
Frank–Kasper Phases of Diblock Copolymer Melts: Self-Consistent Field Results of Two Commonly Used Models


Abstract
1. Introduction
2. Models and Methods
2.1. The DPDC Model and Its SCF Calculations
2.2. The “Standard” Model and Its SCF Calculations
3. Results and Discussion
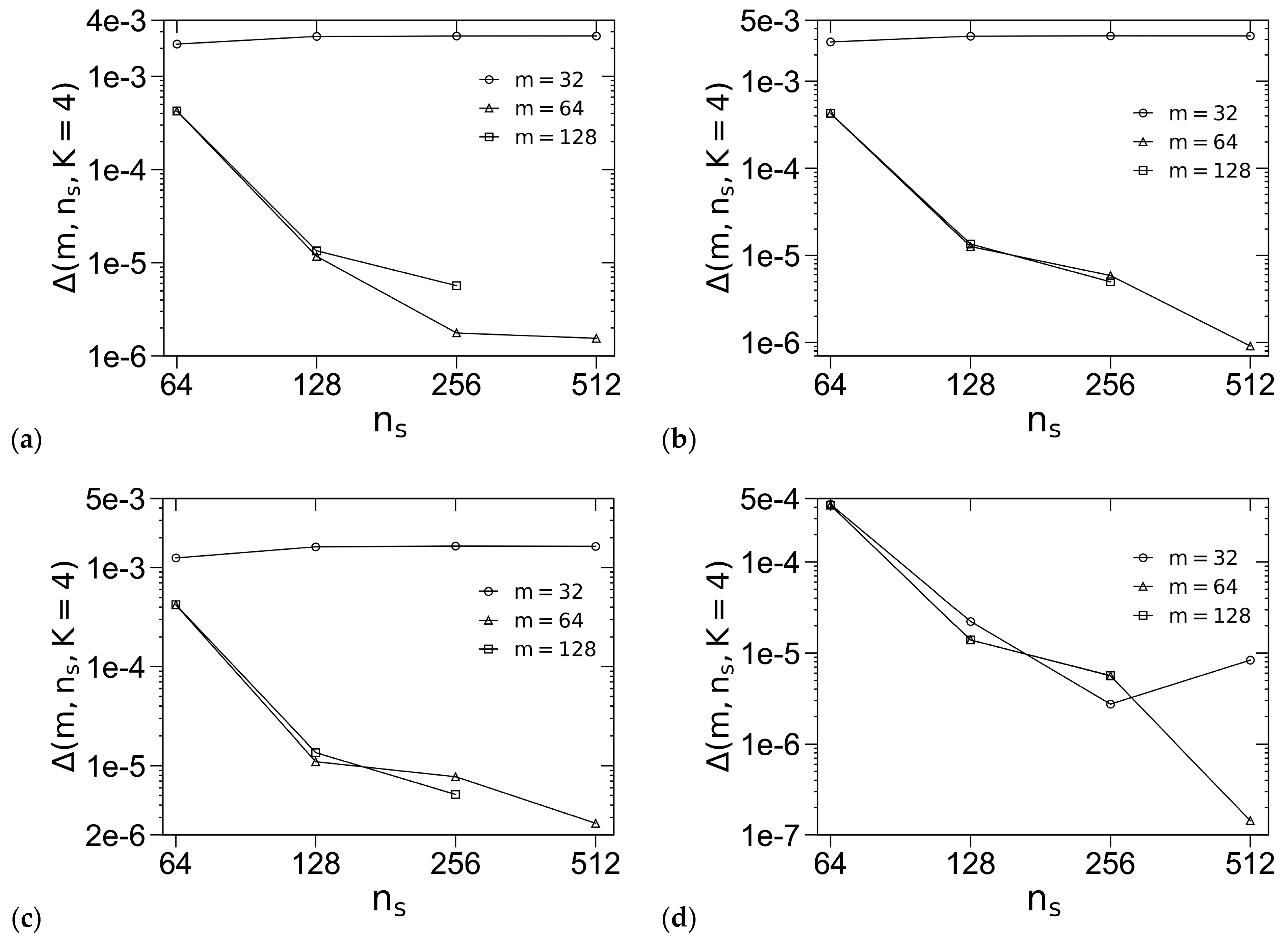
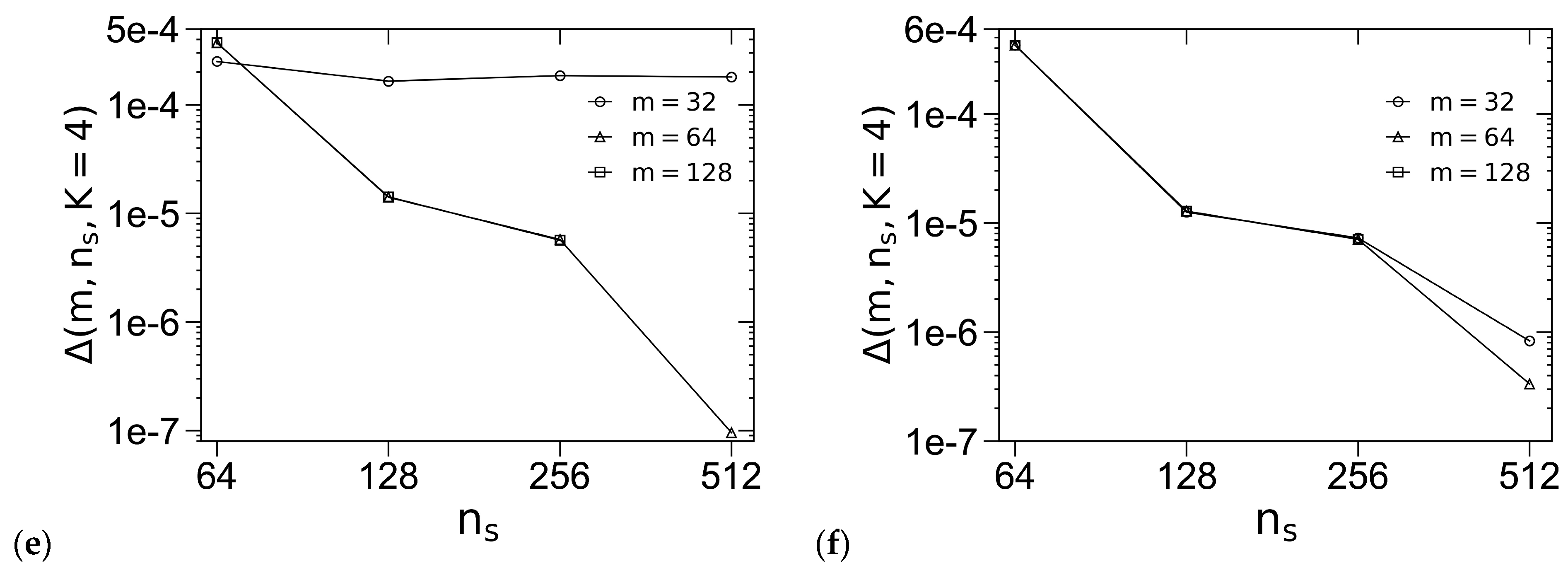
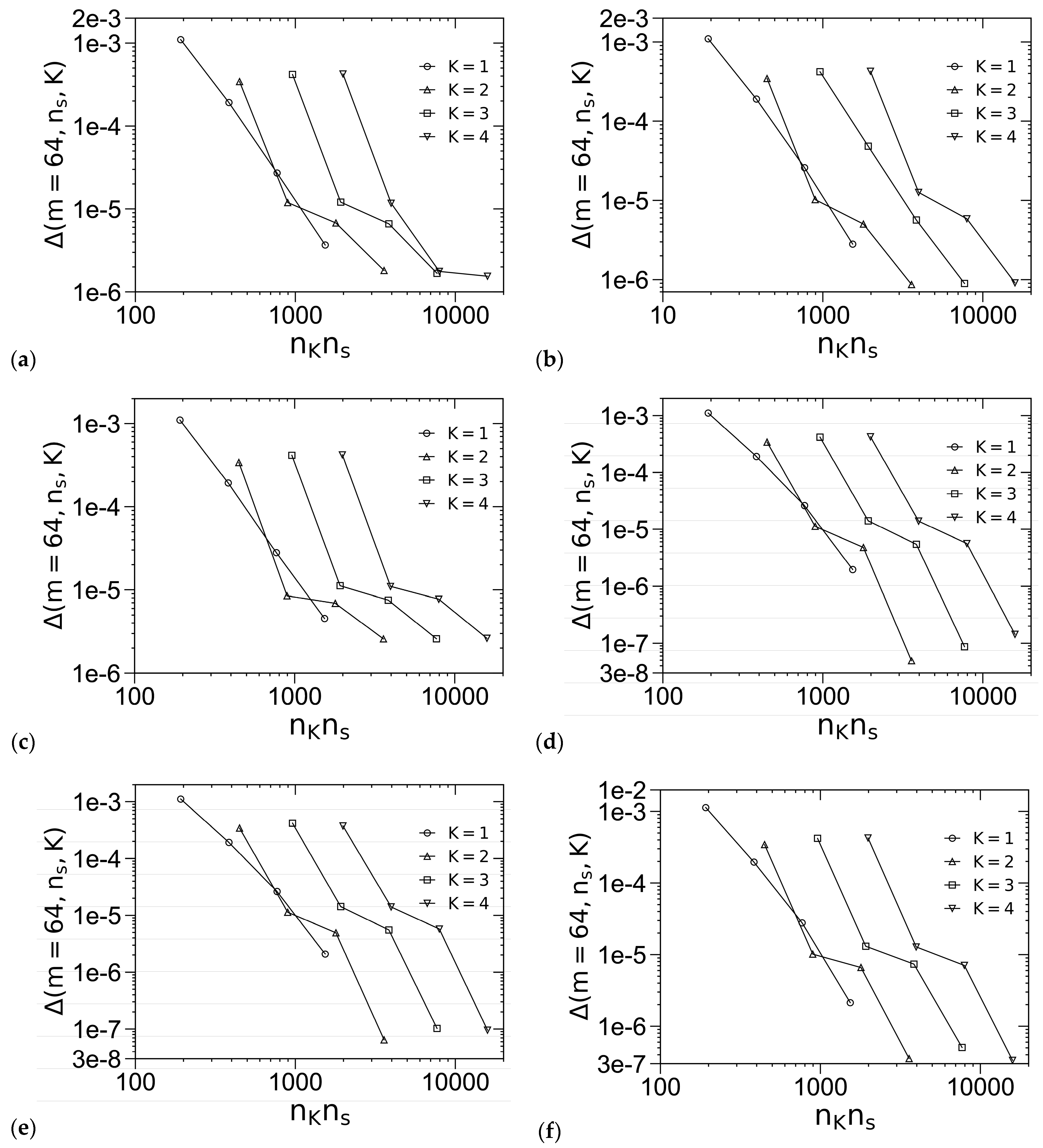
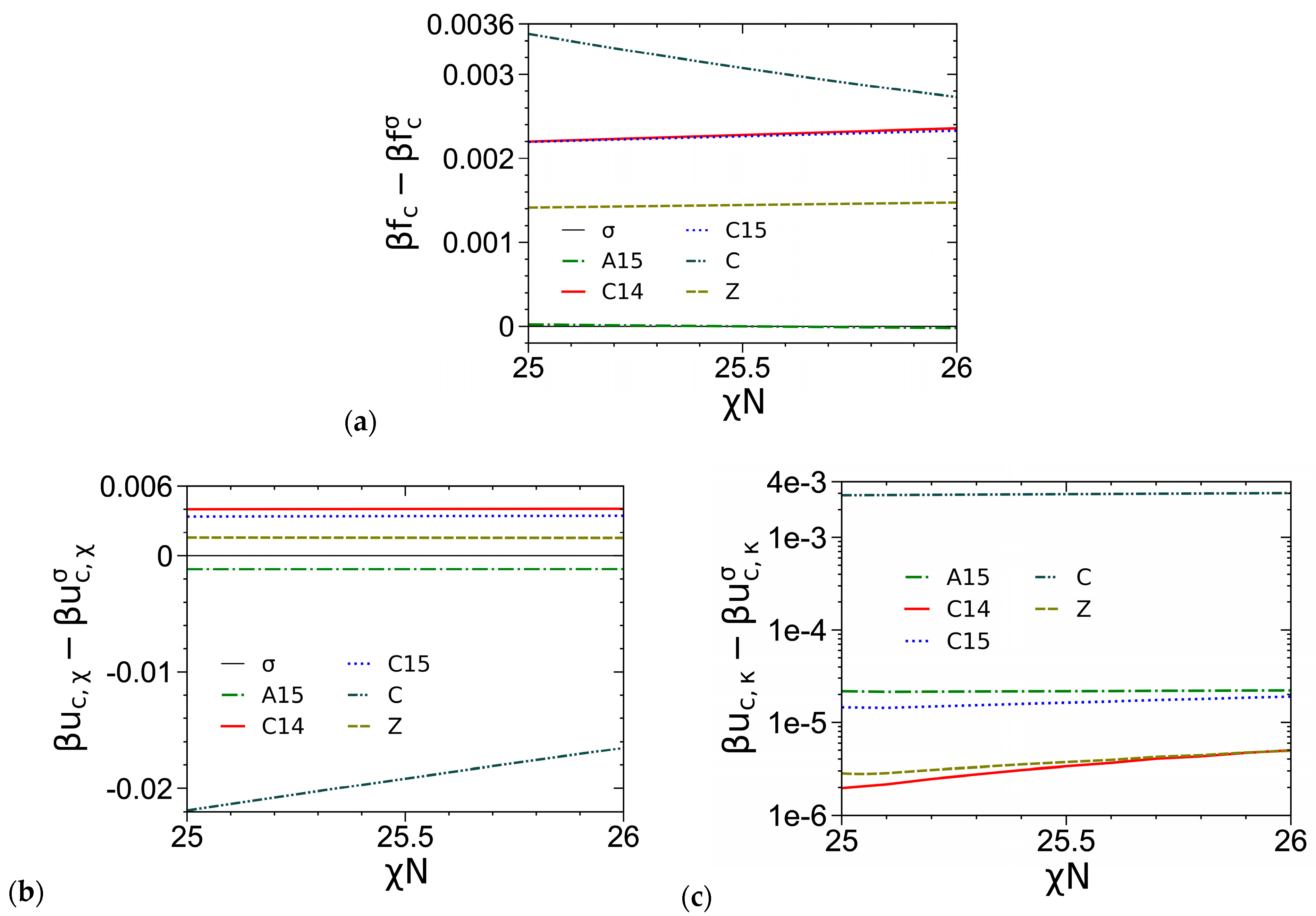
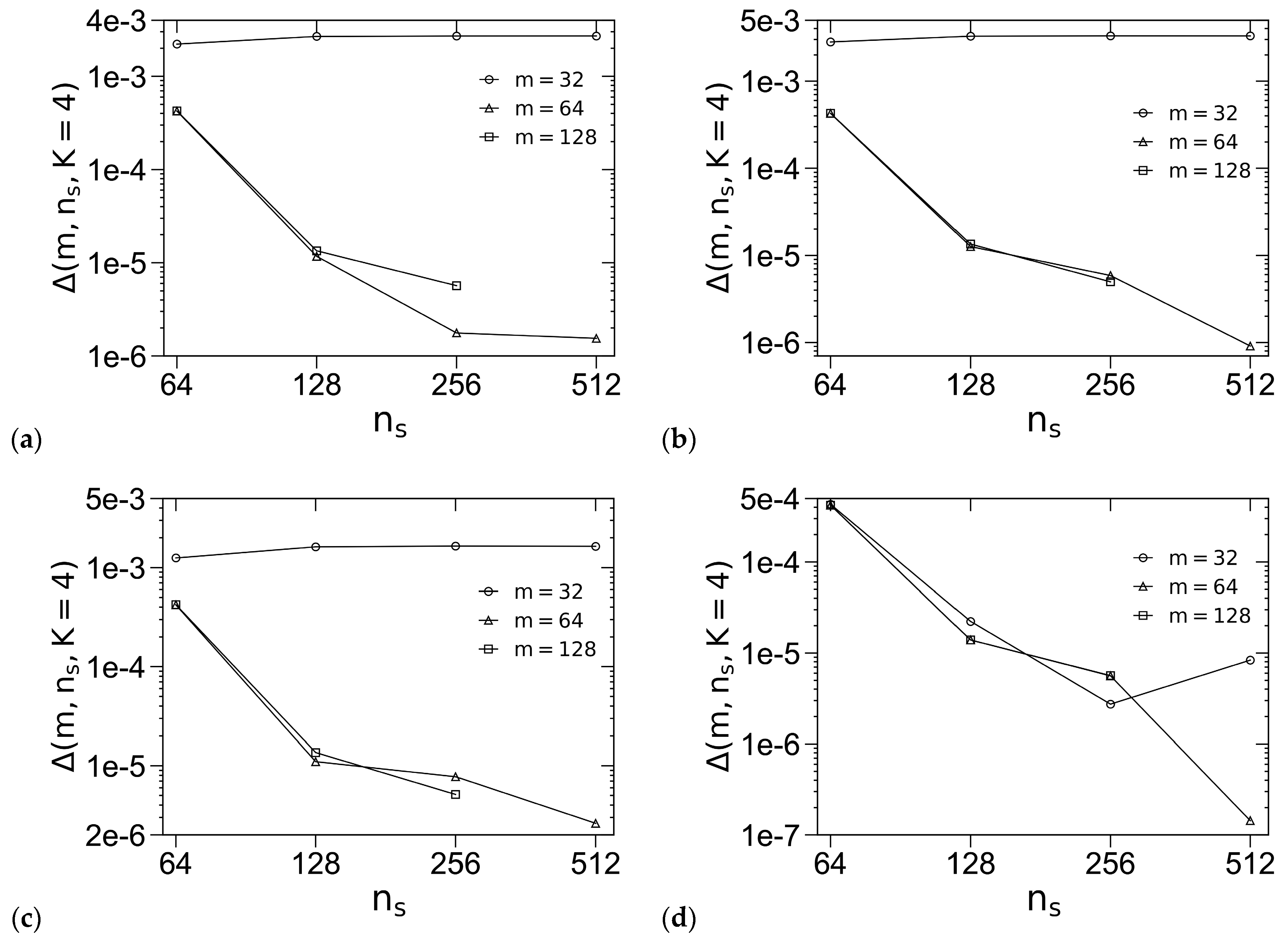
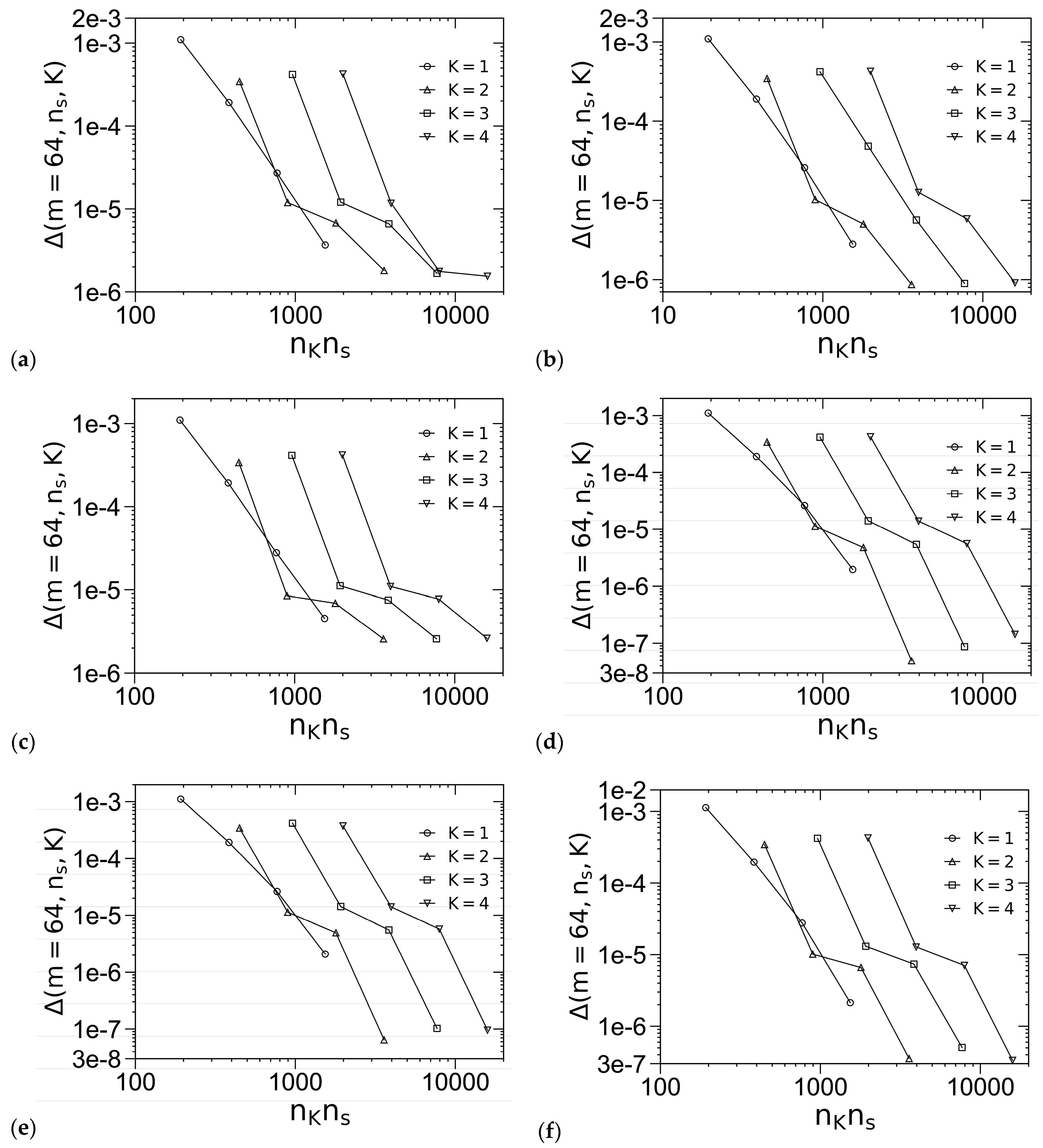
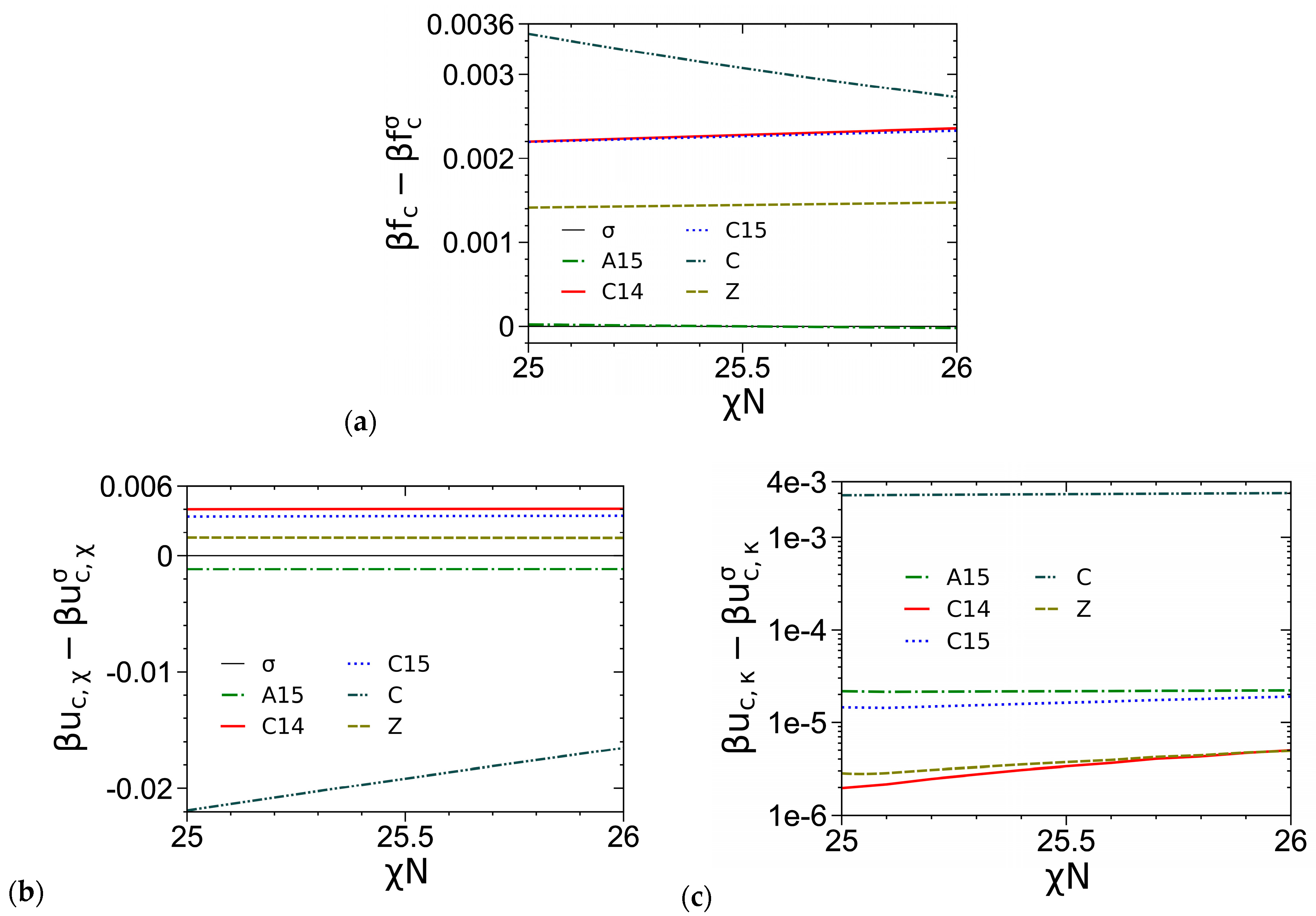
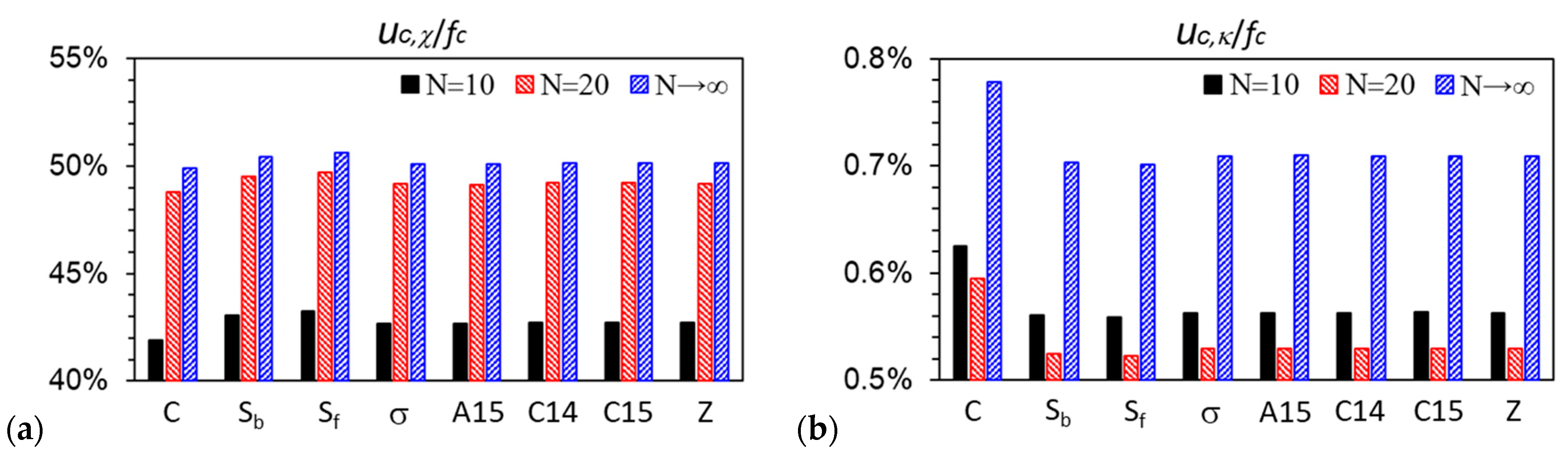
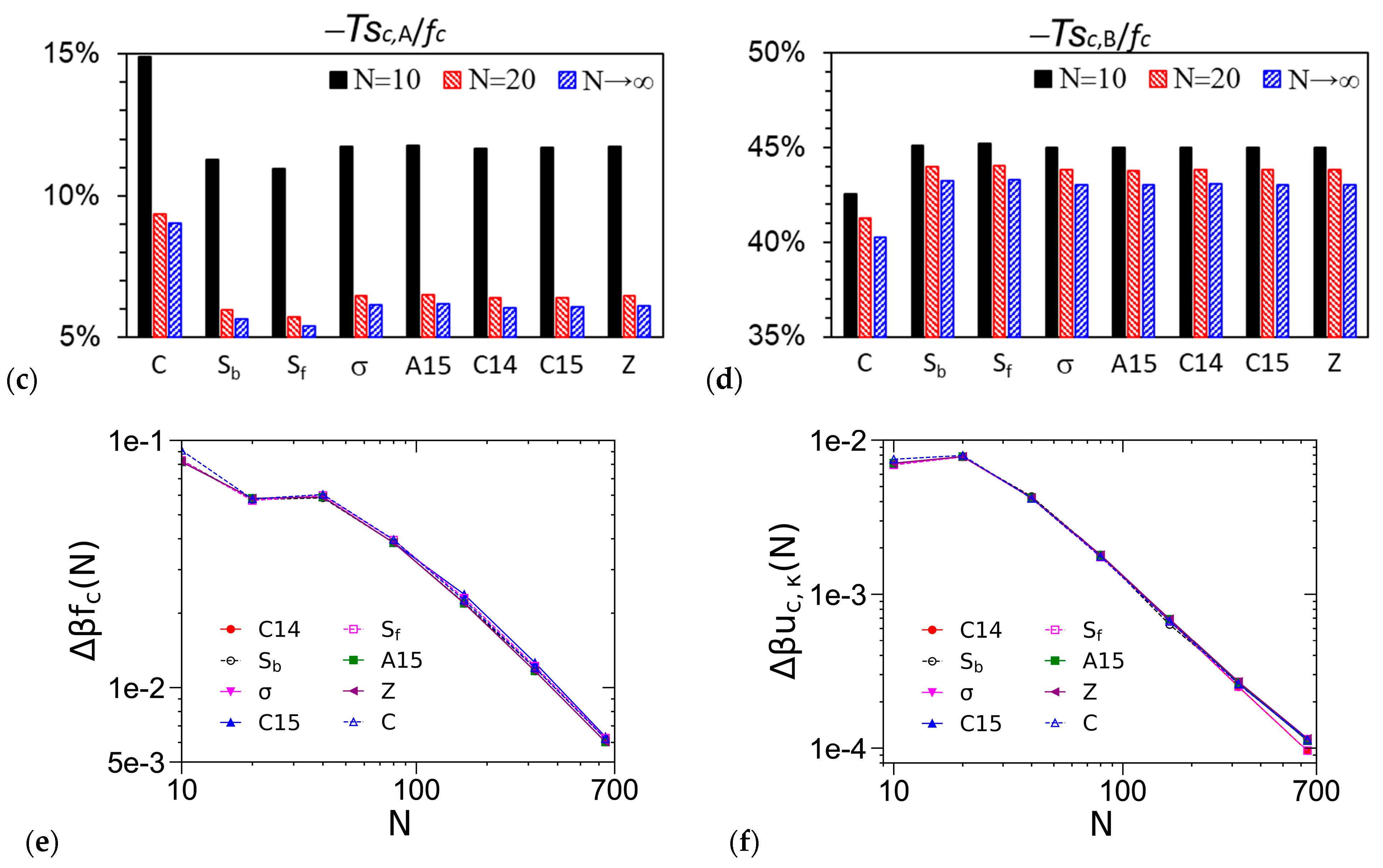
3.1. Unit-Cell Discretization and Accuracy of βfc
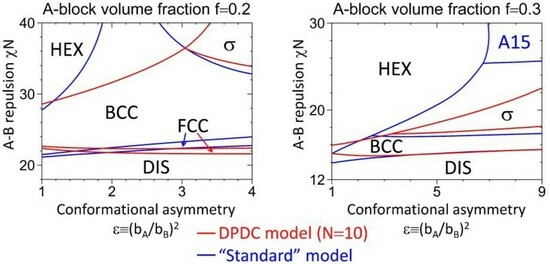
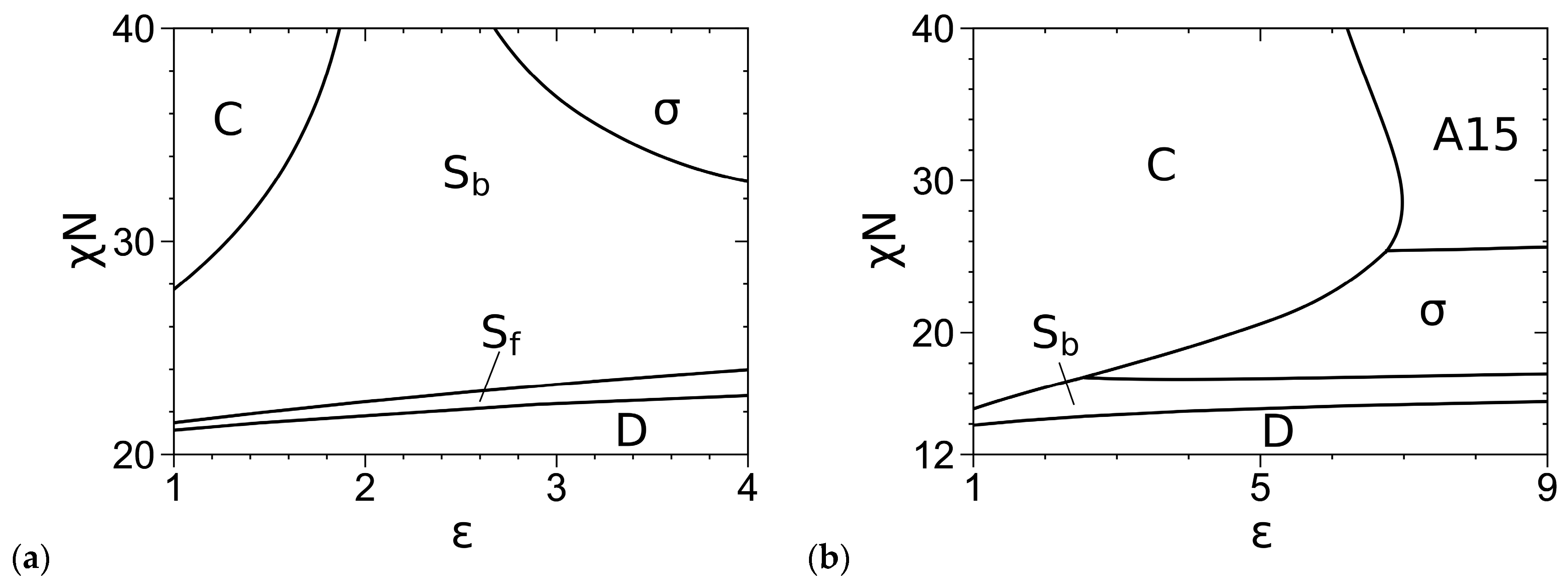
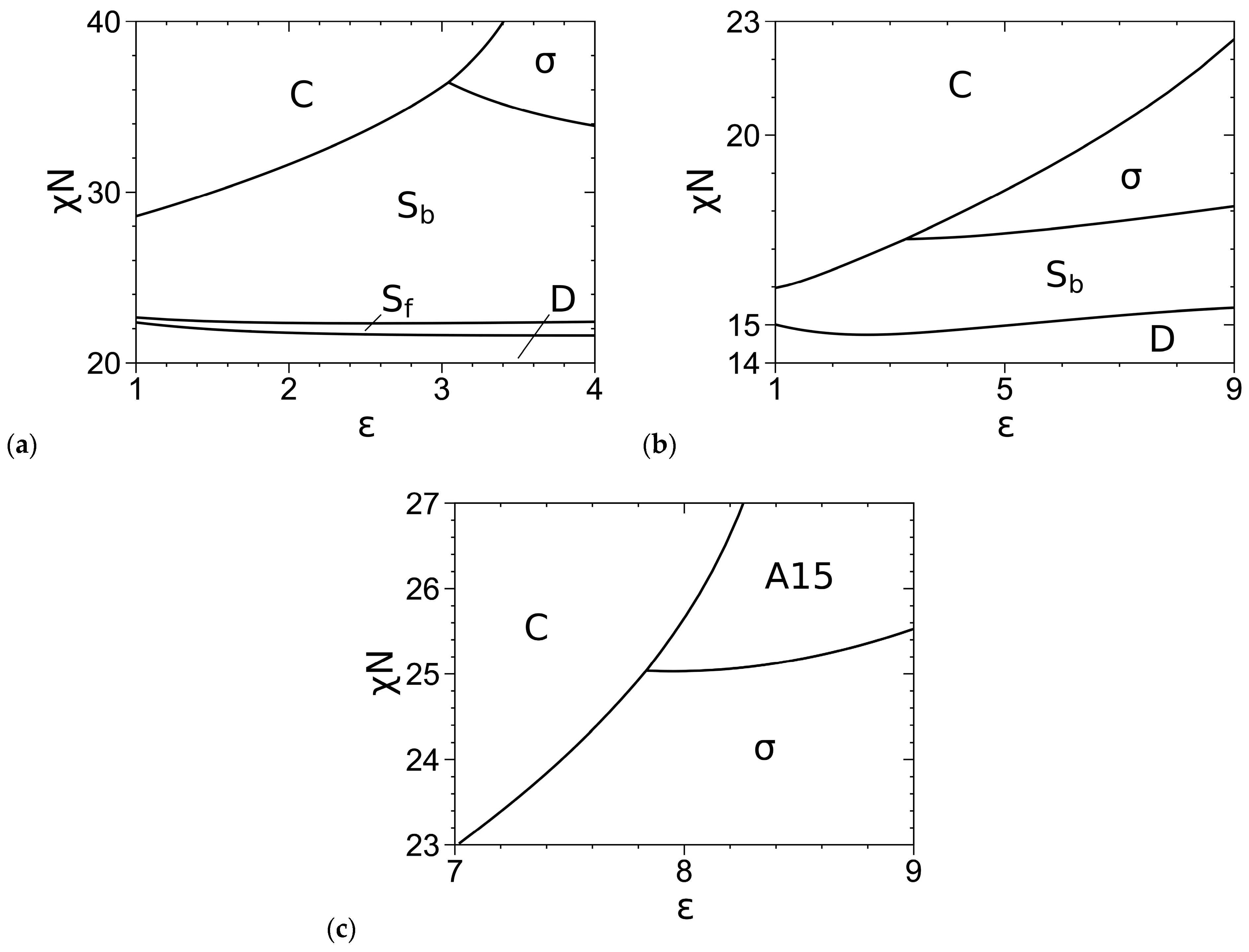
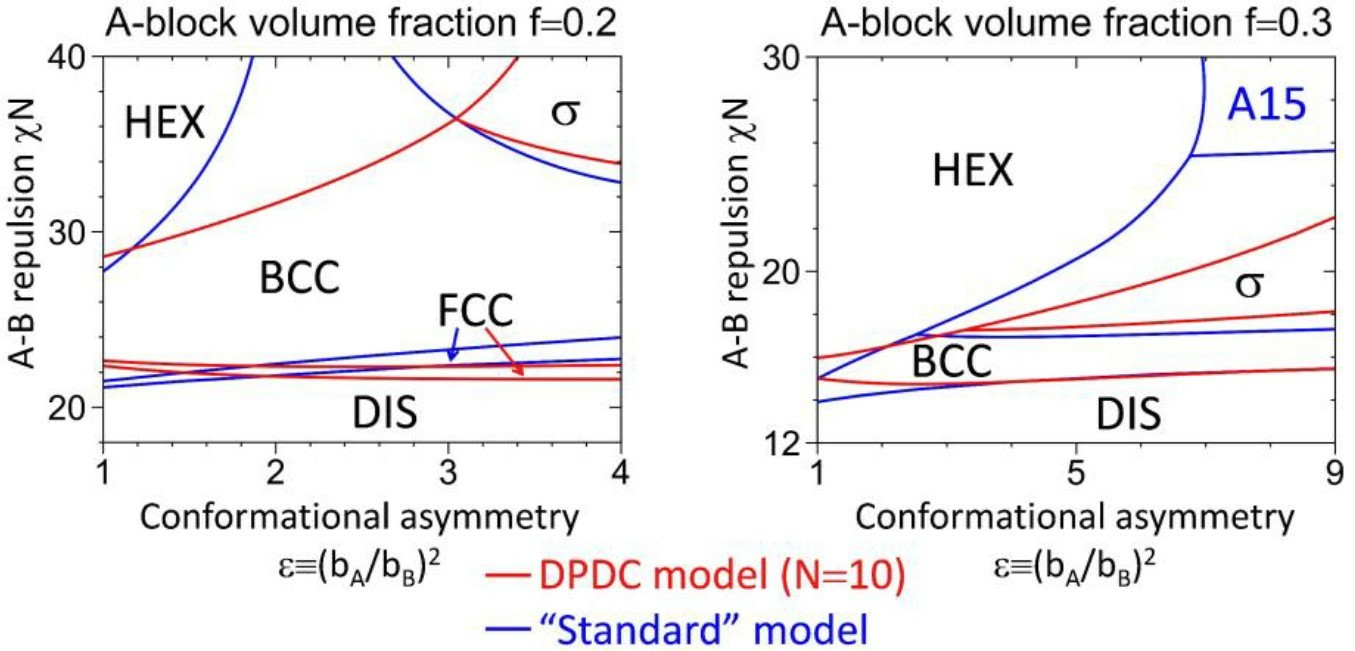
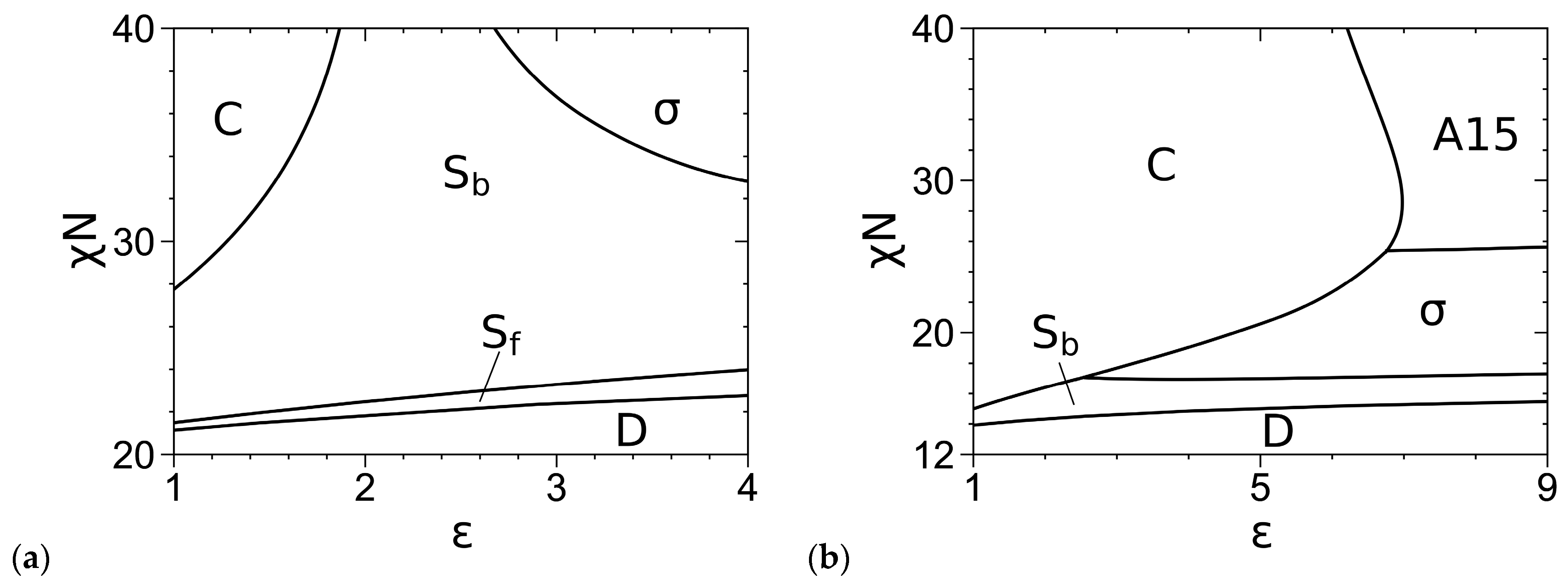
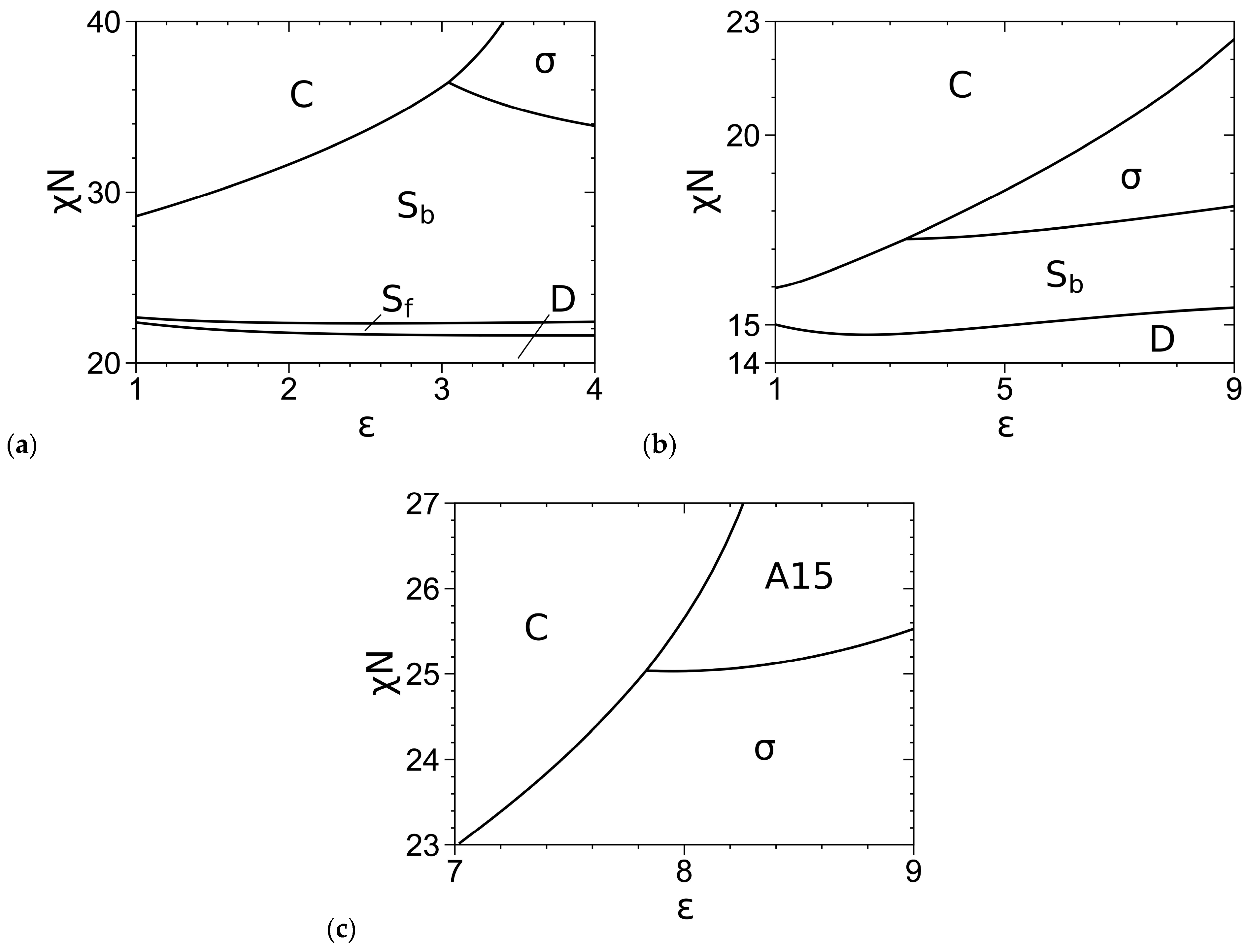
3.2. Phase Diagrams
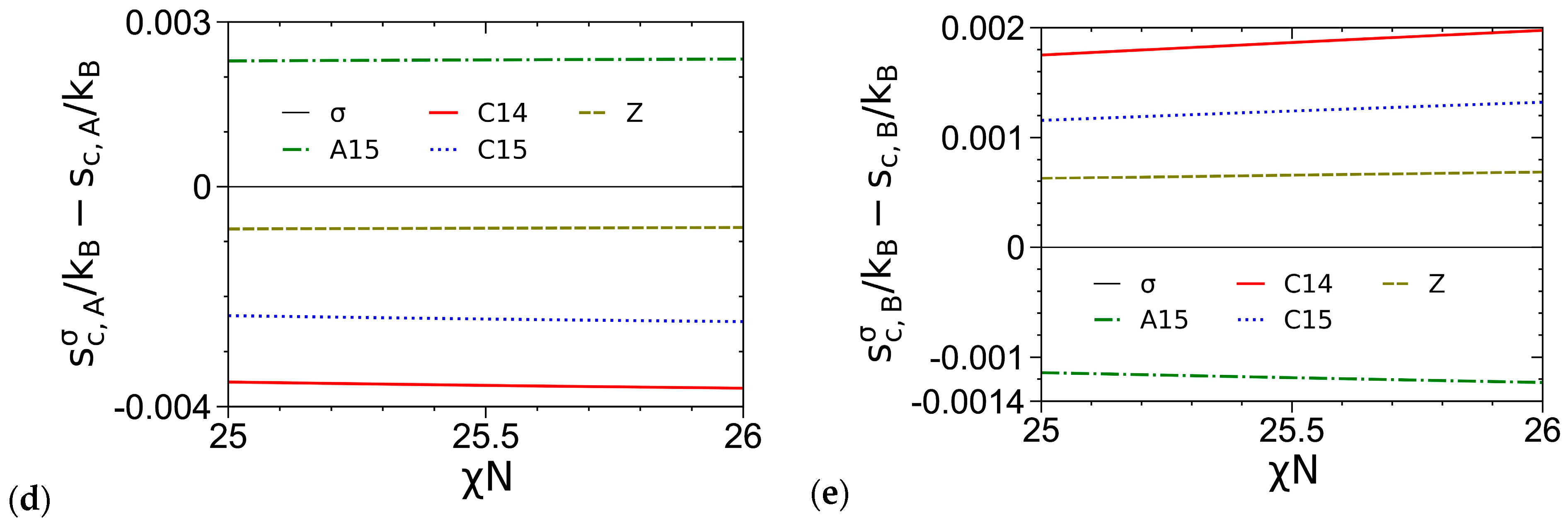
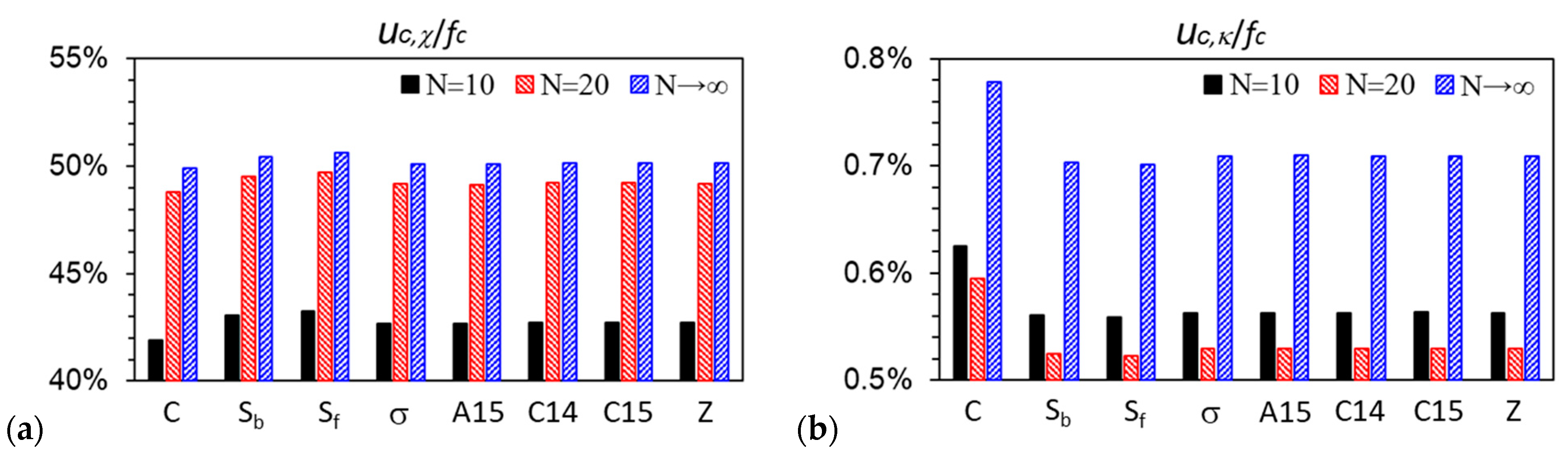
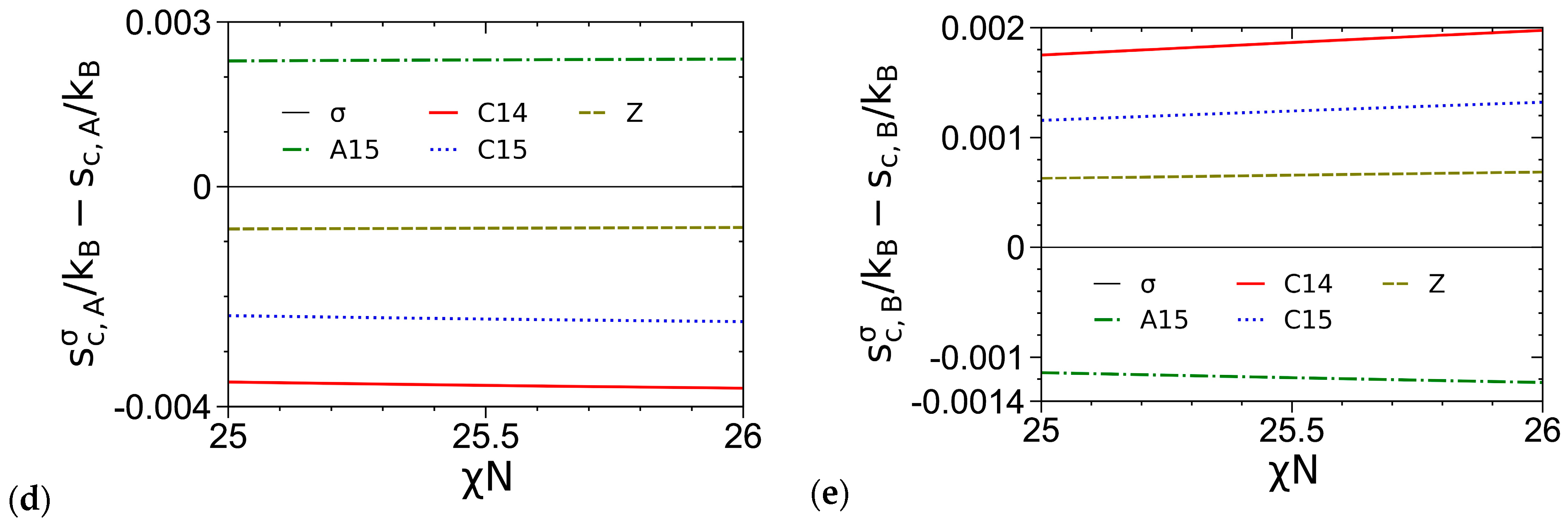
3.3. Curves of βfc and Its Components
3.4. Effects of N in the DPDC Model
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, J.; Wang, Q. Frank–Kasper Phases of Diblock Copolymer Melts Studied with the DPD Model: SCF Results. Macromolecules 2022, 55, 8931–8939, Erratum in Macromolecules 2024, in press. [Google Scholar] [CrossRef]
