
Theoretical Study on the High Polymer Molecular Weight of Heteroatom-Substituted Constrained Geometry Catalyst

 ,
, 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Computational Methods
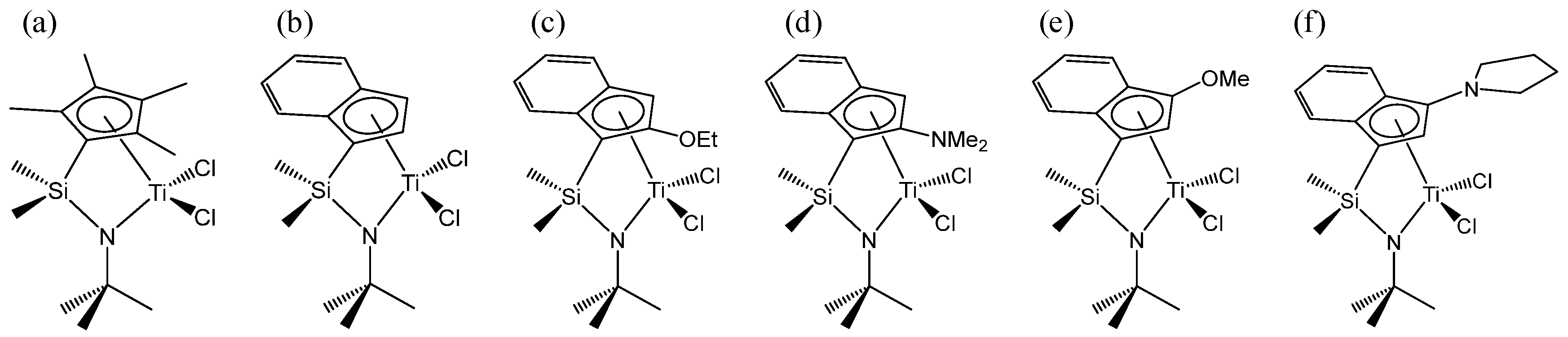
3. Discussion and Results
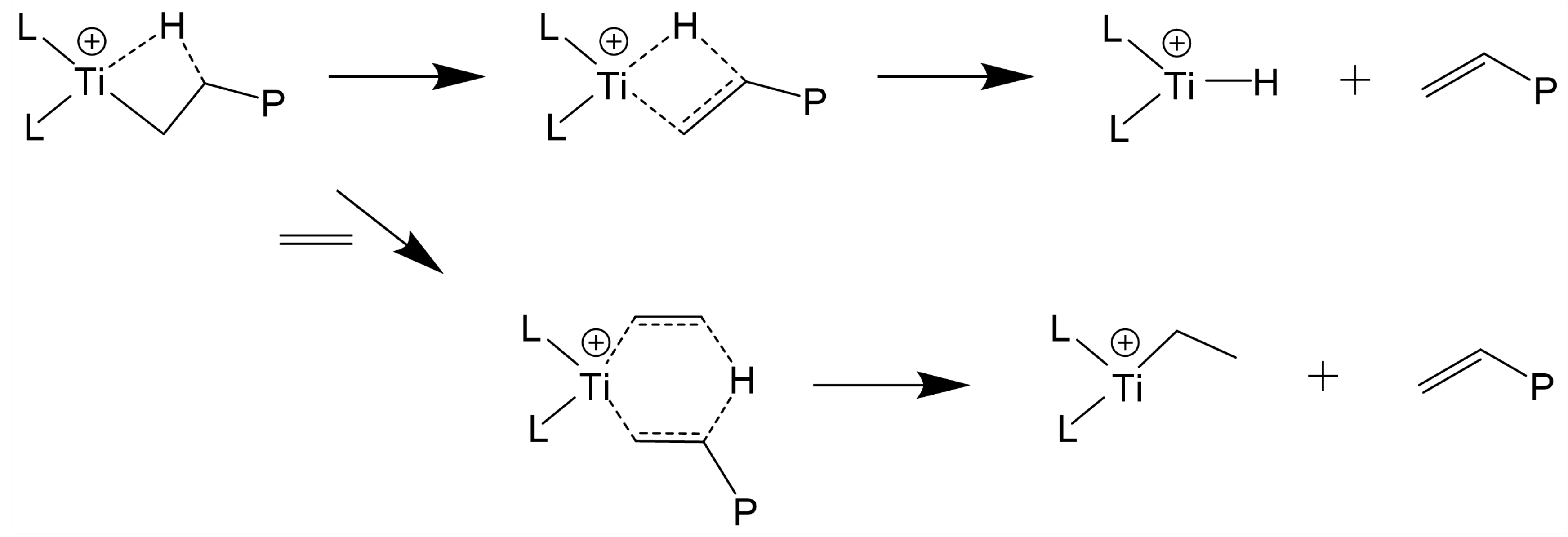
