.21% | 1.935 | −0.095 | |
| 284.77 | 62,767.35 | 1.75 | 116,978.52 | 0.92 | |||||
| O1s | 532.42 | 82,356.19 | 1.57 | 148,456.83 | 0.84 | 22.33% | 0.844 | 0.020 | |
| 15% a | Si2p | 100.83 | 7234.27 | 1.49 | 11,474.71 | 0.18 | 25.78% | 1.000 | 0.000 |
| 102.35 | 25,715.67 | 1.87 | 51,260.25 | 0.82 | |||||
| C1s | 283.51 | 12,468.89 | 1.26 | 16,769.45 | 0.12 | 53.47% | 2.074 | 0.044 | |
| 284.84 | 71,719.00 | 1.68 | 128,095.72 | 0.88 | |||||
| O1s | 532.45 | 81,880.60 | 1.60 | 150,201.29 | 0.80 | 20.75% | 0.805 | −0.019 | |
| 5% b | Si2p | 100.61 | 5895.03 | 1.42 | 8900.28 | 0.15 | 25.07% | 1.000 | 0.000 |
| 102.26 | 25,818.22 | 1.89 | 51,972.14 | 0.85 | |||||
| C1s | 283.37 | 12,287.41 | 1.18 | 15,383.71 | 0.11 | 53.09% | 2.118 | 0.088 | |
| 284.76 | 74,468.33 | 1.60 | 126,702.54 | 0.89 | |||||
| O1s | 532.36 | 85,026.62 | 1.63 | 158,707.76 | 0.87 | 21.83% | 0.871 | 0.047 | |
| 10% b | Si2p | 100.59 | 4233.19 | 1.32 | 5938.89 | 0.09 | 26.89% | 1.000 | 0.000 |
| 102.35 | 27,144.65 | 2.00 | 57,705.79 | 0.91 | |||||
| C1s | 283.35 | 9908.59 | 1.15 | 12,088.14 | 0.09 | 48.70% | 1.811 | −0.219 | |
| 284.77 | 67,028.12 | 1.62 | 115,583.89 | 0.91 | |||||
| O1s | 532.41 | 94,238.89 | 1.60 | 173,925.70 | 0.91 | 24.41% | 0.908 | 0.084 | |
| 15% b | Si2p | 100.51 | 2725.68 | 1.11 | 3232.76 | 0.05 | 27.17% | 1.000 | 0.000 |
| 102.43 | 26,596.30 | 2.20 | 62,273.85 | 0.95 | |||||
| C1s | 283.44 | 8269.65 | 1.16 | 10,248.61 | 0.08 | 46.61% | 1.715 | −0.315 | |
| 284.84 | 64,416.59 | 1.66 | 113,486.00 | 0.92 | |||||
| O1s | 532.54 | 99,166.24 | 1.67 | 188,714.81 | 0.97 | 26.22% | 0.965 | 0.142 | |
| PDMS 5:1 | |||||||||
| Species | Binding Energy (eV) | Intensity (CPS) | FWHM (eV) | Area (CPS•eV) | Area (Norm) | Atomic conc. (%) | Atomic Ratio * | Relation Shift | |
| 0% | Si2p | 100.71 | 597.44 | 1.46 | 928.48 | 0.08 | 20.94% | 1.000 | - |
| 102.20 | 2736.10 | 2.07 | 6030.24 | 0.92 | |||||
| C1s | 283.50 | 14,137.02 | 1.23 | 18,560.86 | 13.40 | 59.33% | 2.833 | - | |
| 284.83 | 73,262.12 | 1.54 | 119,971.03 | 86.60 | |||||
| O1s | 532.37 | 8445.93 | 1.66 | 16,195.07 | 0.83 | 17.30% | 0.826 | - | |
| Sn3d | 486.17 | 5100.06 | 2.08 | 19,964.75 | 1.16 | 24.30% | 1.160 | - | |
| 5% a | Si2p | 100.56 | 5524.30 | 1.42 | 8355.49 | 0.08 | 25.19% | 1.000 | 0.000 |
| 102.22 | 25,720.76 | 1.92 | 52,577.57 | 0.92 | |||||
| C1s | 283.50 | 14,142.34 | 1.23 | 18,576.17 | 13.41 | 52.75% | 2.094 | −0.739 | |
| 284.83 | 73,258.51 | 1.54 | 119,956.16 | 86.59 | |||||
| O1s | 532.41 | 84,872.40 | 1.62 | 158,280.22 | 0.88 | 22.06% | 0.876 | 0.050 | |
| 10% a | Si2p | 100.57 | 5524.30 | 1.42 | 8355.49 | 0.08 | 25.99% | 1.000 | 0.000 |
| 102.22 | 25,720.76 | 1.92 | 52,577.57 | 0.92 | |||||
| C1s | 283.50 | 10,205.15 | 1.26 | 13,657.94 | 10.70 | 50.60% | 1.947 | −0.886 | |