- Reddy, A.; Buckley, M.B.; Arora, A.; Bates, F.S.; Dorfman, K.D.; Grason, G.M. Stable Frank-Kasper phases of self-assembled, soft matter spheres. Proc. Natl. Acad. Sci. USA 2018, 115, 10233–10238. [Google Scholar] [CrossRef]
- Bates, M.W.; Lequieu, J.; Barbon, S.M.; Lewis, R.M.; Delaney, K.T.; Anastasaki, A.; Hawker, C.J.; Fredrickson, G.H.; Bates, C.M. Stability of the A15 phase in diblock copolymer melts. Proc. Natl. Acad. Sci. USA 2019, 116, 13194–13199. [Google Scholar] [CrossRef]
- Vigil, D.L.; Quah, T.; Sun, D.; Delaney, K.T.; Fredrickson, G.H. Self-Consistent Field Theory Predicts Universal Phase Behavior for Linear, Comb, and Bottlebrush Diblock Copolymers. Macromolecules 2022, 55, 4237–4244. [Google Scholar] [CrossRef]
- Leibler, L. Theory of Microphase Separation in Block Copolymers. Macromolecules 1980, 13, 1602–1617. [Google Scholar] [CrossRef]
- Matsen, M.W.; Schick, M. Stable and Unstable Phases of a Diblock Copolymer Melt. Phys. Rev. Lett. 1994, 72, 2660–2663. [Google Scholar] [CrossRef]
- Delaney, K.T.; Fredrickson, G.H. Recent Developments in Fully Fluctuating Field-Theoretic Simulations of Polymer Melts and Solutions. J. Phys. Chem. B 2016, 120, 7615–7634. [Google Scholar] [CrossRef]
- Schulze, M.W.; Lewis, R.M.; Lettow, J.H.; Hickey, R.J.; Gillard, T.M.; Hillmyer, M.A.; Bates, F.S. Conformational Asymmetry and Quasicrystal Approximants in Linear Diblock Copolymers. Phys. Rev. Lett. 2017, 118, 207801. [Google Scholar] [CrossRef]
