2. Methods
Atomistic simulations are performed here to investigate lignin mechanics. The simulations have direct access to detailed microscopic information, which is hard to obtain in experiments, and therefore, can provide unparalleled microscopic insights into the structure–property relationship. Atomistic simulations of the mechanics of plant cell walls, including cellulose, hemicellulose, and lignin, have been performed recently to investigate the mechanical strength of bamboo fibrils [
11] and the hygromechanical mechanisms of wood cell walls [
12]. Atomistic simulations have also been performed to study the effects of heat and moisture on the elastic modulus of model lignin molecules [
13]. However, atomistic simulations on the mechanics of lignin molecules with molecular weights, chemical compositions, and molecular architectures close to experimental samples have not been performed systematically. Furthermore, existing atomistic simulations have focused on the elastic response of lignin, and therefore, simulation work has yet to be extended to cover the plastic mechanics of lignin upon large deformation. Comprehensive simulations of lignin mechanics with rich microscopic features and extended mechanical response regimes are necessary for the interdisciplinary engineering of lignin-based polymeric materials and also distinguish this computational research from previous studies in the field.
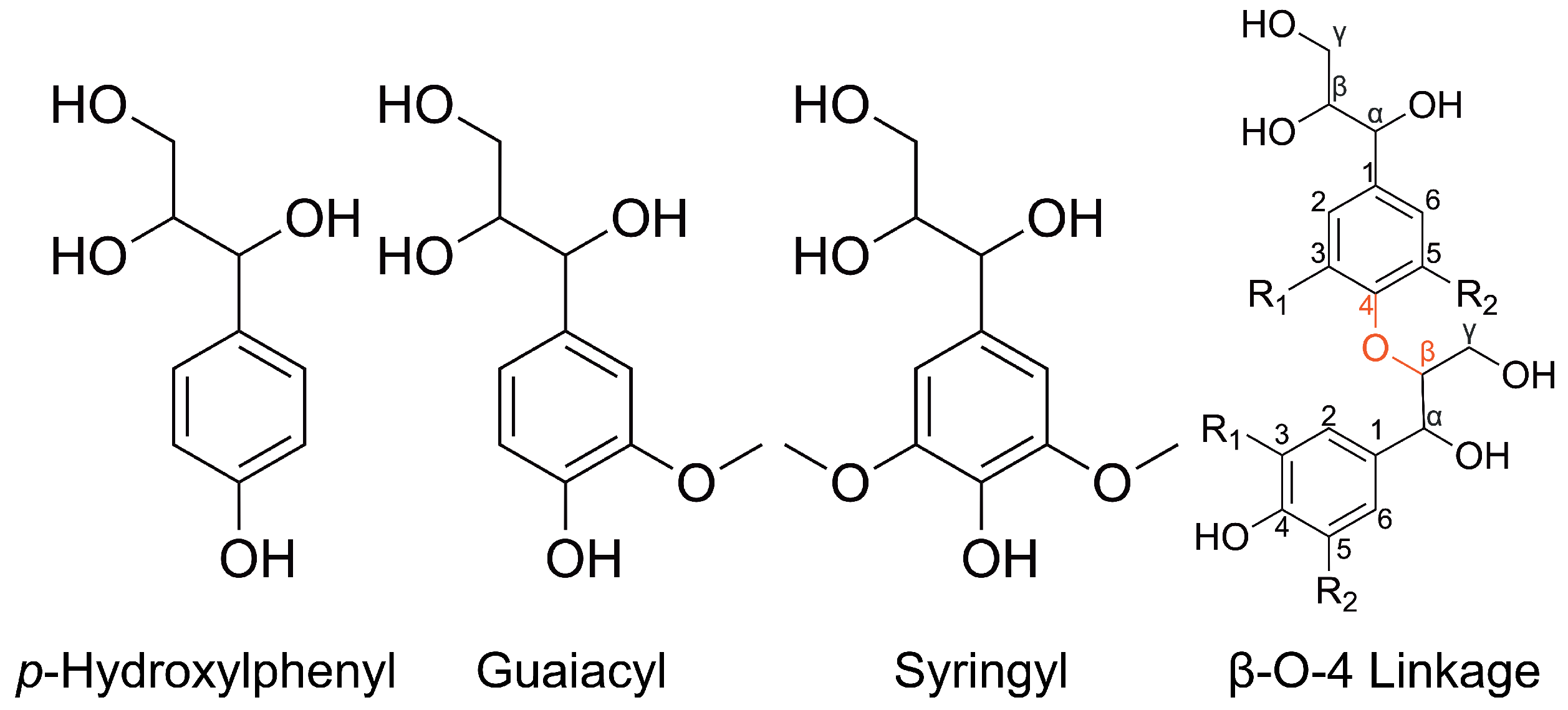
Atomistic simulations of lignin have just emerged, partly because the technical difficulty with modeling the complex structure of a lignin molecule has been addressed only recently. Unlike the well-defined structure of cellulose and hemicellulose, the molecular structure of lignin is described statistically by breaking down a lignin molecule into its constituent basic units, the
p-Hydroxylphenyl (H), guaiacyl (G), and Syringyl (S) lignin monomers, and various linkages between the monomers. For instance, the G monomer and the
-O-4 linkage between two G monomers are the most common lignin monomer and linkage, respectively. The chemical structures of H monomer, G monomer, S monomer, and the
-O-4 linkage between two monomers are shown in
Figure 1. Early atomistic simulations approximated lignin molecules as linear polymers consisting of G monomers and
-O-4 linkages only. In 2018, LigninBuilder was published by Vermaas et al. [
14] and it has greatly facilitated the building of lignin molecules with well-controlled molecular weights, chemical compositions in terms of the ratios of various monomers and linkages, and molecular architectures in terms of the branching coefficient. The molecular features of the atomistic simulation samples created by LigninBuilder closely match those revealed by the analytical chemistry of natural lignin samples. Moreover, these samples have been used to study the conformations and solubility of lignin in different solvents, showing consistency with experimental observations [
15]. The builder allows one to focus on the relation of mechanical properties to lignin’s structure in the simulations rather than the characterization and reproduction of lignin’s structure.
