
Highly Effective Synthetic Polymer-Based Blockers of Non-Specific Interactions in Immunochemical Analyses



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Monomers, Chain Transfer Agent and Polymer Precursors
2.2.1. Synthesis of HPMA
2.2.2. Synthesis of 3-(3-Methacrylamido-propanoyl)thiazolidine-2-thione
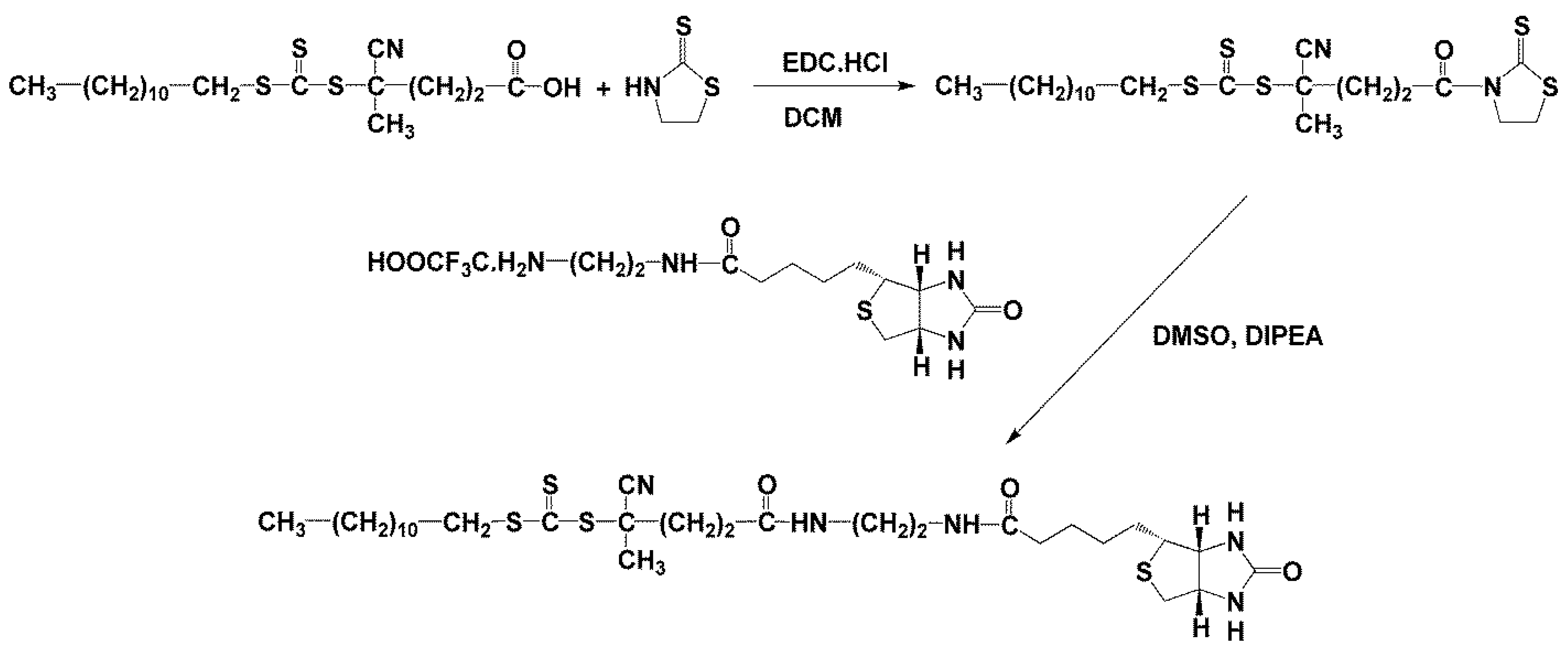
2.2.3. Synthesis of Chain Transfer Agent N-[2-[5-[(3aR,4R,6aS)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno [3,4-d]imidazol-4-yl]pentanoylamino]ethyl]-4-cyano-4-dodecylsulfanylcarbothioylsulfanyl-pentanamide (Dodecyl-trithiocarbonate-ED-biotin)
2.2.4. Synthesis of Copolymer Precursors by RAFT Copolymerisation
2.2.5. Synthesis of Copolymer Precursor by Radical Solution Copolymerisation
2.2.6. Synthesis of Copolymer Precursor by Radical Precipitation Copolymerisation
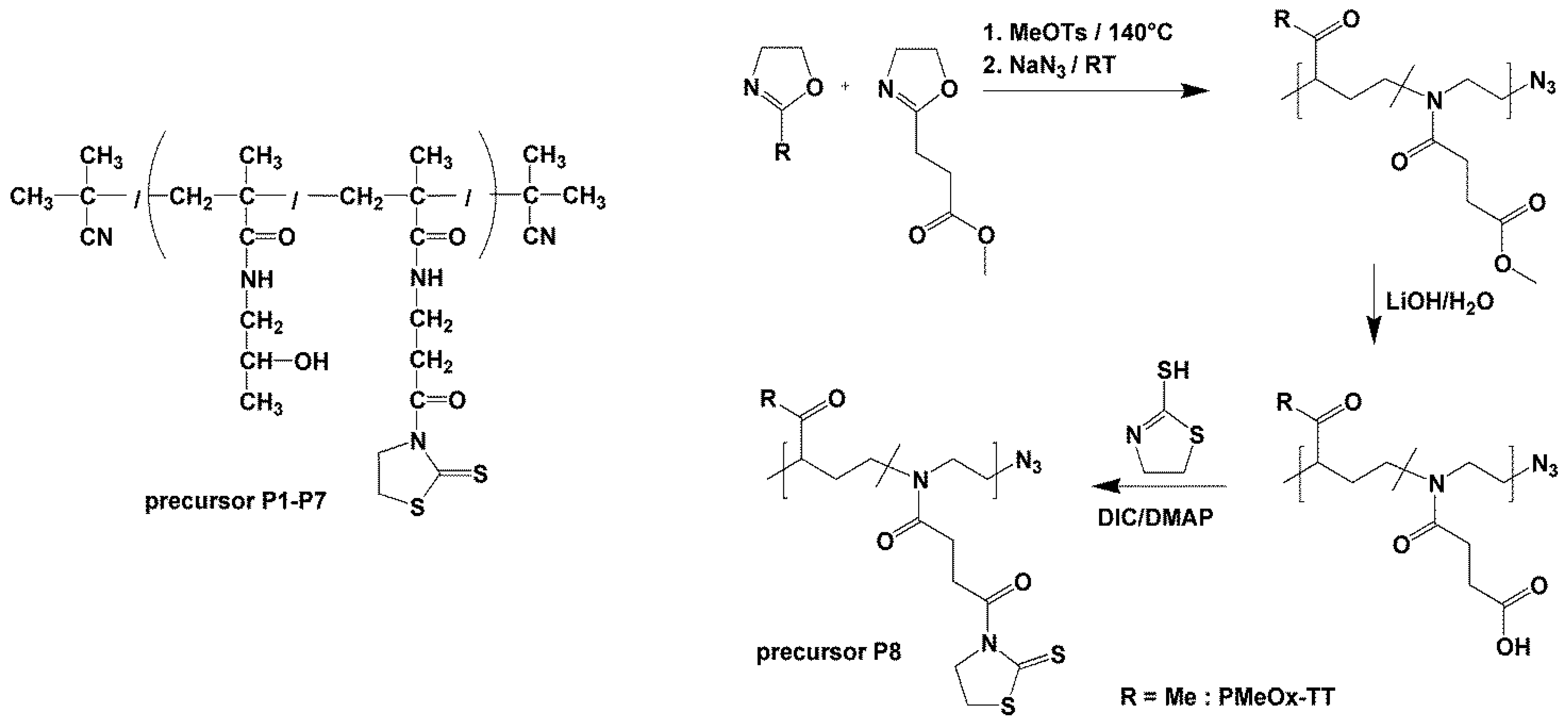
2.2.7. Synthesis of p(MeOx95-stat-(COOH)Ox5)-N3
2.2.8. Synthesis of pMeOx TT (P8)
2.3. Synthesis of Polymer Conjugates
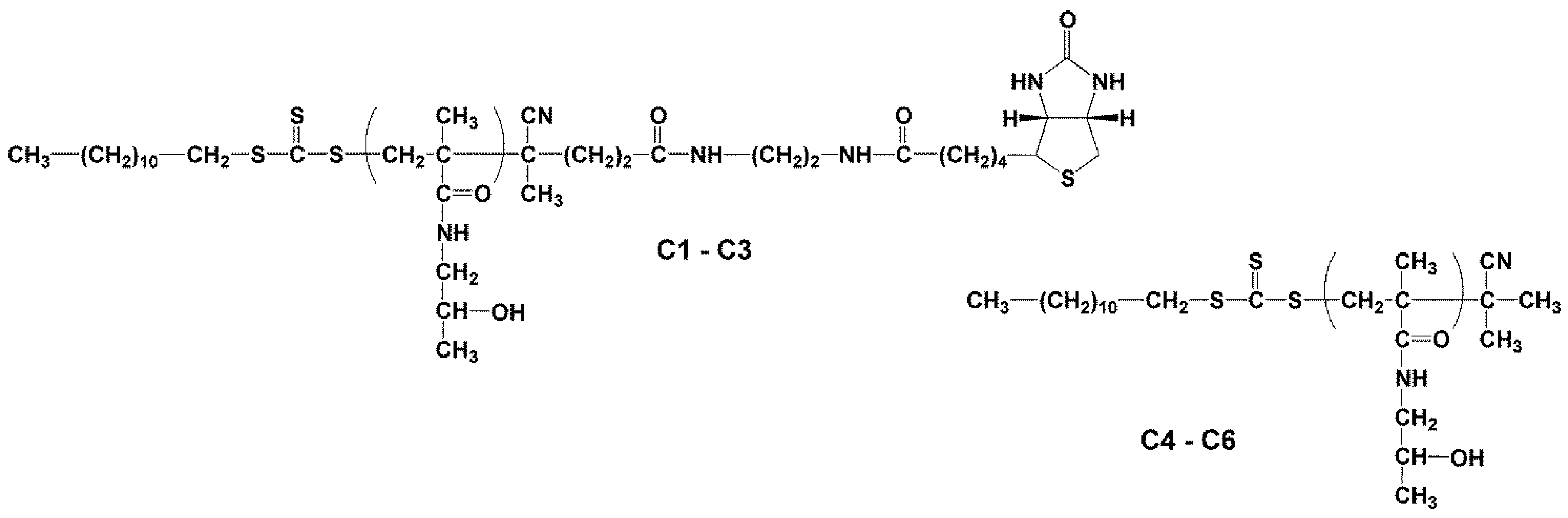
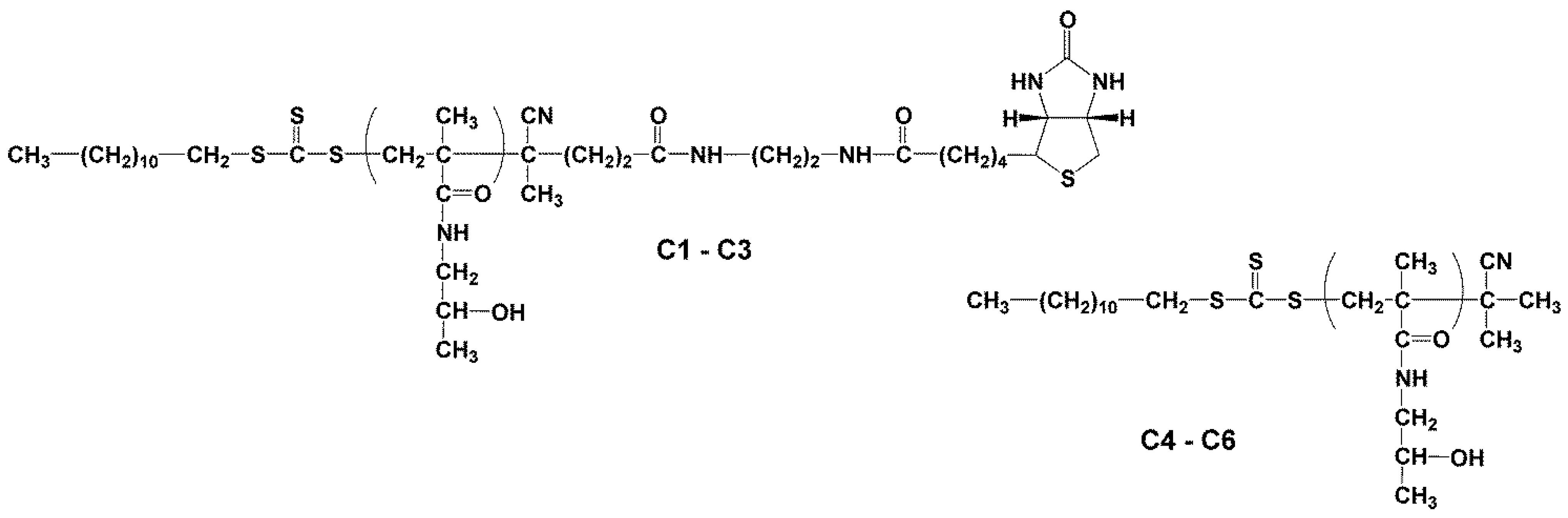
2.3.1. Synthesis of Heterotelechelic Polymer Conjugates C1–C3 Dodecyl-p(HPMA)-ED-biotin Using RAFT Polymerisation
2.3.2. Synthesis of Semitelechelic Dodecyl-p(HPMA) Conjugates C4–C6
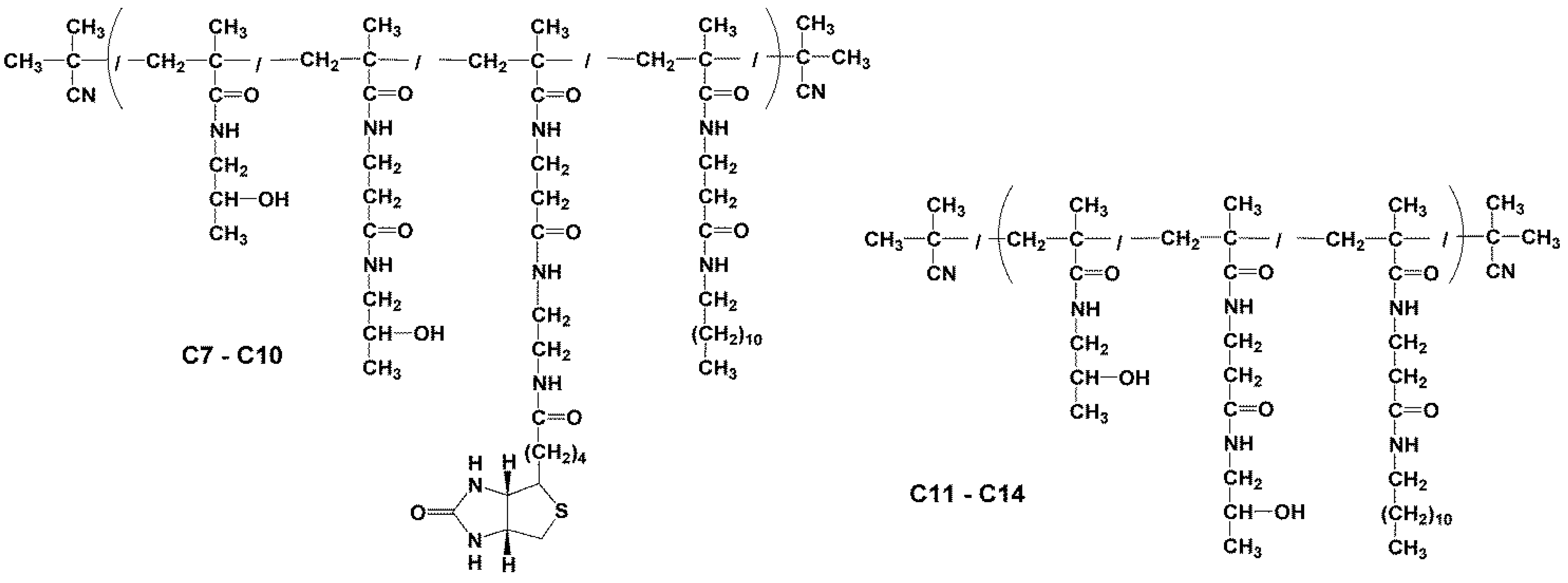
2.3.3. Synthesis of Conjugates p(HPMA-co-Ma-β-Ala-dodecylamin-co-Ma-β-Ala-ED-biotin) (C7–C10)
2.3.4. Synthesis of Polymer Conjugates p(HPMA-co-Ma-β-Ala-dodecylamin) (C11–C14)
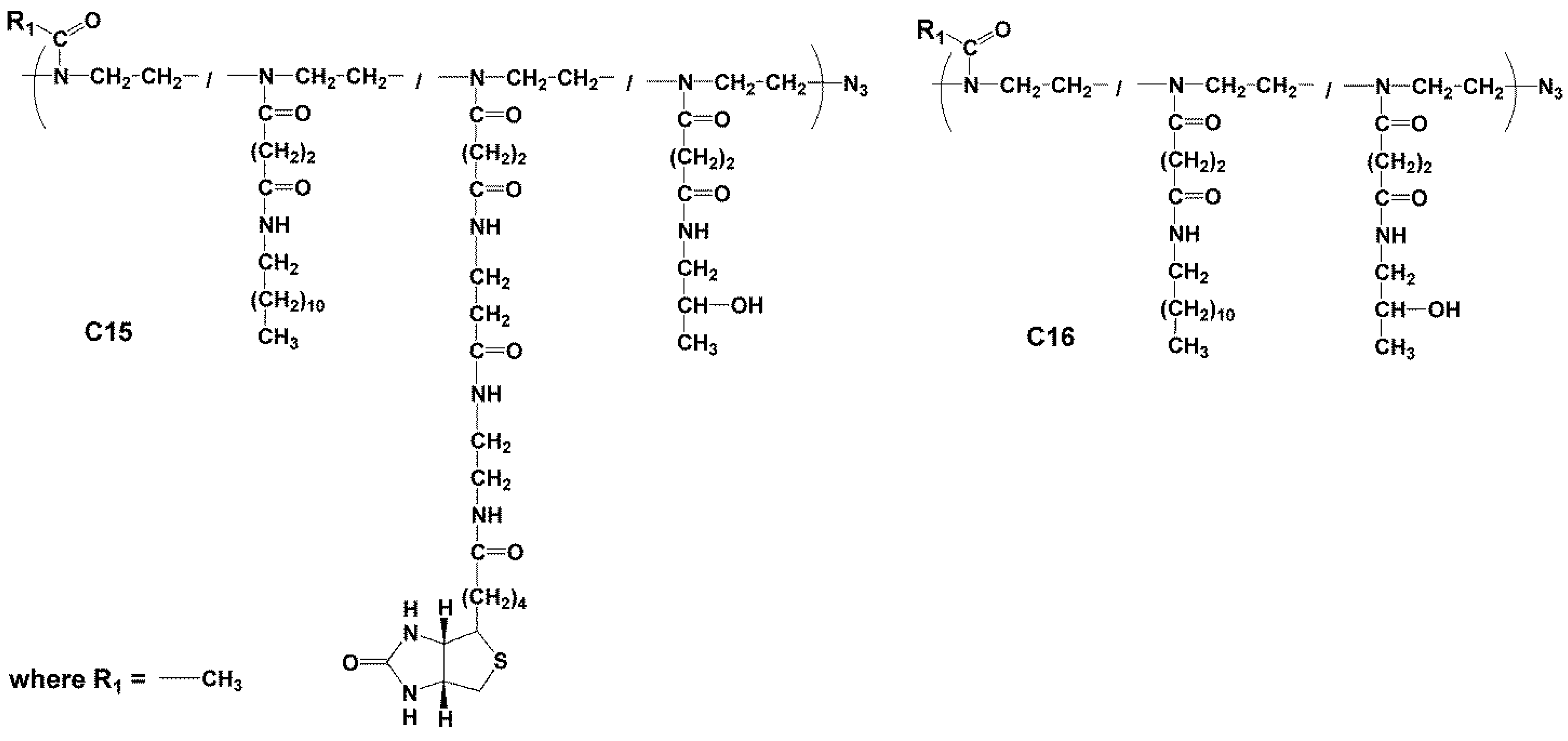
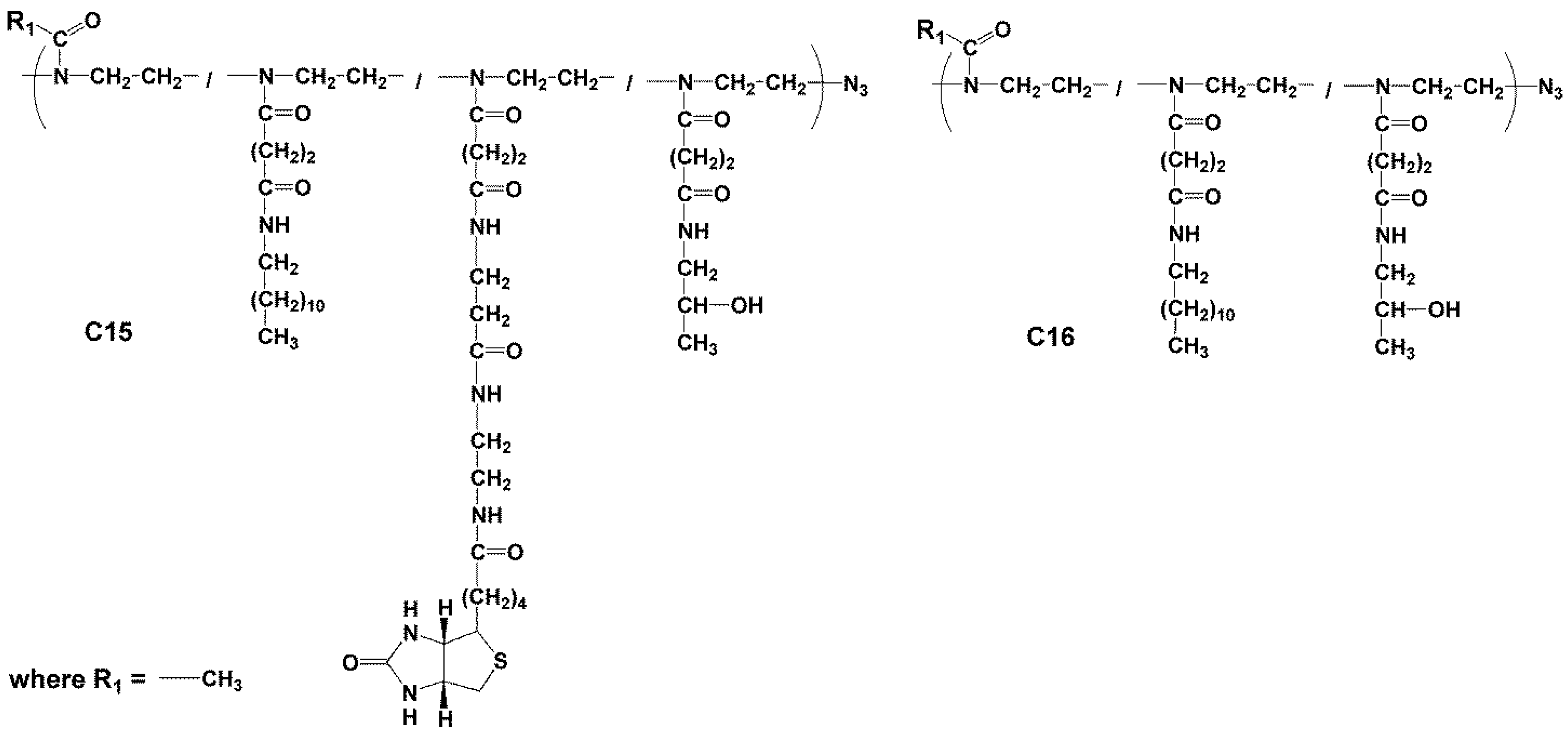
