
Facile Preparation of Cellulose Beads with Tunable Graded Pores and High Mechanical Strength


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
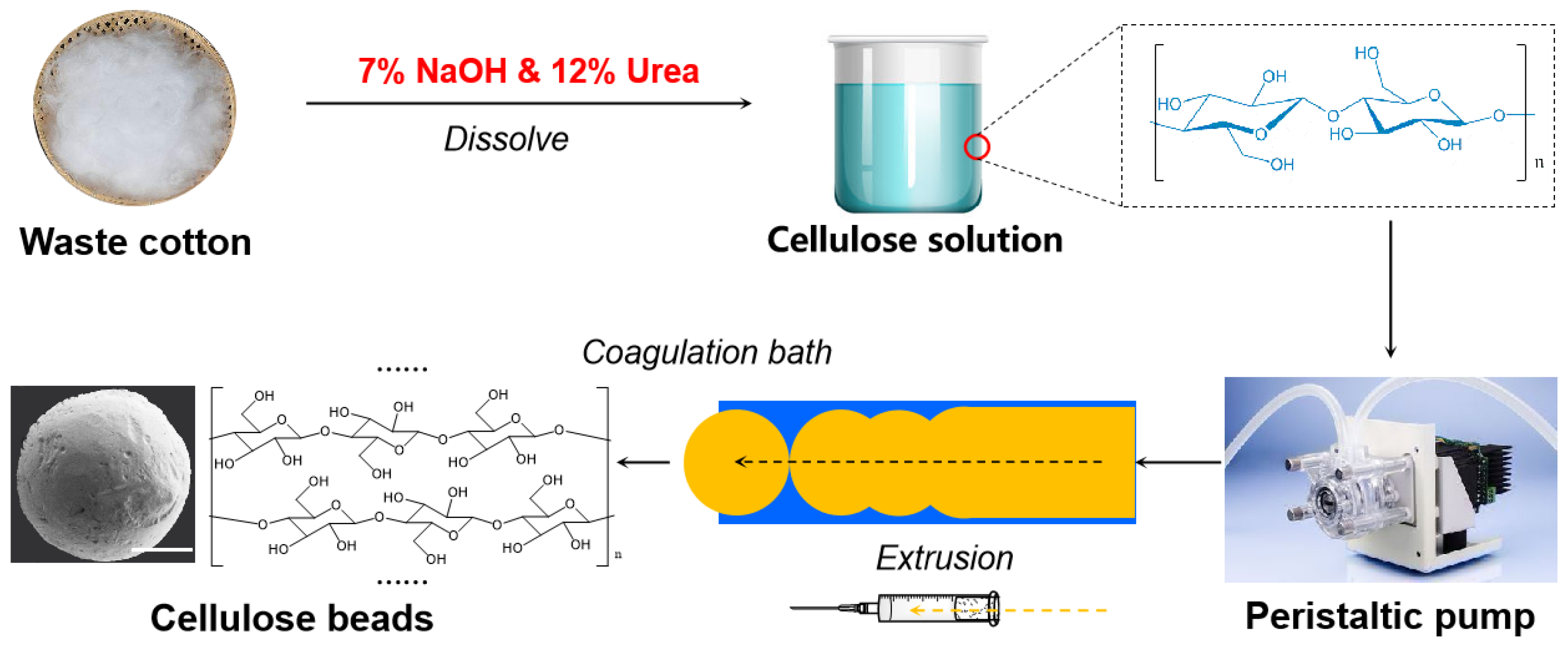
2.2. Preparation of Cellulose Beads with Tunable Pore Size and Structure
2.3. Characterization
2.3.1. Field Emission Scanning Electron Microscope (FE-SEM)
2.3.2. Determination of the Weight, Size, and Shape of the Cellulose Beads
2.3.3. Moisture Content
2.3.4. Porosity
2.3.5. Nitrogen Adsorption Performance
2.3.6. Mechanical Properties
3. Results and Discussion
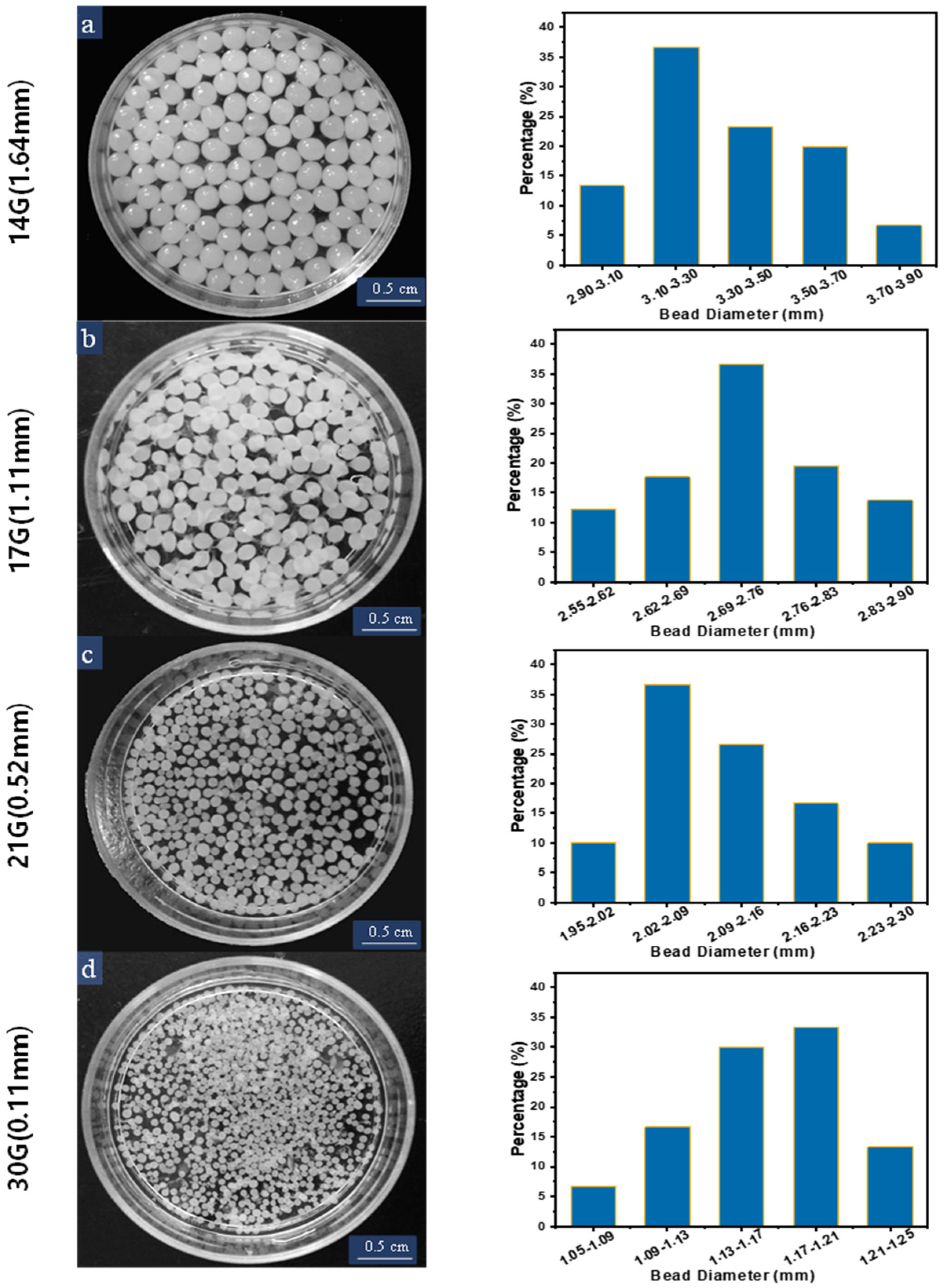
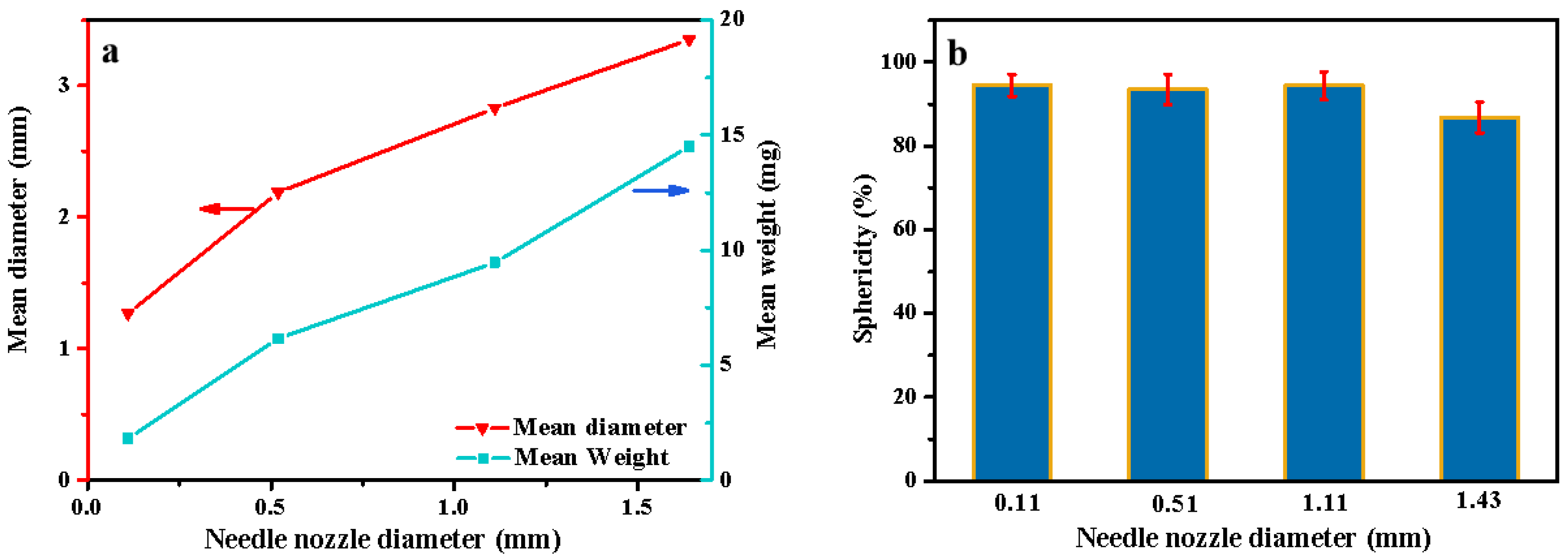
3.1. Size and Shape of the Cellulose Beads
3.2. Physical Properties of the Porous Cellulose Beads
3.3. Effects of Different Reaction Conditions on the Morphology and Core–Shell Structure of the Cellulose Beads
3.3.1. Effect of Cellulose Solution Concentration
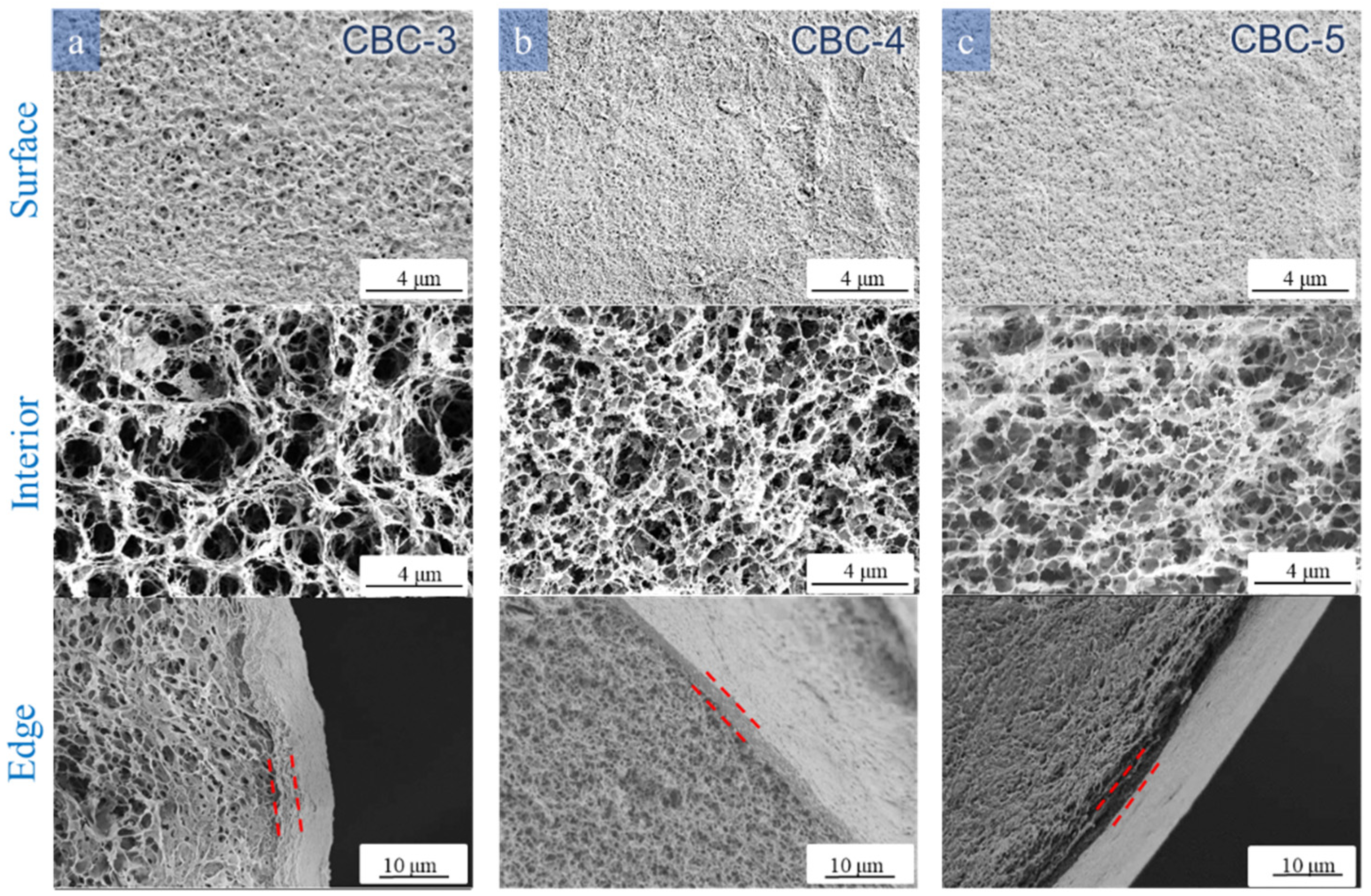
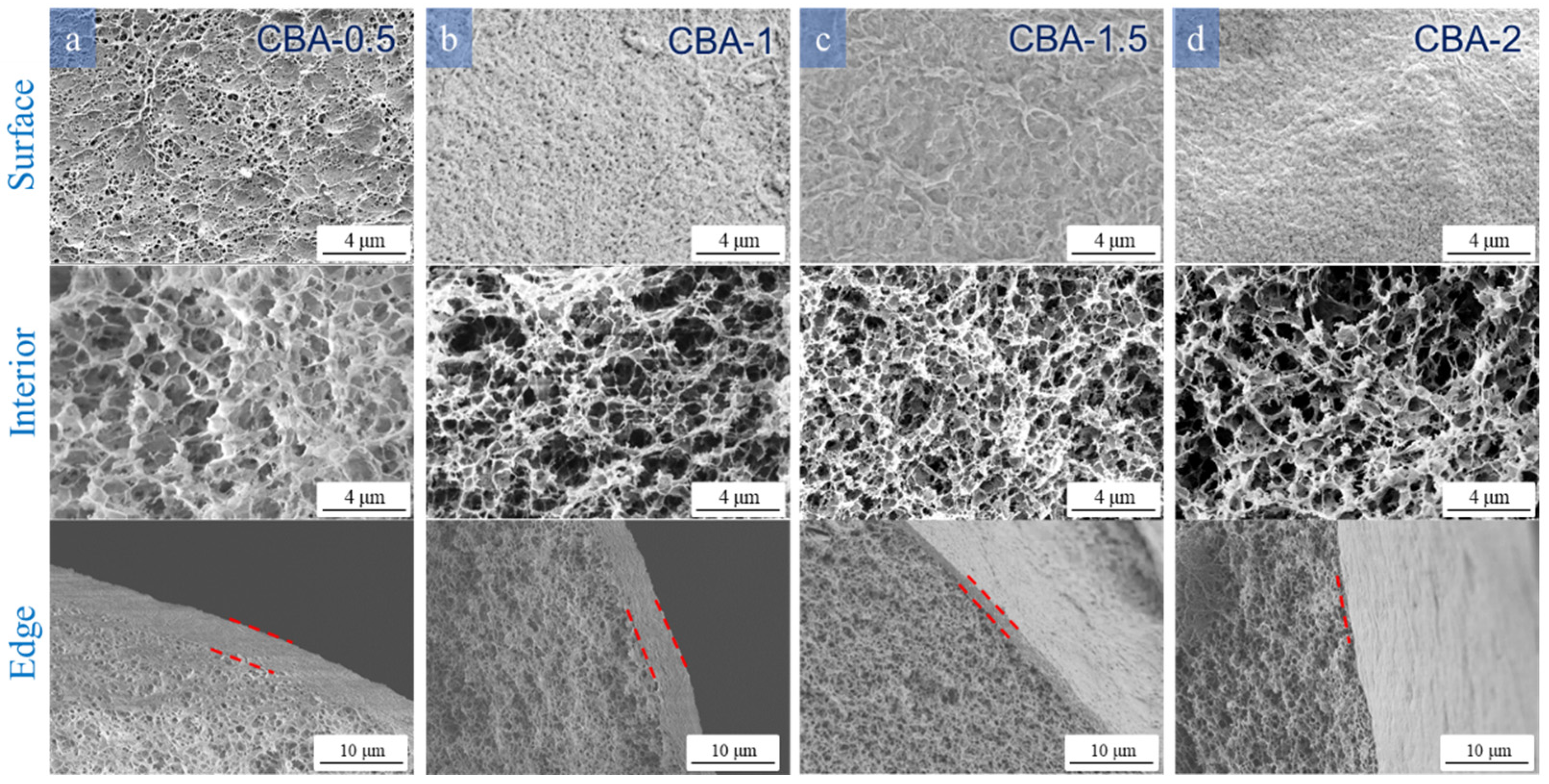
3.3.2. Effect of H2SO4 and Na2SO4 Concentrations in the Coagulation Bath
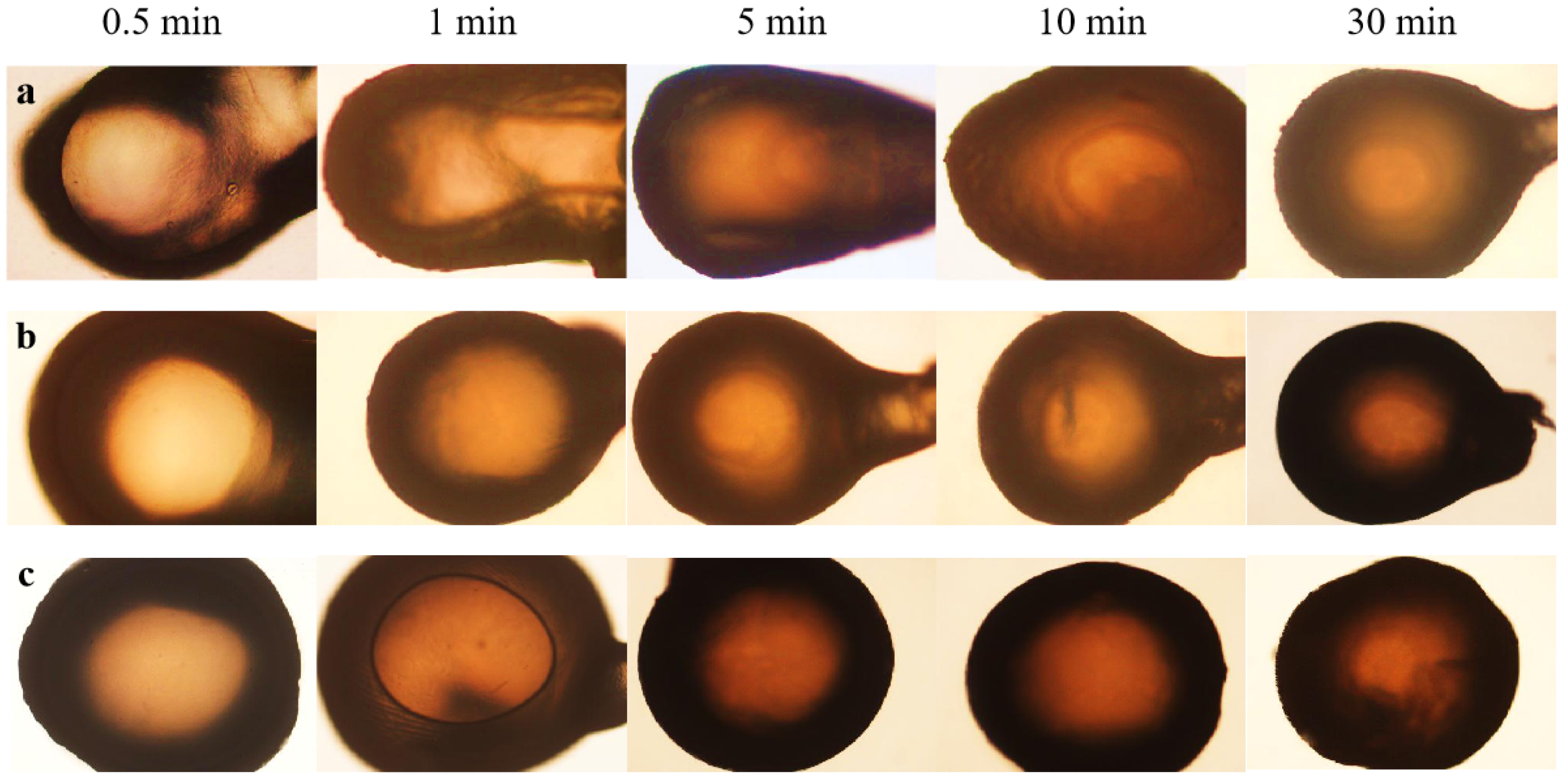

3.3.3. Effect of the Temperature of the Coagulation Bath
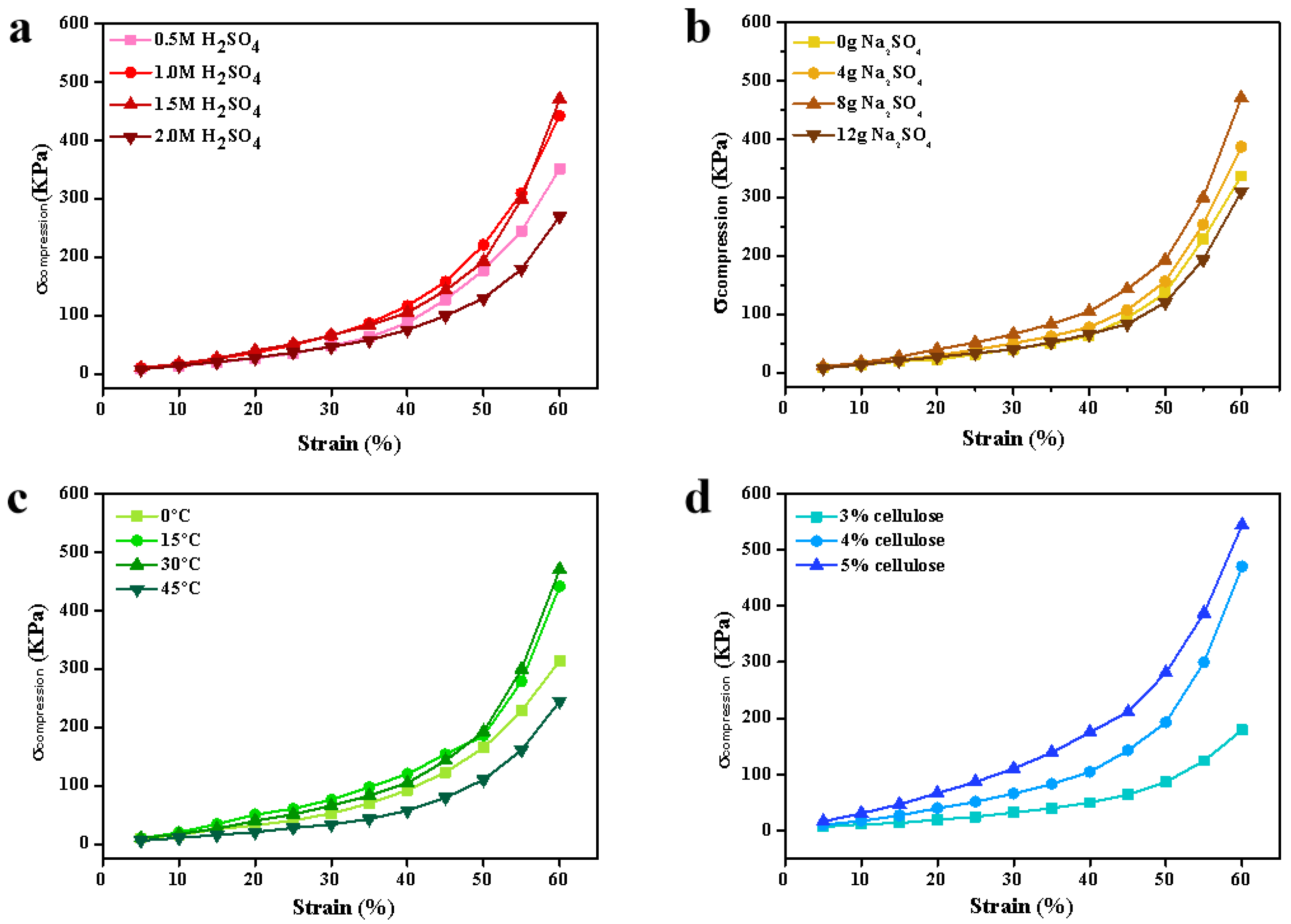
3.4. Mechanical Properties of the Cellulose Beads
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Li, S.; Qiao, L.; Liang, C.; Zhao, L.; Du, K. Boronate-immobilized cellulose nanofiber-reinforced cellulose microspheres for pH-dependent adsorption of glycoproteins. Carbohydr. Polym. 2022, 298, 120068. [Google Scholar] [CrossRef]
- Liu, S.; Sun, Y.; Guo, D.; Lu, R.; Mao, Y.; Ou, H. Porous boronate imprinted microsphere prepared based on new RAFT functioned cellulose nanocrystalline with multiple H-bonding at the emulsion droplet interface for highly specific separation of Naringin. Chem. Eng. J. 2023, 452, 139294. [Google Scholar] [CrossRef]