Three representative lignin samples were simulated with the structural information provided by LigninBuilder [
14]. The three samples were the lignin of miscanthus, birch, and spruce, which represent grass, hardwood, and softwood, respectively. As shown in
Table 1, each sample has a distribution of molecular weights, a unique chemical composition in terms of the fractions of the H, G, and S monomers, and a different branching coefficient that quantifies the fraction of monomers at branching points. The chain number of all three simulation samples is
. Additionally, model lignin samples were produced to further study the molecular weight effects. Each model lignin sample consists of linear chains made of G monomers and
-O-4 linkages (poly-G). As such, the chemical composition is homogeneous and the molecular architecture is purely linear with no branching. Five poly-G samples were created, where the number of G monomers per chain varied from
to
, and the numbers of chains were
, and 200 accordingly.
The classical CHARMM force fields [
16] for molecular dynamics were used in the atomistic simulations of lignin. The potential energy function in CHARMM includes terms describing the individual covalent bonds, the angle-dependent bending of 2 consecutive bonds, the torsion of the dihedral angle of 3 consecutive bonds, and the improper out-of-plane bending of 3 consecutive bonds. As a result, the intra-molecular connectivity is defined. The potential energy function also includes non-bonded terms such as the pairwise interatomic Lennard-Jones (LJ) potentials and the electrostatic interactions between charges carried by the atoms. The original CHARMM force fields have been modified to allow more precise parameterization of the interaction parameters that match the quantum chemistry calculations on lignin as well as the experimental data on lignin’s structure. The parameters of the modified CHARMM force fields are provided by LigninBuilder [
14]. Specifically, Vermaas et al. performed extensive quantum mechanical calculations of the H, G, and S lignin monomers and their various dimer combinations. The calculations generated data for developing comprehensive CHARMM force fields tailored for lignin. Compared to the general CHARMM force fields, a more accurate representation of the interactions and conformations of lignin monomers and dimers was achieved. See the developer’s GitHub repository for more details regarding the modified CHARMM force fields [
17].
All the atomistic simulations were performed using Large-scale Atomic/Molecular Massively Parallel Simulator LAMMPS [
18], which makes use of Message Passing Interface (MPI) for parallel communication and is a free and open-source simulation package. The cut-off radius for the pairwise LJ interaction was 12 Å, and the long-range electrostatic interactions were computed using the particle–particle particle–mesh (PPPM) solver with a precision of
. The timestep for evolving the trajectory of the atomistic simulation was 1 fs.
Figure 2 shows the different stages of the simulations in this work.