- Jeon, S.; Jun, T.; Jo, S.; Ahn, H.; Lee, S.; Lee, B.; Ryu, D.Y. Frank-Kasper Phases Identified in PDMS-b-PTFEA Copolymers with High Conformational Asymmetry. Macromol. Rapid Commun. 2019, 40, 1900259. [Google Scholar] [CrossRef]
- Kim, K.; Arora, A.; Lewis, R.M.; Liu, M.J.; Li, W.H.; Shi, A.C.; Dorfman, K.D.; Bates, F.S. Origins of low-symmetry phases in asymmetric diblock copolymer melts. Proc. Natl. Acad. Sci. USA 2018, 115, 847–854. [Google Scholar] [CrossRef]
- Zhang, C.; Bates, M.W.; Geng, Z.S.; Levi, A.E.; Vigil, D.; Barbon, S.M.; Loman, T.; Delaney, K.T.; Fredrickson, G.H.; Bates, C.M.; et al. Rapid Generation of Block Copolymer Libraries Using Automated Chromatographic Separation. J. Am. Chem. Soc. 2020, 142, 9843–9849. [Google Scholar] [CrossRef]
- Lee, S.; Gillard, T.M.; Bates, F.S. Fluctuations, Order, and Disorder in Short Diblock Copolymers. AICHE J. 2013, 59, 3502–3513. [Google Scholar] [CrossRef]
- Lee, S.; Leighton, C.; Bates, F.S. Sphericity and symmetry breaking in the formation of Frank-Kasper phases from one component materials. Proc. Natl. Acad. Sci. USA 2014, 111, 17723–17731. [Google Scholar] [CrossRef]
- Gillard, T.M.; Lee, S.; Bates, F.S. Dodecagonal quasicrystalline order in a diblock copolymer melt. Proc. Natl. Acad. Sci. USA 2016, 113, 5167–5172. [Google Scholar] [CrossRef]
- Kim, K.; Schulze, M.W.; Arora, A.; Lewis, R.M.; Hillmyer, M.A.; Dorfman, K.D.; Bates, F.S. Thermal processing of diblock copolymer melts mimics metallurgy. Science 2017, 356, 520–523. [Google Scholar] [CrossRef]