| 284.80 | 66,373.00 | 1.61 | 113,936.51 | 89.30 | |||||
| O1s | 532.50 | 87,513.28 | 1.61 | 161,871.71 | 0.90 | 23.41% | 0.901 | 0.075 | |
| 15% a | Si2p | 100.73 | 918.92 | 1.49 | 1456.09 | 0.07 | 25.58% | 1.000 | 0.000 |
| 102.23 | 4952.13 | 2.03 | 10,696.51 | 0.93 | |||||
| C1s | 283.46 | 1798.26 | 1.36 | 2596.39 | 9.56 | 51.86% | 2.027 | −0.806 | |
| 284.81 | 13,236.78 | 1.74 | 24,556.04 | 90.44 | |||||
| O1s | 532.52 | 16,964.46 | 1.59 | 31,657.35 | 0.88 | 22.55% | 0.882 | 0.055 | |
| 5% b | Si2p | 100.59 | 2754.67 | 1.23 | 3605.89 | 0.04 | 24.15% | 1.000 | 0.000 |
| 102.36 | 19,783.53 | 1.97 | 41,467.13 | 0.96 | |||||
| C1s | 283.18 | 3733.70 | 0.96 | 3806.93 | 3.66 | 51.91% | 2.149 | −0.684 | |
| 284.80 | 53,565.28 | 1.76 | 100,322.67 | 96.34 | |||||
| O1s | 532.47 | 69,902.20 | 1.67 | 134,749.28 | 0.99 | 23.94% | 0.991 | 0.165 | |
| 10% b | Si2p | 100.86 | 6752.94 | 1.47 | 10,592.09 | 0.09 | 27.22% | 1.000 | 0.000 |
| 102.37 | 26,917.55 | 1.95 | 55,842.61 | 0.91 | |||||
| C1s | 283.51 | 11,301.88 | 1.22 | 14,633.87 | 10.87 | 50.57% | 1.858 | −0.976 | |
| 284.85 | 65,557.64 | 1.72 | 120,000.53 | 89.13 | |||||
| O1s | 532.45 | 86,180.03 | 1.62 | 159,336.90 | 0.82 | 22.21% | 0.816 | −0.010 | |
| 15% b | Si2p | 100.64 | 4419.27 | 1.32 | 6206.52 | 0.06 | 27.44% | 1.000 | 0.000 |
| 102.35 | 25,854.94 | 2.01 | 553,93.02 | 0.94 | |||||
| C1s | 283.48 | 9092.33 | 1.18 | 11,413.69 | 9.84 | 48.15% | 1.755 | −1.079 | |
| 284.79 | 59,787.62 | 1.64 | 104,542.07 | 90.16 | |||||
| O1s | 532.45 | 88,917.78 | 1.60 | 163,418.89 | 0.89 | 24.41% | 0.890 | 0.063 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cordoba, A.; Cauich-Rodríguez, J.V.; Vargas-Coronado, R.F.; Velázquez-Castillo, R.; Esquivel, K. A Novel In Situ Sol-Gel Synthesis Method for PDMS Composites Reinforced with Silica Nanoparticles. Polymers 2024, 16, 1125. https://doi.org/10.3390/polym16081125
Cordoba A, Cauich-Rodríguez JV, Vargas-Coronado RF, Velázquez-Castillo R, Esquivel K. A Novel In Situ Sol-Gel Synthesis Method for PDMS Composites Reinforced with Silica Nanoparticles. Polymers. 2024; 16(8):1125. https://doi.org/10.3390/polym16081125
Chicago/Turabian StyleCordoba, Aldo, Juan Valerio Cauich-Rodríguez, Rossana Faride Vargas-Coronado, Rodrigo Velázquez-Castillo, and Karen Esquivel. 2024. "A Novel In Situ Sol-Gel Synthesis Method for PDMS Composites Reinforced with Silica Nanoparticles" Polymers 16, no. 8: 1125. https://doi.org/10.3390/polym16081125
APA StyleCordoba, A., Cauich-Rodríguez, J. V., Vargas-Coronado, R. F., Velázquez-Castillo, R., & Esquivel, K. (2024). A Novel In Situ Sol-Gel Synthesis Method for PDMS Composites Reinforced with Silica Nanoparticles. Polymers, 16(8), 1125. https://doi.org/10.3390/polym16081125