3. Results and Discussion
The lignin molecules in a sample need to be packed in a cubic simulation box with periodic boundary conditions for the study of bulk mechanics. This was completed at an artificially high temperature of
K, which sped up the uniform distribution of lignin molecules throughout the simulation box. The temperature in all simulations was controlled by a Nosé–Hoover thermostat with a characteristic damping time 50 fs. Subsequently, the sample was equilibrated at a temperature of
K and pressure of
atm over
million time steps. The pressure was controlled using an additional Nosé–Hoover barostat with a characteristic damping time of 500 fs. After the equilibration, the sample was quenched at
atm from
K to
K. The quenching lasted for 3 million time steps or, equivalently, 3 ns. The glass transition temperature
was determined by extrapolating the high-temperature and low-temperature simulation data of mass density and finding their intersection [
19], as illustrated by the red lines in
Figure 3a. It was determined that
= 363 K, 379 K, and 370 K for the miscanthus, birch, and spruce lignin samples, respectively. At room temperature (
K), the density was 1.23 g/cm
3 for the three samples and comparable with the typical mass density of lignin, which is around 1.3 g/cm
3.
To confirm a sample at
K is in the glassy state, the mean squared displacement (MSD) of lignin monomers as a function of time was computed and compared with the length scale
of a monomer [
19], where
V is the volume of the simulation box containing
lignin monomers. The MSD over 1 ns, which is the time period for a unit strain at a strain rate of 10
−6 fs
−1, is well below the square of a lignin monomer with a size of
, which is around 50 Å
2. As such, the lignin samples have rather limited molecular mobility, as expected for the glassy state, during the deformations at a strain rate of 10
−6 fs
−1 or higher. Discussing further details of the glassy dynamics is out of the scope of this work.
The glassy samples at
K were used in the mechanics simulations. Two deformation modes were simulated. In one mode, uniaxial compression was applied along the
z-direction with a true strain rate of 10
−6 fs
−1, and the pressure along the lateral
x-and
y-directions was maintained at 1 atm using a Nosé–Hoover barostat with a characteristic damping time of 500 fs. The temperature during the compression was kept at
K using a Nosé–Hoover thermostat.
Figure 4 visualizes the poly-G lignin sample with
G monomers per chain under compressive strain, where one lignin chain is highlighted in red, while others are dimmed as the background. In the second mode, tensile loading was applied along the
z-direction. The box expanded at a constant engineering strain rate 10
−4 fs
−1. Meanwhile, the box sizes in the lateral
x- and
y-directions were kept unchanged to introduce a tri-axial stress condition necessary for the modeling of cavitation and mechanical failure.
The stress–strain curves for the three lignin samples under uniaxial compression are shown in
Figure 5a. All curves exhibit an initial linear elastic regime, a yield peak, and then a plastic flow with ultimate strain hardening as the strain increases. These features resemble the mechanical response of amorphous glassy polymers under uniaxial compression [
19]. From the initial linear elastic regime, Young’s modulus
E is determined. The stress–strain curve at strains below
is fit to a linear function, with the slope being
E. The fitting results are
GPa,
GPa, and
GPa, for the miscanthus, birch, and spruce lignin samples, respectively. All values fall in the range of
GPa–6.7 GPa from the experiments on real lignin samples [
20]. The modulus is comparable to that of synthetic thermoplastics such as polystyrene but higher than that of synthetic elastomers by more than one order of magnitude. This result is consistent with the existence of lignin samples as rigid powders on a macroscopic scale.
The plastic flow stress
in the post-yield and pre-strain-hardening regime is determined as the average stress for
.
GPa,
GPa, and
GPa for the miscanthus, birch, and spruce lignin samples, respectively. As shown in
Figure 5b,c, additional simulations at higher deformation rates demonstrate that both
E and
are in the regime, where they show only weak logarithmic dependencies on the strain rate, as expected for the glassy state [
21].