- Barbon, S.M.; Song, J.-A.; Chen, D.; Zhang, C.; Lequieu, J.; Delaney, K.T.; Anastasaki, A.; Rolland, M.; Fredrickson, G.H.; Bates, M.W.; et al. Architecture Effects in Complex Spherical Assemblies of (AB)n-Type Block Copolymers. ACS Macro Lett. 2020, 9, 1745–1752. [Google Scholar] [CrossRef]
- Uddin, M.H.; Rodriguez, C.; López-Quintela, A.; Leisner, D.; Solans, C.; Esquena, J.; Kunieda, H. Phase Behavior and Microstructure of Poly(oxyethylene)−Poly(dimethylsiloxane) Copolymer Melt. Macromolecules 2003, 36, 1261–1271. [Google Scholar] [CrossRef]
- Mueller, A.J.; Lindsay, A.P.; Jayaraman, A.; Lodge, T.P.; Mahanthappa, M.K.; Bates, F.S. Quasicrystals and Their Approximants in a Crystalline–Amorphous Diblock Copolymer. Macromolecules 2021, 54, 2647–2660. [Google Scholar] [CrossRef]
- Jeon, S.; Jun, T.; Jeon, H.I.; Ahn, H.; Lee, S.; Lee, B.; Ryu, D.Y. Various Low-Symmetry Phases in High-χ and Conformationally Asymmetric PDMS-b-PTFEA Copolymers. Macromolecules 2021, 54, 9351–9360. [Google Scholar] [CrossRef]
- Lee, S.; Bluemle, M.J.; Bates, F.S. Discovery of a Frank-Kasper sigma Phase in Sphere-Forming Block Copolymer Melts. Science 2010, 330, 349–353. [Google Scholar] [CrossRef]
- Lindsay, A.P.; Jayaraman, A.; Peterson, A.J.; Mueller, A.J.; Weigand, S.; Almdal, K.; Mahanthappa, M.K.; Lodge, T.P.; Bates, F.S. Reevaluation of Poly(ethylene-alt-propylene)-block-Polydimethylsiloxane Phase Behavior Uncovers Topological Close-Packing and Epitaxial Quasicrystal Growth. ACS Nano 2021, 15, 9453–9468. [Google Scholar] [CrossRef]