2.3.5. Synthesis of Polymer Conjugate C15
2.3.6. Synthesis of Conjugate C16—Poly(2-methyl-oxazolin-co-N’-dodecyl-N-ethyl-butanediamide-co-N’-2-hydroxypropyl-N-ethyl-butanediamide)
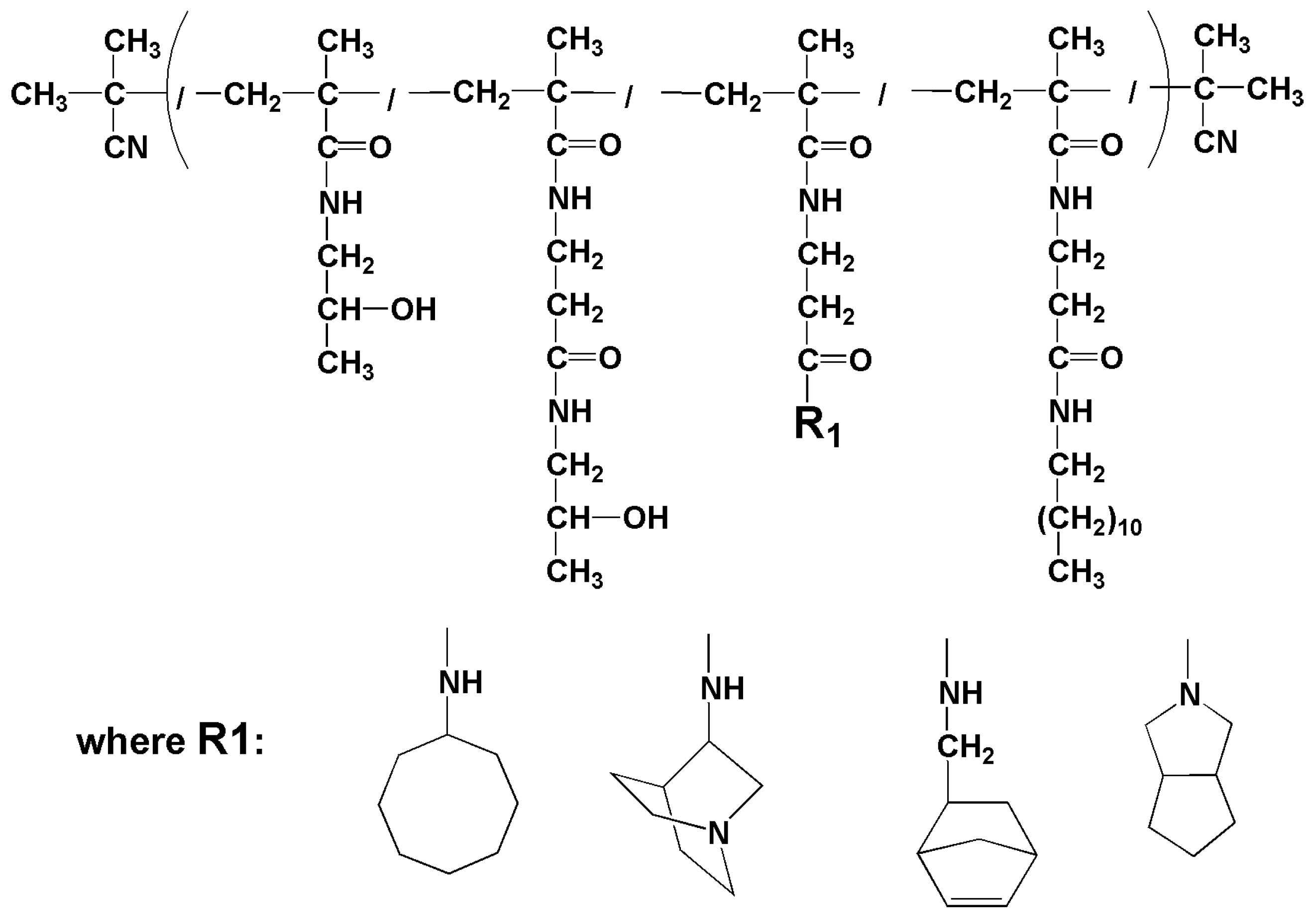
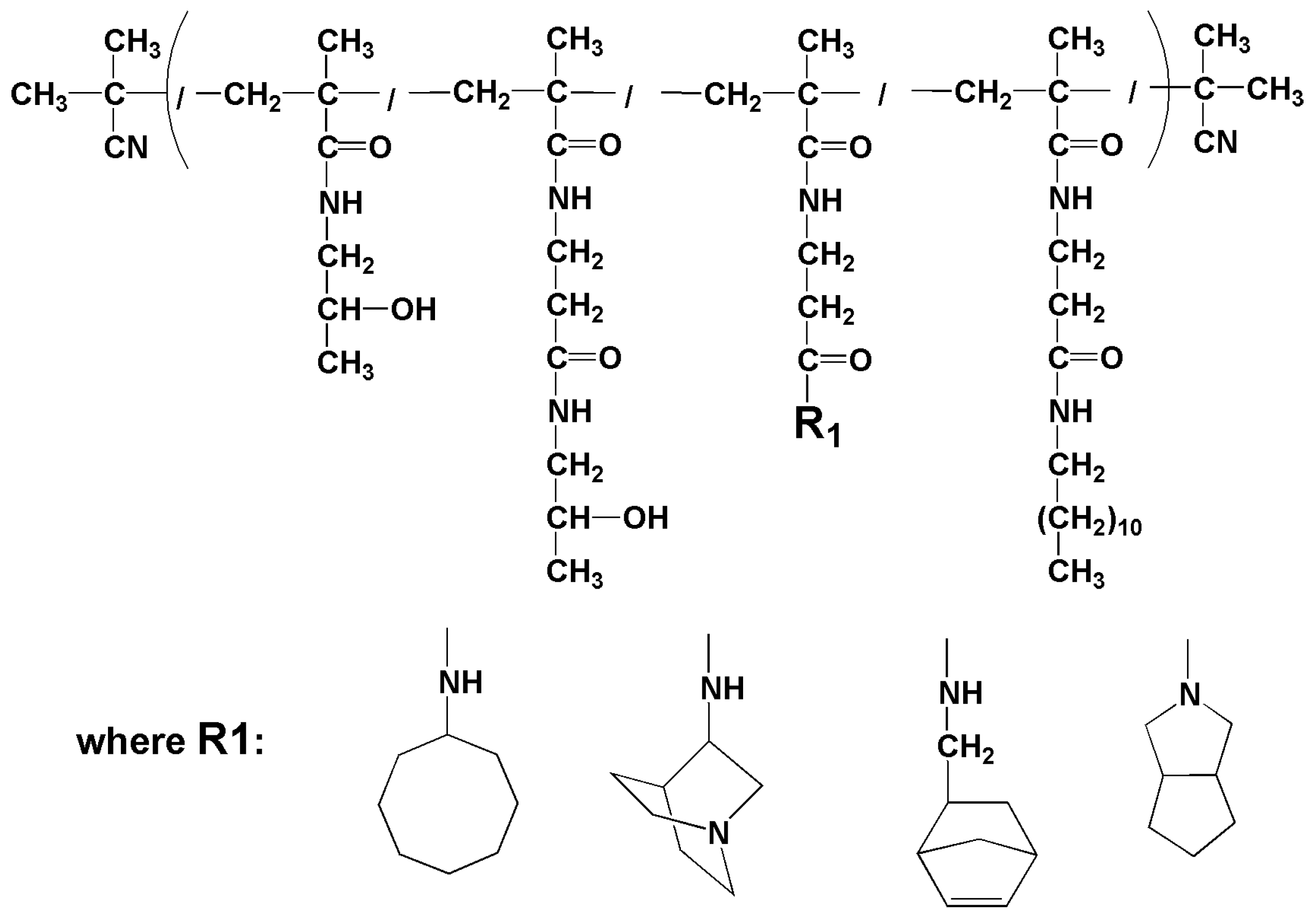
2.3.7. Synthesis of Polymer Conjugate C17—p(HPMA-co-Ma-β-Ala-dodecylamine-co-Ma-β-Ala-aminocyklooktane)
2.3.8. Synthesis of Polymer Conjugate C18—p(HPMA-co-Ma-β-Ala-dodecyl-amin-co-Ma-β-Ala-norbornen-2-methylamin)
2.3.9. Synthesis of Polymer Conjugate C19—p(HPMA-co-Ma-β-Ala-dodecyl-amin-co-Ma-β-Ala-aminoquinuclidin)
2.3.10. Synthesis of Polymer Conjugate C20—p(HPMA-co-Ma-β-Ala-dodecyl-amin-co-Ma-β-Ala- 3-azabicyclo [3,3,0]octane)
2.4. Characterisation of Polymer Precursors and Polymer Conjugates
2.5. Evaluation in Diagnostic Methods
2.5.1. Preparation of Antibody-Biotin Conjugate
2.5.2. Preparation of BSA-Biotin Conjugate
2.5.3. Preparation of Horseradish-Peroxidase Conjugate
2.5.4. Sorption of Biotinylated Conjugates
2.5.5. Sorption of Non-Biotinylated Conjugates
2.5.6. ELISA of Human Thyroid Stimulating Hormone (hTSH) in Serum
2.5.7. System with BSA
2.5.8. System with Conjugates C8 and C14
- -
- 50 µL of calibrator or sample + 150 µL of IgG-HRP conjugate solution was pipetted into each well
- -
- 120 min incubation at laboratory temperature without shaking
- -
- the solution was aspirated and washed 3 times with washing solution
- -
- 200 µL TMB was added and incubated for 10 min at room temperature in the dark
- -
- 50 µL of 2 M HCl was added and the optical density was measured at 450 nm (OD450)