3.1. β-H Elimination
3.2. β-H Transfer to Monomer
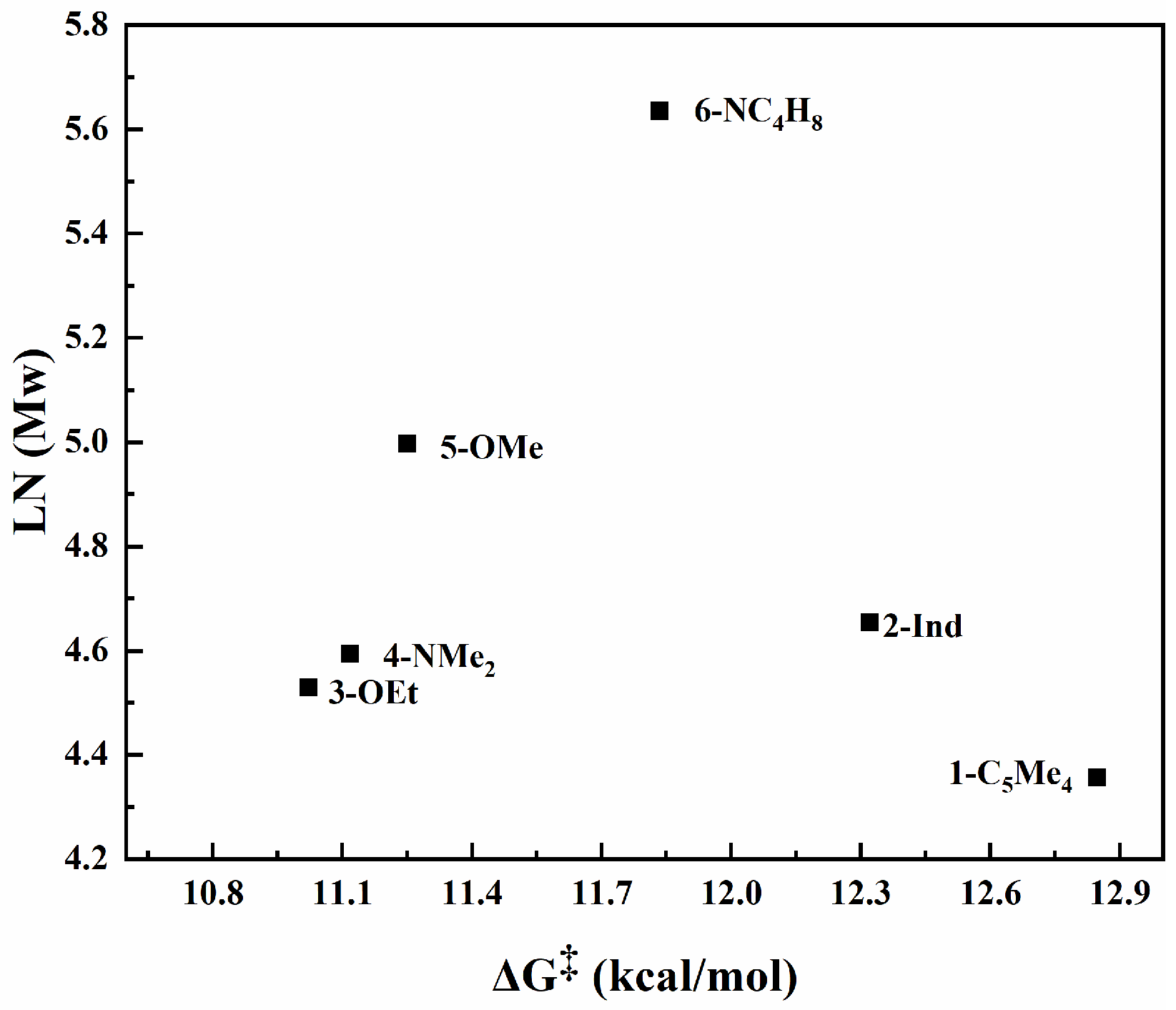
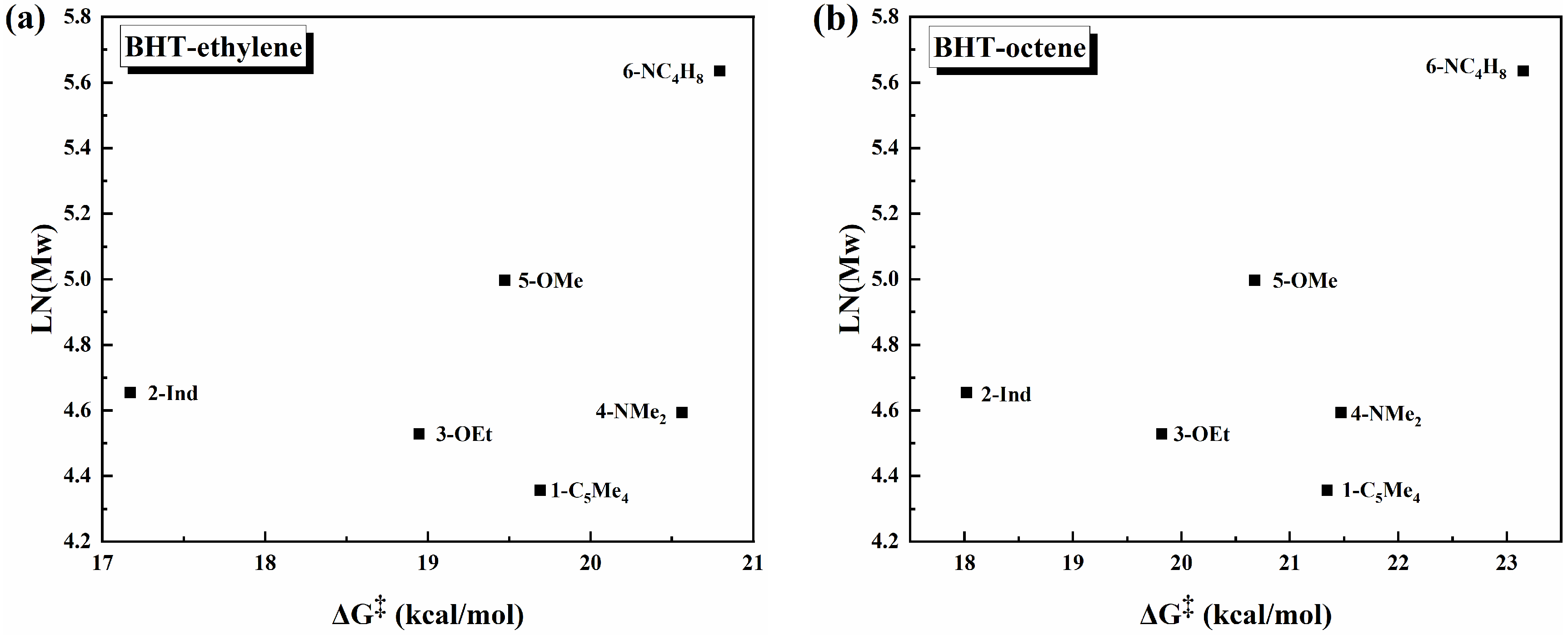
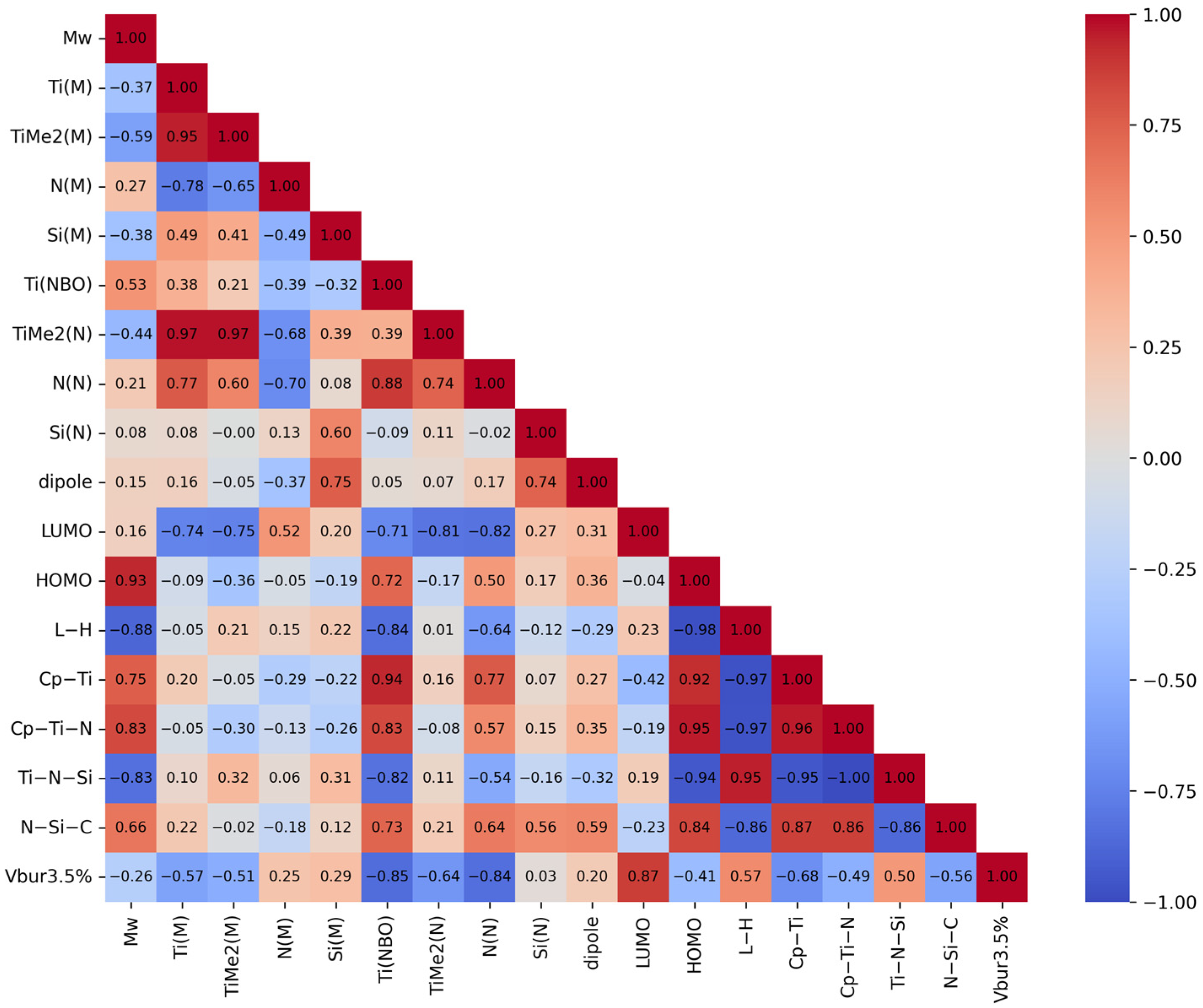
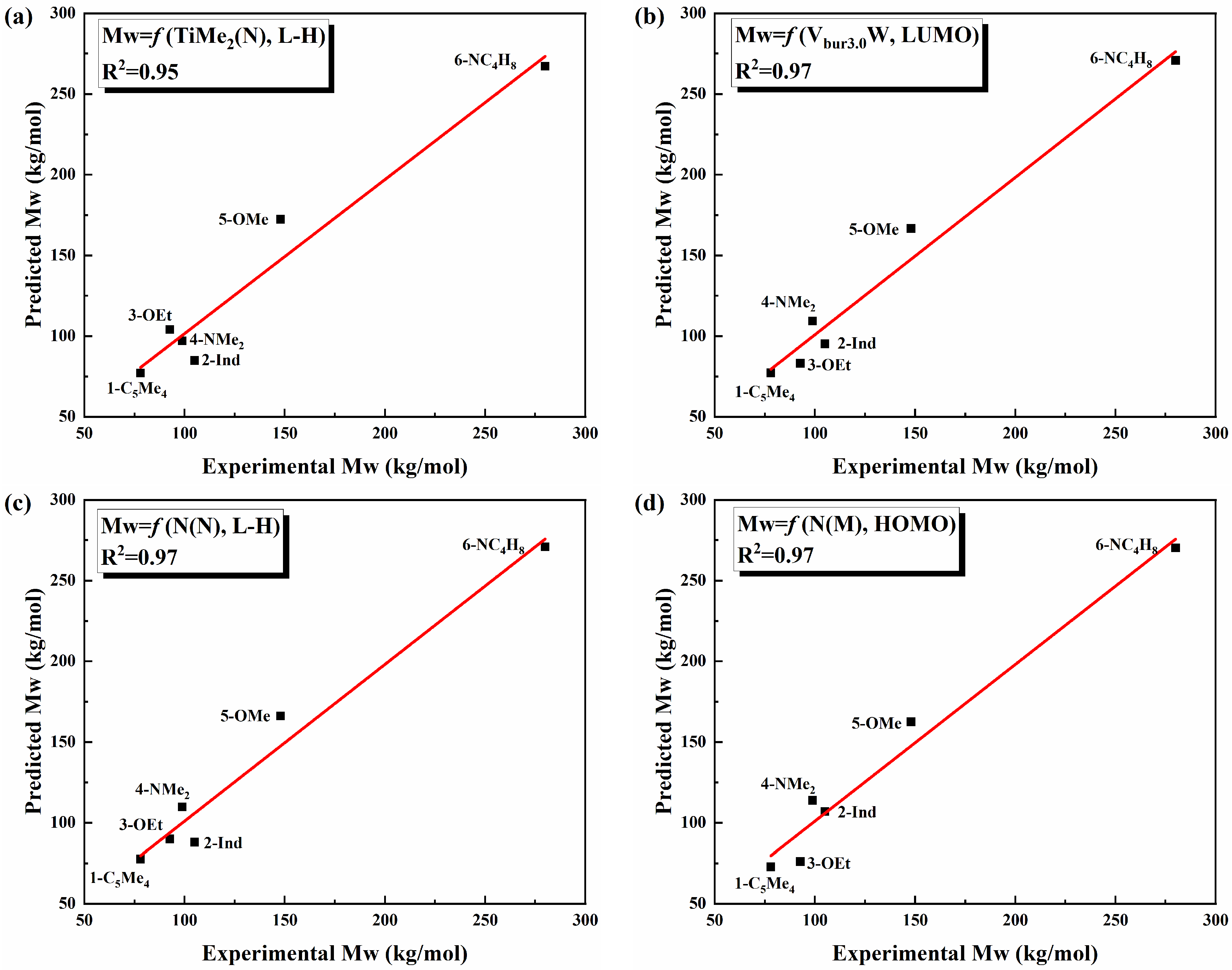
3.3. QSPR Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Li, F.; Liu, W. Progress in the