- Jeon, S.; Jun, T.; Jo, S.; Kim, K.; Lee, B.; Lee, S.; Ryu, D.Y. Modifying Frank–Kasper Mesophases by Modulating Chain Configuration in PDMS-b-PTFEA Copolymers. Macromolecules 2022, 55, 8049–8057. [Google Scholar] [CrossRef]
- Zhou, D.; Xu, M.; Ma, Z.; Gan, Z.; Tan, R.; Wang, S.; Zhang, Z.; Dong, X.H. Precisely Encoding Geometric Features into Discrete Linear Polymer Chains for Robust Structural Engineering. J. Am. Chem. Soc. 2021, 143, 18744–18754. [Google Scholar] [CrossRef]
- Zhou, D.; Xu, M.; Ma, Z.; Gan, Z.; Zheng, J.; Tan, R.; Dong, X.-H. Discrete Diblock Copolymers with Tailored Conformational Asymmetry: A Precise Model Platform to Explore Complex Spherical Phases. Macromolecules 2022, 55, 7013–7022. [Google Scholar] [CrossRef]
- Collanton, R.P.; Dorfman, K.D. Interfacial geometry in particle-forming phases of diblock copolymers. Phys. Rev. Mater. 2022, 6, 015602. [Google Scholar] [CrossRef]
- Fredrickson, G.H. (Chemical Engineering, University of California, Santa Barbara, CA, USA). Private Communication, 2023.
- Matsen, M.W. Self-Consistent Field Theory for Melts of Low-Molecular-Weight Diblock Copolymer. Macromolecules 2012, 45, 8502–8509. [Google Scholar] [CrossRef]