3. Results and Discussions
3.1. Synthesis of Polymer Precursors
3.2. Synthesis of Polymer Conjugates
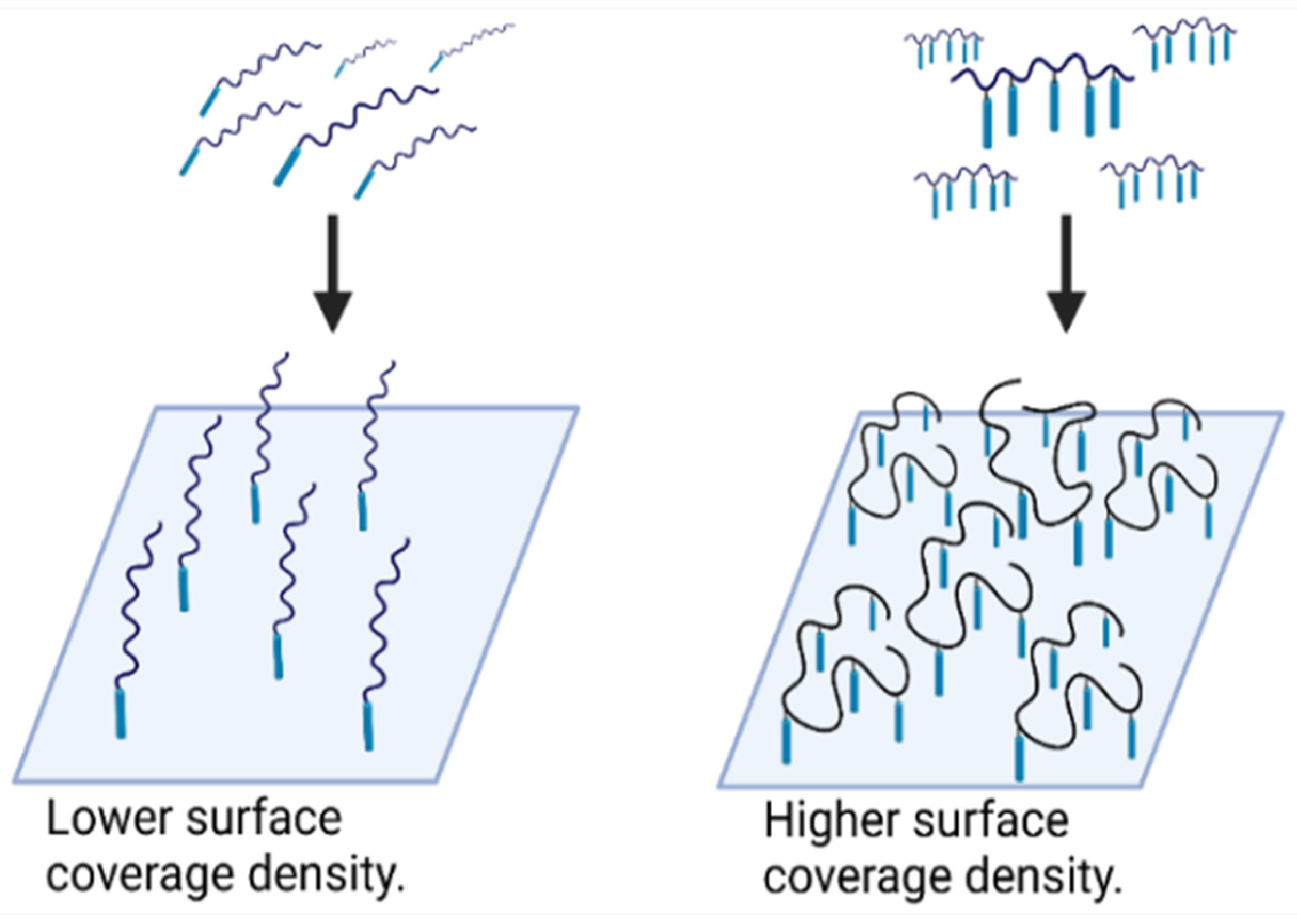
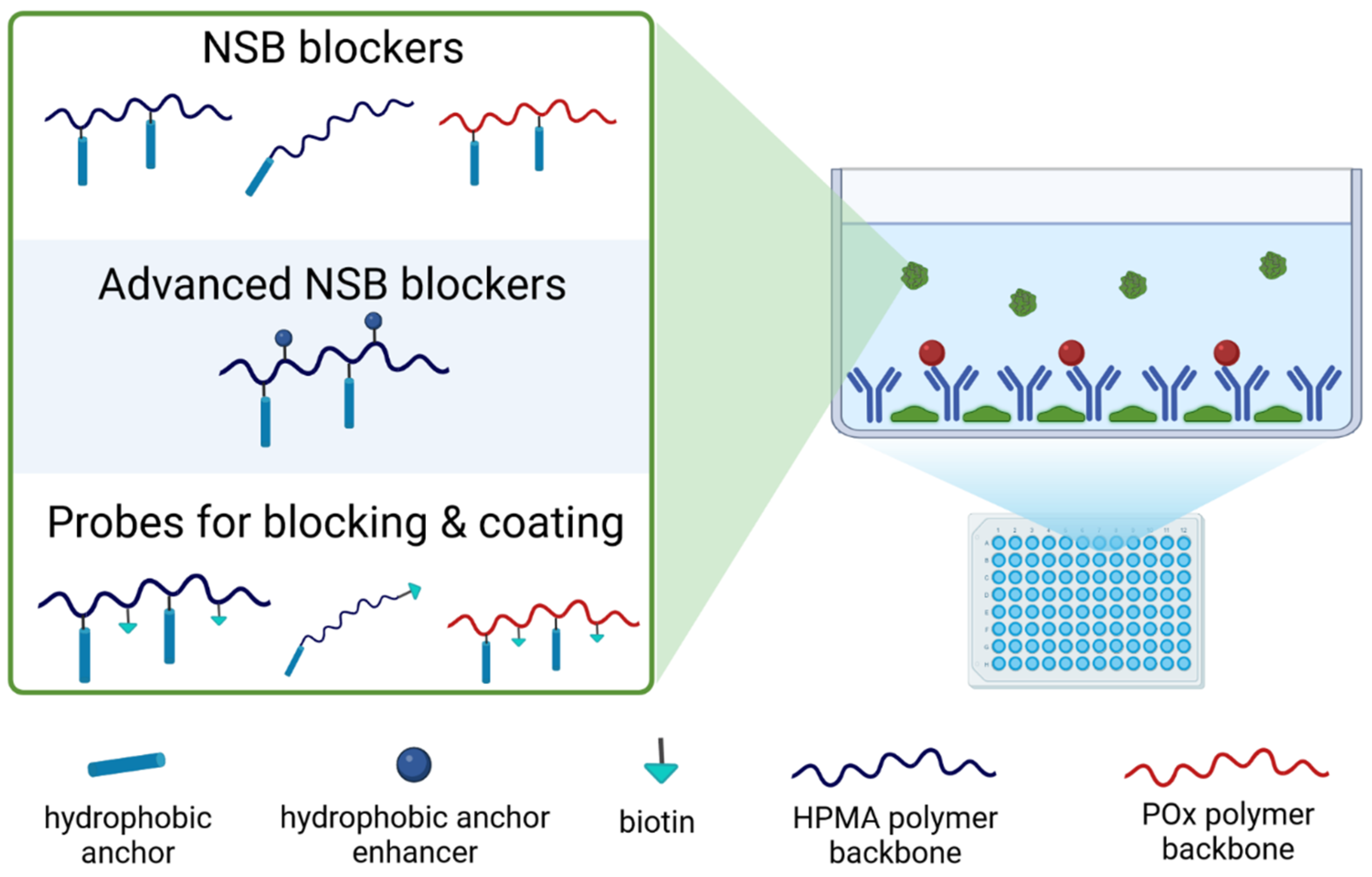
3.3. NSB Blockers
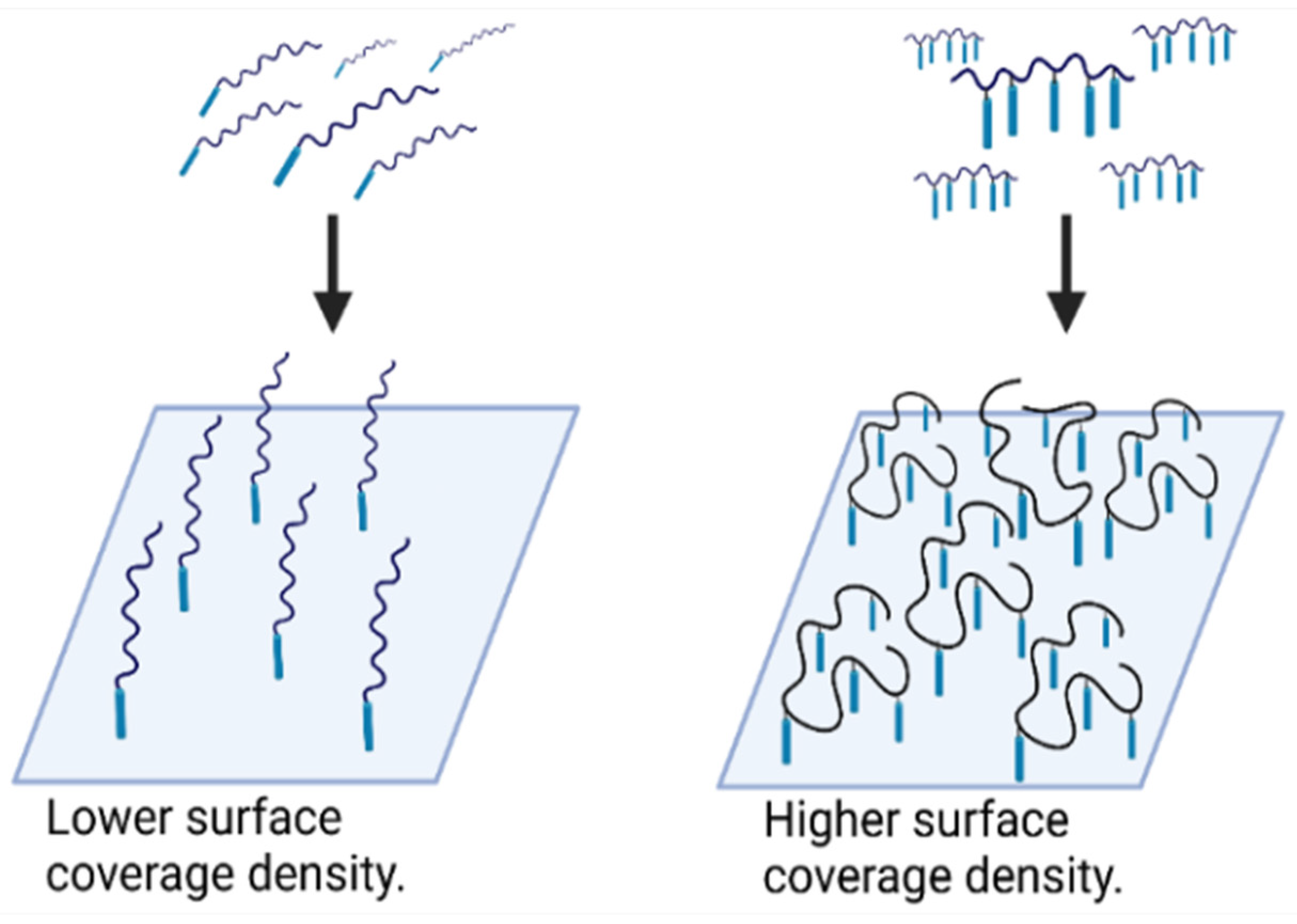
3.4. Probes for Blocking and Coating
3.5. Advanced NSB Blockers
3.6. Competitive Assay with BSA for Biotinylated Conjugates
3.7. The Ability of Non-Biotinylated Conjugates as NSB Reagents
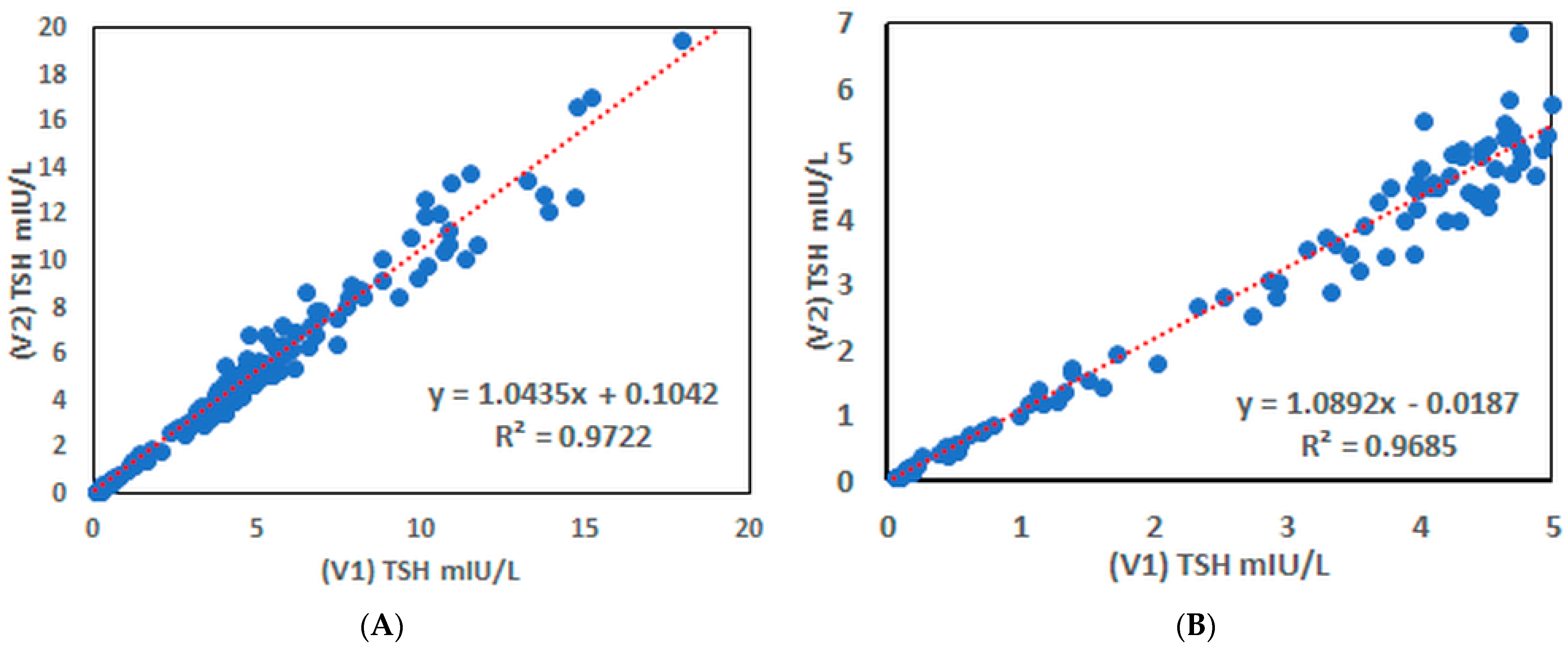
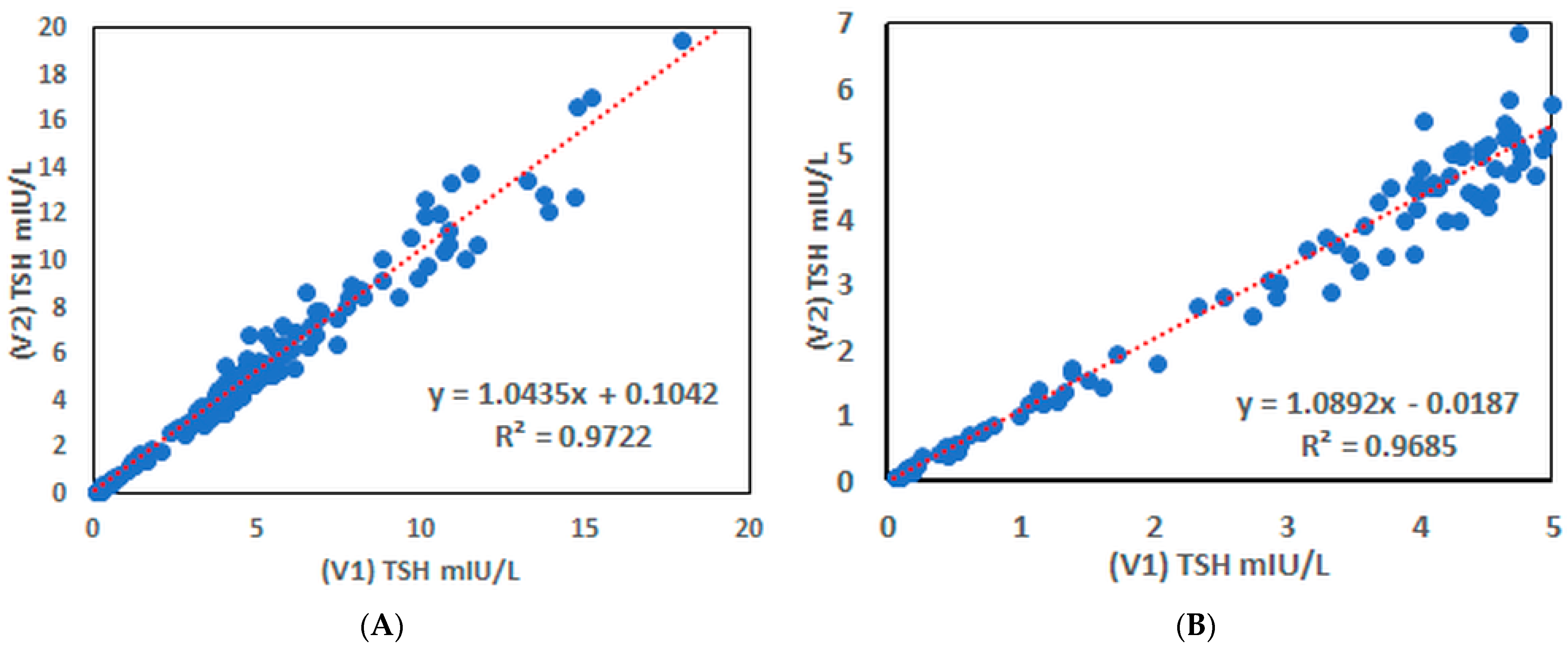
3.8. Determination of hTSH in Human Serum Samples
3.9. Advanced Polymer Blocker Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Steinitz, M. Quantitation of the blocking effect of Tween 20 and bovine serum albumin in ELISA microwells. Anal. Biochem. 2000, 282, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Wedege, E.; Svenneby, G. Effects of the Blocking-agents Bovine Serum-albumin and Tween 20 in Different Buffers on Immunoblotting of Brain Proteins and Marker Proteins. J. Immunol. Methods 1986, 88, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Robertson, P.W.; Whybin, L.R.; Cox, J. Reduction in Non-specific Binding in Enzyme Immunoassays Using Casein Hydrolysate in Serum Diluents. J. Immunol. Methods 1985, 76, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Rodda, D.J.; Yamazaki, H. Poly(vinyl alcohol) as a blocking agent in enzyme immunoassays. Immunol. Investig. 1994, 23, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Gardas, A.; Lewartowska, A. Coating of proteins to polystyrene ELISA plates in the presence of detergents. J. Immunol. Methods 1988, 106, 251–255. [Google Scholar] [CrossRef]