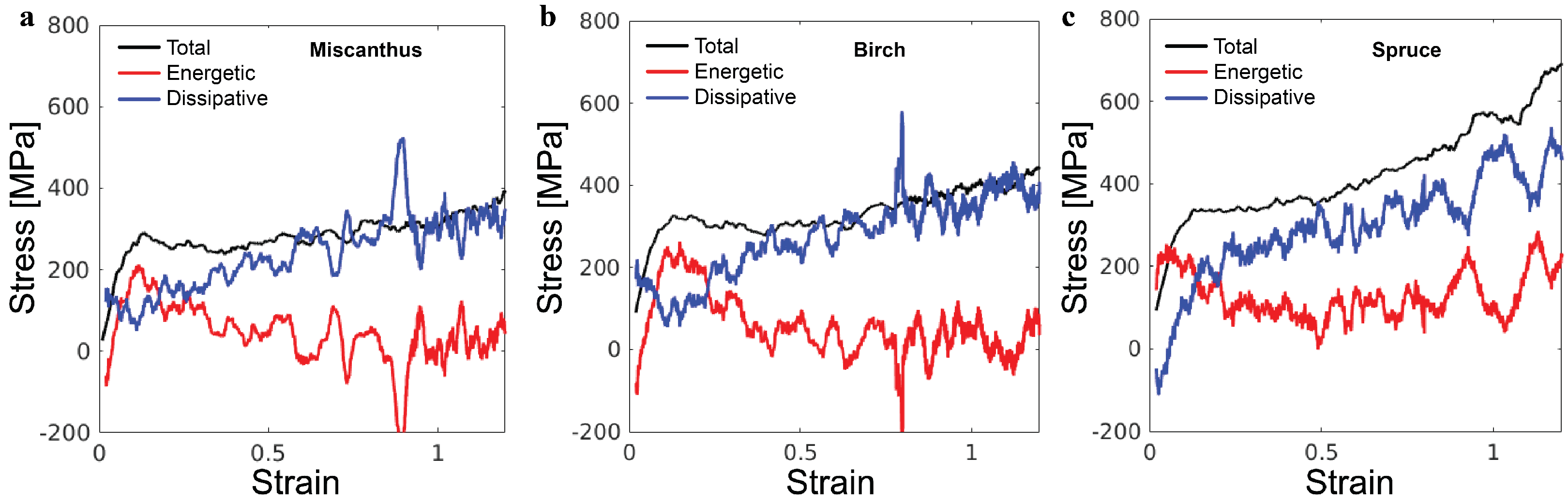
The simulations enable the decomposition of the overall stress
to an energetic component
and a dissipative component
. This decomposition follows the first law of thermodynamics, which states that work
w and heat transaction
q are two equivalent ways of increasing the internal energy
u. From
, the heat dissipated out of the system is
, and therefore,
[
22]. In the simulations, the dissipation of heat is realized via the thermostat. The black lines in
Figure 6 show the total stress
for the three lignin samples. The red lines are the corresponding
, which is computed as the gradient of internal energy density with respect to strain. The blue lines are the corresponding
. In all cases, while
contributes significantly to the initial elastic regime and the yielding,
is the major component for the stress rise in the strain-hardening regime. Such decomposition of the overall stress closely matches the behavior of synthetic polymer glasses in coarse-grained molecular simulations, demonstrating the manifestation of the generic glassy polymer mechanics in the lignin samples [
19]. As such, our extensive knowledge of glassy polymer mechanics may be used to guide tailoring of the mechanics of lignin-based materials.