- Sandhu, P.; Zong, J.; Yang, D.; Wang, Q. On the comparisons between dissipative particle dynamics simulations and self-consistent field calculations of diblock copolymer microphase separation. J. Chem. Phys. 2013, 138, 194904. [Google Scholar] [CrossRef] [PubMed]
- Lequieu, J. Combining particle and field-theoretic polymer models with multi-representation simulations. J. Chem. Phys. 2023, 158, 244902. [Google Scholar] [CrossRef] [PubMed]
- Fredrickson, G.H.; Ganesan, V.; Drolet, F. Field-theoretic computer simulation methods for polymers and complex fluids. Macromolecules 2002, 35, 16–39. [Google Scholar] [CrossRef]
- Wang, Q.; Yin, Y. Fast off-lattice Monte Carlo simulations with “soft” repulsive potentials. J. Chem. Phys. 2009, 130, 104903. [Google Scholar] [CrossRef]
- Dorfman, K.D. Frank–Kasper Phases in Block Polymers. Macromolecules 2021, 54, 10251–10270. [Google Scholar] [CrossRef]
- Liu, Y.; Lei, H.; Guo, Q.-Y.; Liu, X.; Li, X.; Wu, Y.; Li, W.; Zhang, W.; Liu, G.; Yan, X.-Y.; et al. Spherical Packing Superlattices in Self-Assembly of Homogenous Soft Matter: Progresses and Potentials. Chin. J. Polym. Sci. 2023, 41, 607–620. [Google Scholar] [CrossRef]
- Available online: https://github.com/dmorse/pscfpp (accessed on 23 December 2023).
- Arora, A.; Qin, J.; Morse, D.C.; Delaney, K.T.; Fredrickson, G.H.; Bates, F.S.; Dorfman, K.D. Broadly Accessible Self-Consistent Field Theory for Block Polymer Materials Discovery. Macromolecules 2016, 49, 4675–4690. [Google Scholar] [CrossRef]