- Lim, C.S.; Krishnan, G.; Sam, C.K.; Ng, C.C. On optimizing the blocking step of indirect enzyme-linked immunosorbent assay for Epstein-Barr virus serology. Clin. Chim. Acta 2013, 415, 158–161. [Google Scholar] [CrossRef]
- Huber, D.; Rudolf, J.; Ansari, P.; Galler, B.; Fuhrer, M.; Hasenhindl, C.; Baumgartner, S. Effectiveness of natural and synthetic blocking reagents and their application for detecting food allergens in enzyme-linked immunosorbent assays. Anal. Bioanal. Chem. 2009, 394, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, N.; Bade, S.; Rockendorf, N.; Ramaker, K.; Frey, A. Polyethylene glycol-conjugated alkylamines—A novel class of surfactants for the saturation of immunoassay solid phase surfaces. Talanta 2020, 211, 120741. [Google Scholar] [CrossRef]
- Fujimoto, N.; Rockendorf, N.; Bade, S.; Ramaker, K.; Frey, A. Novel synthetic blocking reagents. EP2261662B1, 15 December 2010. [Google Scholar]
- Studentsov, Y.Y.; Schiffman, M.; Strickler, H.D.; Ho, G.Y.F.; Pang, Y.Y.S.; Schiller, J.; Herrero, R.; Burk, R.D. Enhanced enzyme-linked immunosorbent assay for detection of antibodies to virus-like particles of human papillomavirus. J. Clin. Microbiol. 2002, 40, 1755–1760. [Google Scholar] [CrossRef] [PubMed]
- Bouten, P.J.M.; Hertsen, D.; Vergaelen, M.; Monnery, B.D.; Boerman, M.A.; Goossens, H.; Catak, S.; van Hest, J.C.M.; Van Speybroeck, V.; Hoogenboom, R. Accelerated living cationic ring-opening polymerization of a methyl ester functionalized 2-oxazoline monomer. Polym. Chem. 2015, 6, 514–518. [Google Scholar] [CrossRef]