- Gutierrez-Climente, R.; Gomez-Caballero, A.; Guerreiro, A.; Garcia-Mutio, D.; Unceta, N.; Goicolea, M.A.; Barrio, R.J. Molecularly imprinted nanoparticles grafted to porous silica as chiral selectors in liquid chromatography. J. Chromatogr. A 2017, 1508, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yang, Z.; Hao, J.; Ding, F.; Li, Z.; Ren, X. Hollow nanosphere-doped bacterial cellulose and polypropylene wound dressings: Biomimetic nanocatalyst mediated antibacterial therapy. Chem. Eng. J. 2022, 432, 134309. [Google Scholar] [CrossRef]
- Zhu, H.; Zhu, E.; Xie, Y.; Liu, D.; Hu, Y.; Shi, Z.; Xiong, C.; Yang, Q. Hydrangea-like nanocellulose microspheres with high dye adsorption and drug encapsulation prepared by emulsion method. Carbohydr. Polym. 2022, 296, 134309. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Abdalkarim, S.Y.H.; Yu, H.-Y.; Asad, R.A.M.; Ge, D.; Zhou, Y. Thermo-sensitive composite microspheres incorporating cellulose nanocrystals for regulated drug release kinetics. Carbohydr. Polym. 2023, 301, 120350. [Google Scholar] [CrossRef] [PubMed]
- Kovářík, T.; Hájek, J.; Hervert, T.; Deshmukh, K.; Pola, M.; Jansa, Z.; Beneš, J.; Svoboda, M. Silica-based geopolymer spherical beads: Influence of viscosity on porosity architecture. Cem. Concr. Compos. 2021, 124, 104261. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, Y.; Huo, H.; Hu, Y.; Xu, X.; Wang, P.; Yang, Y.; Lin, K. Synthesis of three-dimensional nitrogen doped meso/macroporous carbon beads for heterogeneous catalytic solvent-free oxidation of ethylbenzene. Carbon 2020, 158, 226–237. [Google Scholar] [CrossRef]
- Li, H.; Kruteva, M.; Dulle, M.; Wang, Z.; Mystek, K.; Ji, W.; Pettersson, T.; Wågberg, L. Understanding the Drying Behavior of Regenerated Cellulose Gel Beads: The Effects of Concentration and Nonsolvents. ACS Nano 2022, 16, 2608–2620. [Google Scholar] [CrossRef]