- Helfand, E.; Tagami, Y. Theory of Interface Between Immiscible Polymers. J. Polym. Sci. B Polym. Lett. 1971, 9, 741–746. [Google Scholar] [CrossRef]
- Tzeremes, G.; Rasmussen, K.K.; Lookman, T.; Saxena, A. Efficient computation of the structural phase behavior of block copolymers. Phys. Rev. E 2002, 65, 041806. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, A.; Qin, J.; Morse, D.C. Linear response and stability of ordered phases of block copolymer melts. Macromolecules 2008, 41, 942–954. [Google Scholar] [CrossRef]
- Press, W.H.; Teukolsky, S.A.; Vetterling, W.T.; Flannery, B.P. Numerical Recipes in C: The Art of Scientific Computing; Cambridge University Press: Cambridge, NY, USA, 1992; Chapter 4.3. [Google Scholar]
- Matsen, M.W. Fast and accurate SCFT calculations for periodic block-copolymer morphologies using the spectral method with Anderson mixing. Eur. Phys. J. E 2009, 30, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Morse, D.C.; Bates, F.S.; Dorfman, K.D. Accelerating self-consistent field theory of block polymers in a variable unit cell. J. Chem. Phys. 2017, 146, 244902. [Google Scholar] [CrossRef] [PubMed]
- Press, W.H.; Teukolsky, S.A.; Vetterling, W.T.; Flannery, B.P. Numerical Recipes in C: The Art of Scientific Computing; Cambridge University Press: Cambridge, NY, USA, 1992; Chapter 9.2. [Google Scholar]