- Fairbanks, B.D.; Thissen, H.; Maurdev, G.; Pasic, P.; White, J.F.; Meagher, L. Inhibition of Protein and Cell Attachment on Materials Generated from <i>N</i>-(2-Hydroxypropyl) Acrylamide. Biomacromolecules 2014, 15, 3259–3266. [Google Scholar] [CrossRef] [PubMed]
- Šubr, V.; Ulbrich, K. Synthesis and properties of new N-(2-hydroxypropyl)methacrylamide copolymers containing thiazolidine-2-thione reactive groups. React. Funct. Polym. 2006, 66, 1525–1538. [Google Scholar] [CrossRef]
- Ishitake, K.; Satoh, K.; Kamigaito, M.; Okamoto, Y. Stereogradient Polymers Formed by Controlled/Living Radical Polymerization of Bulky Methacrylate Monomers. Angew. Chem. Int. Ed. 2009, 48, 1991–1994. [Google Scholar] [CrossRef] [PubMed]
- Perrier, S.; Takolpuckdee, P.; Mars, C.A. Reversible addition-fragmentation chain transfer polymerization: End group modification for functionalized polymers and chain transfer agent recovery. Macromolecules 2005, 38, 2033–2036. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Precursor | Polymerisation | Mw [g.mol−1] | Mn [g.mol−1] | Ð | TT Content mol% |
|---|---|---|---|---|---|
| P1 | RAFT | 74,000 | 58,000 | 1.25 | 13.8 |
| P2 | RAFT | 52,000 | 51,000 | 1.02 | 9.6 |
| P3 | RAFT | 49,000 | 40,000 | 1.23 | 11.6 |
| P4 | RAFT | 26,000 | 25,000 | 1.04 | 10.3 |
| P5 | RAFT | 11,000 | 10,000 | 1.09 | 8.1 |
| P6 | Solution FRP | 38,000 | 18,000 | 2.05 | 12.8 |