- Zeng, H.; Sun, S.; Xu, K.; Zhao, W.; Hao, R.; Zhang, J.; Li, D. Adsorption of As(V) by magnetic alginate-chitosan porous beads based on iron sludge. J. Clean. Prod. 2022, 359, 132117. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, Z.; Li, Y.; Zou, D. Three-in-one multifunctional luminescent metal-organic gels/sodium alginate beads for high-performance adsorption and detection of chlortetracycline hydrochloride, and high-security anti-counterfeiting. Chem. Eng. J. 2023, 452, 139194. [Google Scholar] [CrossRef]
- Ramachandran, J.; Serrano, J.M.; Liu, T.; Cho, J.; Arias-Monje, P.J.; Lu, M.; Kirmani, M.H.; Elliott, J.; Jang, S.S.; Liu, G.; et al. Porous carbon fibers from gel-spun polyacrylonitrile and poly(methyl methacrylate)-block-poly(acrylonitrile). Carbon 2022, 192, 332–346. [Google Scholar] [CrossRef]
- Rol, F.; Belgacem, M.N.; Gandini, A.; Bras, J. Recent advances in surface-modified cellulose nanofibrils. Prog. Polym. Sci. 2019, 88, 241–264. [Google Scholar] [CrossRef]
- Razzak, A.; Khiari, R.; Moussaoui, Y.; Belgacem, M.N. Cellulose Nanofibers from Schinus molle: Preparation and Characterization. Molecules 2022, 27, 6738. [Google Scholar] [CrossRef] [PubMed]
- Alves, L.; Medronho, B.; Antunes, F.E.; Fernández-García, M.P.; Ventura, J.; Araújo, J.P.; Romano, A.; Lindman, B. Unusual extraction and characterization of nanocrystalline cellulose from cellulose derivatives. J. Mol. Liq. 2015, 210, 106–112. [Google Scholar] [CrossRef]
- Kihlman, M.; Medronho, B.F.; Romano, A.L.; Germgård, U.; Lindman, B. Cellulose Dissolution in an Alkali Based Solvent: Influence of Additives and Pretreatments. J. Braz. Chem. Soc. 2013, 24, 295–303. [Google Scholar] [CrossRef]