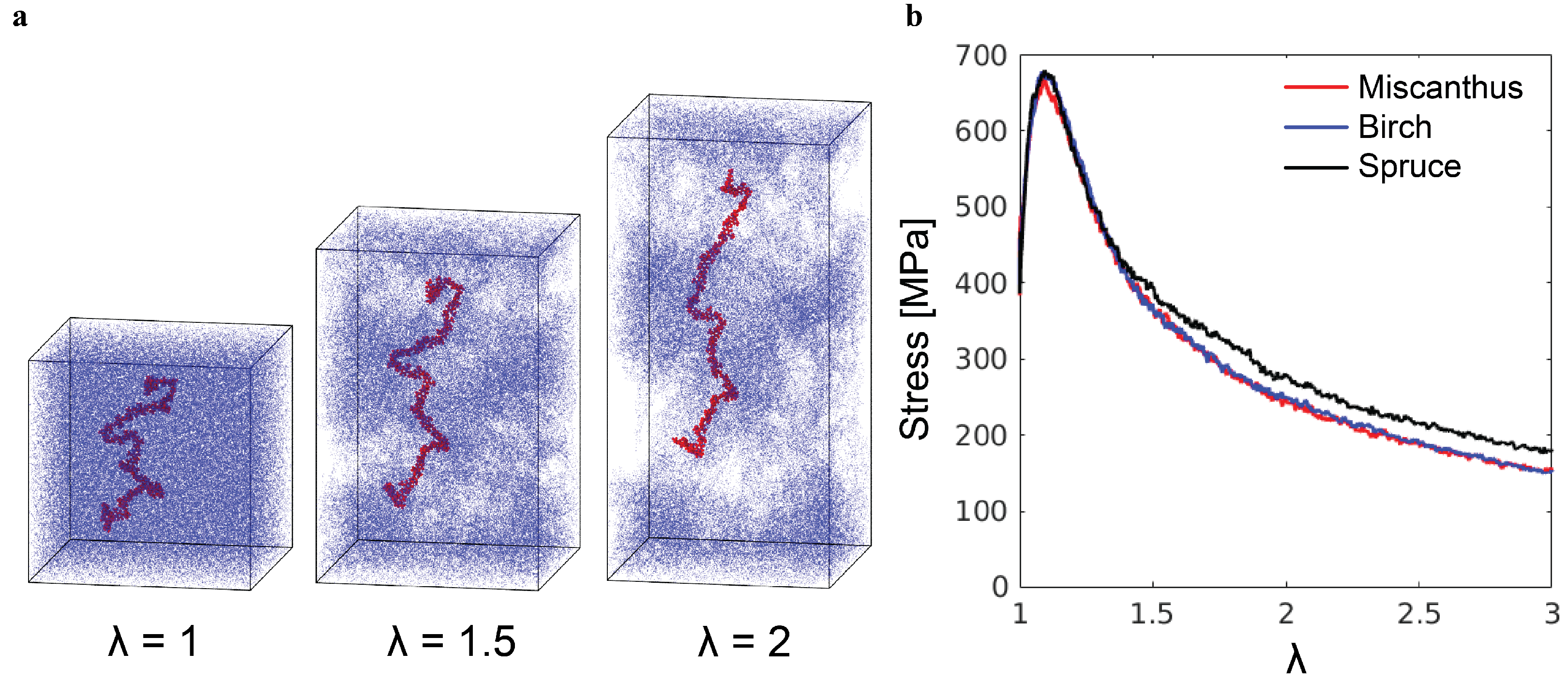
The tensile loading results in a qualitatively different mechanical response, as shown in
Figure 7. After the initial linear elastic regime and the yield peak, the tensile stress decreases with increasing stretch
, where
and
are the initial and current box sizes in the
z-direction. The stress drop is a consequence of cavitation and catastrophic chain pullout, as illustrated by the snapshots in
Figure 7a. The stretch in the simulations is the local stretch of lignin molecules, rather than the stretch of a lignin sample in experiments. On the macroscopic scale, a lignin sample exists in the powder form and does not possess any measurable stretchability. Such brittleness is related to the microscopic failure mechanism revealed by the simulations here.
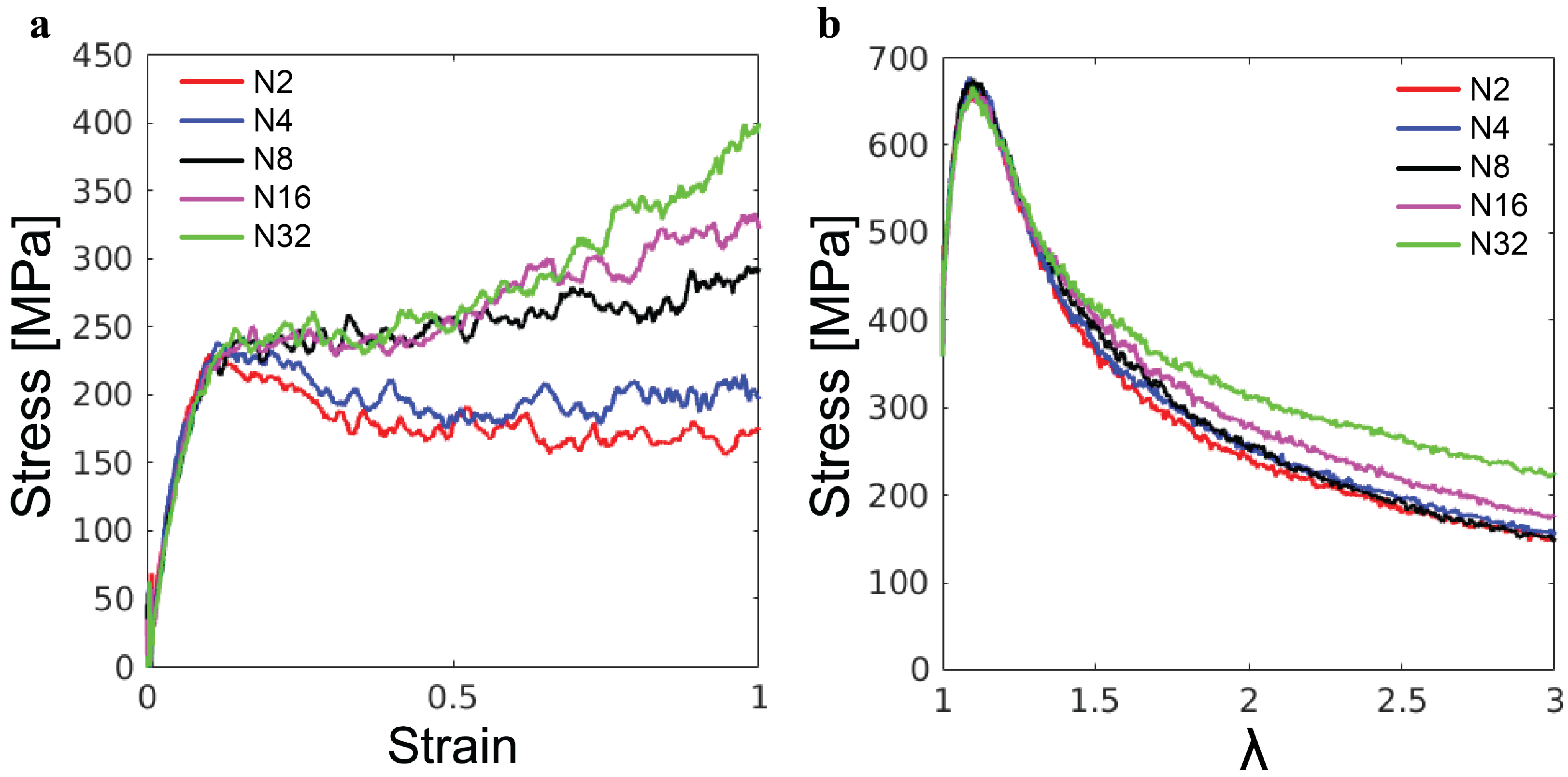
The simulations of the poly-G samples with varying chain lengths
N further reveal the effects of molecular weight on the mechanical response of lignin. As shown in
Figure 8a, for the uniaxial compression, the initial elastic and yield stress both exhibit almost no dependence on
N. By contrast,
N-dependencies are observed in the post-yield regime. For
and
, there is a slight drop in stress following the yield peak, indicating strain softening, but eventually, the stress stabilizes in the steady state. For
, there is no strain softening and the plastic flow is almost independent of
N for compressive strain up to
. However, the ultimate strain hardening becomes stronger as
N increases. Based on our understanding of strain hardening in synthetic polymer glasses, the strain hardening for larger
N is stronger because longer molecules need more plastic rearrangements of their conformations to accommodate the same global strain [
19]. The different molecular weights of the lignin samples in
Figure 8a may explain their different strain-hardening behaviors. The enhancement of strain hardening from miscanthus to birch and then to spruce is related to the increase in the average molecular weight from
kDa to
kDa and then to
kDa (see
Table 1).
The tensile stress–stretch curves for the poly-G samples are shown in
Figure 8b. As in the compression test, there is almost no
N-dependence of the initial elastic response and yielding. The post-yield tensile stress for larger local stretching of molecules increases with
N, as it is harder to separate longer poly-G chains from each other. For synthetic polymer glass, it has been shown that the molecular weight has to be sufficiently high to allow the formation of entanglements and thus arrest the catastrophic chain pullout [
21,
23]. Clearly, up to
, the poly-G chains are not sufficiently long for the emergence of any entanglements. Comparing
Figure 8b and
Figure 7b, one may explain the higher tensile stress of the spruce lignin sample at large
as the result of its higher molecular weight compared to the other lignin samples. For the mechanics under both compression and tension, the mechanics of lignin samples are anticipated to be affected by their chemical composition and branching coefficients as well, which needs further study in the future to clarify.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}