| P7 | Precipitation FRP | 41,000 | 20,000 | 2.08 | 11.8 |
| P8 | P-MeOx | 12,400 | 10,100 | 1.13 | 3.12 |
| Conjugates | Mw g.mol−1 | Mn g.mol−1 | Ð | Dodecyl Content wt% | Biotin Content wt% |
|---|---|---|---|---|---|
| C1 | 58,000 | 52,000 | 1.12 | 0.36 | 0.29 |
| C2 | 22,000 | 19,000 | 1.18 | 0.97 | 0.67 |
| C3 | 18,000 | 17,500 | 1.01 | 1.05 | 0.74 |
| C4 | 11,000 | 11,000 | 1.02 | 1.7 | - |
| C5 | 30,000 | 29,000 | 1.02 | 0.64 | - |
| C6 | 58,000 | 57,000 | 1.02 | 0.32 | - |
| Conjugates | Precursor | Mw g.mol−1 | Mn g.mol−1 | Ð | DDA wt% | DDA per Chain | Biotin wt% |
|---|---|---|---|---|---|---|---|
| C7 | P1 | 95,000 | 77,000 | 1.23 | 1.19 | 5.0 | 2.24 |
| C8 | P3 | 59,000 | 57,000 | 1.04 | 1.61 | 5.0 | 2.26 |
| C9 | P4 | 42,000 | 34,000 | 1.24 | 2.77 | 5.0 | 2.91 |
| C10 | P6 | 52,000 | 36,000 | 1.46 | 2.84 | 5.5 | 1.94 |
| C11 | P3 | 66,000 | 61,000 | 1.08 | 2.77 | 9.0 | - |
| C12 | P4 | 43,000 | 35,000 | 1.23 | 1.55 | 2.0 | - |
| C13 | P5 | 16,000 | 15,000 | 1.08 | 2.50 | 2.0 | - |
| C14 | P6 | 34,000 | 20,000 | 1.67 | 1.83 | 2.0 | - |
| C15 a | P8 | 14,000 | 12,300 | 1.14 | 1.24 | 0.8 | 1.03 |
| C16 a | P8 | 14,000 | 12,300 | 1.14 | 0.94 | 0.6 | - |
| Conjugates | Precursor | Mw g.mol−1 | Mn g.mol−1 | Ð | DDA wt% | CO wt% | NOR wt% | AQ wt% | BA wt% |
|---|---|---|---|---|---|---|---|---|---|
| C17 (F3-CO) | P6 | 43,000 | 30,000 | 1.43 | 1.03 | 0.85 | - | - | |
| C18 (F3-NOR) | P6 | 43,000 | 30,000 | 1.43 | 0.96 | - | 2.3 | - | |
| C19 (F3-AQ) | P7 | 39,000 | 27,000 | 1.44 | 1.05 | - | - | 0.87 | |