- Wan, C.; Jiao, Y.; Wei, S.; Zhang, L.; Wu, Y.; Li, J. Functional nanocomposites from sustainable regenerated cellulose aerogels: A review. Chem. Eng. J. 2019, 359, 459–475. [Google Scholar] [CrossRef]
- Acharya, S.; Liyanage, S.; Parajuli, P.; Rumi, S.S.; Shamshina, J.L.; Abidi, N. Utilization of Cellulose to Its Full Potential: A Review on Cellulose Dissolution, Regeneration, and Applications. Polymers 2021, 13, 4344. [Google Scholar] [CrossRef] [PubMed]
- Naserifar, S.; Koschella, A.; Heinze, T.; Bernin, D.; Hasani, M. Investigation of cellulose dissolution in morpholinium-based solvents: Impact of solvent structural features on cellulose dissolution. RSC Adv. 2023, 13, 18639–18650. [Google Scholar] [CrossRef]
- Chen, Y.; Yu, H.-Y.; Li, Y. Highly Efficient and Superfast Cellulose Dissolution by Green Chloride Salts and Its Dissolution Mechanism. ACS Sustain. Chem. Eng. 2020, 8, 18446–18454. [Google Scholar] [CrossRef]
- Yang, J.; Medronho, B.; Lindman, B.; Norgren, M. Simple One Pot Preparation of Chemical Hydrogels from Cellulose Dissolved in Cold LiOH/Urea. Polymers 2020, 12, 373. [Google Scholar] [CrossRef]
- Yang, Q.; Fujisawa, S.; Saito, T.; Isogai, A. Improvement of mechanical and oxygen barrier properties of cellulose films by controlling drying conditions of regenerated cellulose hydrogels. Cellulose 2012, 19, 695–703. [Google Scholar] [CrossRef]
- Fauziyah, M.a.; Widiyastuti, W.; Balgis, R.; Setyawan, H. Production of cellulose aerogels from coir fibers via an alkali–urea method for sorption applications. Cellulose 2019, 26, 9583–9598. [Google Scholar] [CrossRef]
- Tu, H.; Li, X.; Liu, Y.; Luo, L.; Duan, B.; Zhang, R. Recent progress in regenerated cellulose-based fibers from alkali/urea system via spinning process. Carbohydr. Polym. 2022, 296, 119942. [Google Scholar] [CrossRef] [PubMed]