- Zong, J.; Wang, Q. On the order-disorder transition of compressible diblock copolymer melts. J. Chem. Phys. 2015, 143, 184903. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.F. The theory of polymer solutions at intermediate concentration. Proc. Phys. Soc. 1966, 88, 265–280. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| This Work | Ref. [3] | |
|---|---|---|
| f = 0.2 and χN = 40 | ||
| C/Sb | ε = 1.874 | ε = 1.882 |
| Sb/σ | ε = 2.667 | ε = 2.722 |
| f = 0.2 and ε = 9 | ||
| D/Sf | χN = 24.225 | χN = 24.155 |
| Sf/Sb | χN = 26.261 | χN = 26.177 |
| Sb/σ | χN = 30.746 | χN = 30.483 |
| f = 0.3 and χN = 40 | ||
| C/A15 | ε = 6.210 | ε = 6.249 |
| f = 0.3 and ε = 9 | ||
| Sb/σ | χN = 17.303 | χN = 17.257 |
| σ/A15 | χN = 25.629 | χN = 25.629 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, J.; Wang, Q. Frank–Kasper Phases of Diblock Copolymer Melts: Self-Consistent Field Results of Two Commonly Used Models. Polymers 2024, 16, 372. https://doi.org/10.3390/polym16030372
He J, Wang Q. Frank–Kasper Phases of Diblock Copolymer Melts: Self-Consistent Field Results of Two Commonly Used Models. Polymers. 2024; 16(3):372. https://doi.org/10.3390/polym16030372
Chicago/Turabian StyleHe, Juntong, and Qiang Wang. 2024. "Frank–Kasper Phases of Diblock Copolymer Melts: Self-Consistent Field Results of Two Commonly Used Models" Polymers 16, no. 3: 372. https://doi.org/10.3390/polym16030372
APA StyleHe, J., & Wang, Q. (2024). Frank–Kasper Phases of Diblock Copolymer Melts: Self-Consistent Field Results of Two Commonly Used Models. Polymers, 16(3), 372. https://doi.org/10.3390/polym16030372