| C20 (F3-BA) | P7 | 52,000 | 28,000 | 1.74 | 1.27 | - | - | - | 3.3 + |
| Conjugate C4 | Conjugate C5 | Conjugate C13 | Conjugate C14 | BSA | |
|---|---|---|---|---|---|
| 0.0125 g/L | 0.125 g/L | ||||
| OD450 | |||||
| 1.357 | 2.471 | 1.748 | 1.821 | 1.304 | |
| 1.437 | 2.416 | 1.624 | 1.747 | 1.288 | |
| 1.446 | 2.485 | 1.587 | 1.872 | 1.227 | |
| 1.403 | 2.485 | 1.656 | 1.843 | 1.227 | |
| Mean | 1.411 | 2.464 | 1.654 | 1.821 | 1.261 |
| C.V., % * | 2.86 | 1.34 | 4.17 | 2.93 | 3.22 |
| Calibrator TSH, mIU/L | System with BSA (V1) | System with Conjugates C8 and C14 (V2) | ||
|---|---|---|---|---|
| Mean OD450 | C.V. *, % | Mean OD450 | C.V. *, % | |
| 0 | 0.043 | 2.64 | 0.041 | 0.17 |
| 0.1 | 0.048 | 1.18 | 0.051 | 1.96 |
| 0.5 | 0.082 | 0.52 | 0.093 | 1.06 |
| 2.5 | 0.269 | 2.50 | 0.330 | 3.92 |
| 10 | 0.899 | 3.30 | 1.123 | 1.53 |
| 50 | 3.416 | 1.51 | 3.827 | 1.83 |
| BSA 1.0 g/L | Conjugate | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C8 | C10 | C11 | C12 | C14 | C15 | C16 | C17 | C18 | C19 | C20 | ||
| 0.1 g/L | ||||||||||||
| OD450 | ||||||||||||
| 0.699 | 0.992 | 0.732 | 0.850 | 0.530 | 0.687 | 0.987 | 1.090 | 0.592 | 0.805 | 0.895 | 0.521 | |
| 0.555 | 0.967 | 0.626 | 0.881 | 0.527 | 0.664 | 1.118 | 1.156 | 0.652 | 0.800 | 0.851 | 0.540 | |
| 0.879 | 1.051 | 0.625 | 1.026 | 0.634 | 0.698 | 1.138 | 1.062 | 0.565 | 0.789 | 0.811 | 0.527 | |
| 1.012 | 1.034 | 0.697 | 0.938 | 0.618 | 0.699 | 1.025 | 1.048 | 0.592 | 0.665 | 0.738 | 0.536 | |
| 0.995 | 1.003 | 0.639 | 0.920 | 0.539 | 0.682 | 1.016 | 1.066 | 0.688 | 0.640 | 0.712 | 0.512 | |