- Boujemaoui, A.; Carlsson, L.; Malmström, E.; Lahcini, M.; Berglund, L.; Sehaqui, H.; Carlmark, A. Facile Preparation Route for Nanostructured Composites: Surface-Initiated Ring-Opening Polymerization of ε-Caprolactone from High-Surface-Area Nanopaper. ACS Appl. Mater. Interfaces 2012, 4, 3191–3198. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fu, Q.; Yu, S.; Yan, M.; Berglund, L. Optically Transparent Wood from a Nanoporous Cellulosic Template: Combining Functional and Structural Performance. Biomacromolecules 2016, 17, 1358–1364. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Prinsen, P.; Wang, H.; Bai, Z.; Wang, H.; Luque, R.; Xuan, J. Macroporous materials: Microfluidic fabrication, functionalization and applications. Chem. Soc. Rev. 2017, 46, 855–914. [Google Scholar] [CrossRef]
- Shi, W.; Ching, Y.C.; Chuah, C.H. Preparation of aerogel beads and microspheres based on chitosan and cellulose for drug delivery: A review. Int. J. Biol. Macromol. 2021, 170, 751–767. [Google Scholar] [CrossRef]
- López-Iglesias, C.; Barros, J.; Ardao, I.; Gurikov, P.; Monteiro, F.J.; Smirnova, I.; Alvarez-Lorenzo, C.; García-González, C.A. Jet Cutting Technique for the Production of Chitosan Aerogel Microparticles Loaded with Vancomycin. Polymers 2020, 12, 273. [Google Scholar] [CrossRef]
- Han, C.; Ma, L.; Tang, T.; Deng, J.; Luo, G. Microdroplet-based synthesis of polymethylsilsesquioxane microspheres with controllable size, surface morphology, and internal structure. Chem. Eng. Sci. 2022, 262, 118054. [Google Scholar] [CrossRef]
- Meng, R.; Liu, L.; Jin, Y.; Luo, Z.; Gao, H.; Yao, J. Recyclable carboxylated cellulose beads with tunable pore structure and size for highly efficient dye removal. Cellulose 2019, 26, 8963–8969. [Google Scholar] [CrossRef]
- Buchtová, N.; Budtova, T. Cellulose aero-, cryo- and xerogels: Towards understanding of morphology control. Cellulose 2016, 23, 2585–2595. [Google Scholar] [CrossRef]
- Medronho, B.; Lindman, B. Competing forces during cellulose dissolution: From solvents to mechanisms. Curr. Opin. Colloid Interface Sci. 2014, 19, 32–40. [Google Scholar] [CrossRef]
- Isobe, N.; Kimura, S.; Wada, M.; Kuga, S. Mechanism of cellulose gelation from aqueous alkali-urea solution. Carbohydr. Polym. 2012, 89, 1298–1300. [Google Scholar] [CrossRef]
- Wang, W.; Li, F.; Yu, J.; Zhou, J.; Wang, H. Effects of coagulation conditions on structure and properties of cellulose-based fibers from aqueous NaOH solvent. Carbohydr. Polym. 2017, 164, 118–126. [Google Scholar] [CrossRef]
- Song, J.; Guo, J.; Zhang, S.; Gong, Y. Properties of cellulose/Antarctic krill protein composite fibers prepared in different coagulation baths. Int. J. Biol. Macromol. 2018, 114, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Saljoughi, E.; Amirilargani, M.; Mohammadi, T. Effect of poly(vinyl pyrrolidone) concentration and coagulation bath temperature on the morphology, permeability, and thermal stability of asymmetric cellulose acetate membranes. J. Appl. Polym. Sci. 2008, 111, 2537–2544. [Google Scholar] [CrossRef]
- Fan, H.; Hartshorn, C.; Buchheit, T.; Tallant, D.; Assink, R.; Simpson, R.; Kissel, D.J.; Lacks, D.J.; Torquato, S.; Brinker, C.J. Modulus–density scaling behaviour and framework architecture of nanoporous self-assembled silicas. Nat. Mater. 2007, 6, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Mariano, M.; Bernardes, J.d.S.; Strauss, M. Mold heat conductance as drive force for tuning freeze-casted nanocellulose foams microarchitecture. MatL 2018, 225, 167–170. [Google Scholar] [CrossRef]