| 1.004 | 1.003 | 0.681 | 0.970 | 0.572 | 0.720 | 1.140 | 1.091 | 0.550 | 0.717 | 0.770 | 0.527 | |
| 0.737 | 0.990 | 0.619 | 0.963 | 0.570 | 0.651 | 1.143 | 1.090 | 0.611 | 0.665 | 0.698 | 0.517 | |
| 0.862 | 0.984 | 0.666 | 0.967 | 0.584 | 0.680 | 1.093 | 1.036 | 0.543 | 0.626 | 0.732 | 0.519 | |
| Mean | 0.843 | 1.003 | 0.661 | 0.939 | 0.599 | 0.685 | 1.083 | 1.080 | 0.570 | 0.714 | 0.776 | 0.525 |
| C.V. *, % | 19.77 | 2.73 | 6.14 | 5.90 | 6.95 | 3.12 | 5.87 | 3.43 | 8.40 | 10.51 | 9.05 | 1.79 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šubr, V.; Kostka, L.; Plicka, J.; Sedláček, O.; Etrych, T. Highly Effective Synthetic Polymer-Based Blockers of Non-Specific Interactions in Immunochemical Analyses. Polymers 2024, 16, 758. https://doi.org/10.3390/polym16060758
Šubr V, Kostka L, Plicka J, Sedláček O, Etrych T. Highly Effective Synthetic Polymer-Based Blockers of Non-Specific Interactions in Immunochemical Analyses. Polymers. 2024; 16(6):758. https://doi.org/10.3390/polym16060758
Chicago/Turabian StyleŠubr, Vladimír, Libor Kostka, Jan Plicka, Ondřej Sedláček, and Tomáš Etrych. 2024. "Highly Effective Synthetic Polymer-Based Blockers of Non-Specific Interactions in Immunochemical Analyses" Polymers 16, no. 6: 758. https://doi.org/10.3390/polym16060758
APA StyleŠubr, V., Kostka, L., Plicka, J., Sedláček, O., & Etrych, T. (2024). Highly Effective Synthetic Polymer-Based Blockers of Non-Specific Interactions in Immunochemical Analyses. Polymers, 16(6), 758. https://doi.org/10.3390/polym16060758