- Ferreira, F.V.; Souza, L.P.; Martins, T.M.M.; Lopes, J.H.; Mattos, B.D.; Mariano, M.; Pinheiro, I.F.; Valverde, T.M.; Livi, S.; Camilli, J.A.; et al. Nanocellulose/bioactive glass cryogels as scaffolds for bone regeneration. Nanoscale 2019, 11, 19842–19849. [Google Scholar] [CrossRef]
- Pankongadisak, P.; Ruktanonchai, U.R.; Supaphol, P.; Suwantong, O. Gelatin scaffolds functionalized by silver nanoparticle-containing calcium alginate beads for wound care applications. Polym. Adv. Technol. 2017, 28, 849–858. [Google Scholar] [CrossRef]
- Harada, N.; Mitsukami, Y.; Uyama, H. Preparation and characterization of water-swellable hydrogel-forming porous cellulose beads. Polymer 2021, 215, 123381. [Google Scholar] [CrossRef]
- Ghavimi, S.A.A.; Lungren, E.S.; Faulkner, T.J.; Josselet, M.A.; Wu, Y.; Sun, Y.; Pfeiffer, F.M.; Goldstein, C.L.; Wan, C.; Ulery, B.D. Inductive co-crosslinking of cellulose nanocrystal/chitosan hydrogels for the treatment of vertebral compression fractures. Int. J. Biol. Macromol. 2019, 130, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Blachechen, L.S.; Fardim, P.; Petri, D.F.S. Multifunctional Cellulose Beads and Their Interaction with Gram Positive Bacteria. Biomacromolecules 2014, 15, 3440–3448. [Google Scholar] [CrossRef]
- Cellulose hydrogel with tunable shape and mechanical properties: From rigid cylinder to soft scaffold. Int. J. Biol. Macromol. 2018, 117, 625–631. [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Preparation Conditions | Porosity (%) | Water Content (%) | SSA (m2 g−1) | Apparent Density (g/m3) | ||||
|---|---|---|---|---|---|---|---|---|
| Sample Code | Ccellulose (wt%) | Coagulation Condition | T (°C) | |||||
| CH2SO4 (M L−1) | CNa2SO4 (g L−1) | |||||||
| CBC-3 | 3 | 1.5 | 80 | 30 | 96.16 | 94.07 | 87.17 | 0.073 |
| CBC-4 | 4 | 1.5 | 80 | 30 | 95.85 | 93.11 | 97.34 | 0.078 |
| CBC-5 | 5 | 1.5 | 80 | 30 | 93.38 | 91.36 | 99.87 | 0.105 |
| CBA-0.5 | 4 | 0.5 | 80 | 30 | 94.85 | 92.41 | 64.66 | 0.076 |
| CBA-1 | 4 | 1 | 80 | 30 | 95.21 | 92.89 | 94.46 | 0.071 |
| CBA-1.5 | 4 | 1.5 | 80 | 30 | 95.85 | 93.11 | 97.34 | 0.078 |
| CBA-2 | 4 | 2 | 80 | 30 | 95.18 | 92.87 | 84.84 | 0.075 |
| CBS-0 | 4 | 1.5 | 0 | 30 | 94.67 | 92.89 | 60.74 | 0.071 |
| CBS-4 | 4 | 1.5 | 40 | 30 | 95.13 | 92.98 | 92.26 | 0.073 |
| CBS-8 | 4 | 1.5 | 80 | 30 | 95.85 | 93.11 | 97.34 | 0.078 |
| CBS-12 | 4 | 1.5 | 120 | 30 | 94.45 | 93.72 | 80.45 | 0.079 |
| CBT-0 | 4 | 1.5 | 80 | 0 | 95.06 | 93.27 | 86.42 | 0.069 |
| CBT-15 | 4 | 1.5 | 80 | 15 | 96.18 | 92.58 | 84.93 | 0.075 |
| CBT-30 | 4 | 1.5 | 80 | 30 | 95.85 | 93.11 | 97.34 | 0.078 |
| CBT-45 | 4 | 1.5 | 80 | 45 | 93.66 | 91.04 | 58.66 | 0.069 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, R.; Liu, L.; Su, X.; Gong, W.; Luo, X.; Gao, H. Facile Preparation of Cellulose Beads with Tunable Graded Pores and High Mechanical Strength. Polymers 2024, 16, 725. https://doi.org/10.3390/polym16060725
Meng R, Liu L, Su X, Gong W, Luo X, Gao H. Facile Preparation of Cellulose Beads with Tunable Graded Pores and High Mechanical Strength. Polymers. 2024; 16(6):725. https://doi.org/10.3390/polym16060725
Chicago/Turabian StyleMeng, Ranjv, Lin Liu, Xiuping Su, Wenli Gong, Xiaolei Luo, and Huiying Gao. 2024. "Facile Preparation of Cellulose Beads with Tunable Graded Pores and High Mechanical Strength" Polymers 16, no. 6: 725. https://doi.org/10.3390/polym16060725
APA StyleMeng, R., Liu, L., Su, X., Gong, W., Luo, X., & Gao, H. (2024). Facile Preparation of Cellulose Beads with Tunable Graded Pores and High Mechanical Strength. Polymers, 16(6), 725. https://doi.org/10.3390/polym16060725