catalyst for ethylene/α-olefin copolymerization at high temperature. Can. J. Chem. Eng. 2023, 101, 4992–5019. [Google Scholar] [CrossRef]
- Sun, M.; Xiao, Y.; Liu, K.; Yang, X.; Liu, P.; Jie, S.; Hu, J.; Shi, S.; Wang, Q.; Lim, K.H.; et al. Synthesis and characterization of polyolefin thermoplastic elastomers: A review. Can. J. Chem. Eng. 2023, 101, 4886–4906. [Google Scholar] [CrossRef]
- Mohite, A.S.; Rajpurkar, Y.D.; More, A.P. Bridging the gap between rubbers and plastics: A review on thermoplastic polyolefin elastomers. Polym. Bull. 2022, 79, 1309–1343. [Google Scholar] [CrossRef]
- Ge, Y.; Hu, L.; Zhang, L.; Fu, Q.; Xu, G.; Xing, Z.; Huang, L.; Zhou, W.; Chen, Y. Polyolefin elastomer as the anode interfacial layer for improved mechanical and air stabilities in nonfullerene solar cells. ACS Appl. Mater. Interfaces 2020, 12, 10706–10716. [Google Scholar] [CrossRef]
- Ren, Q.; Fan, J.; Zhang, Q.; Yi, J.; Feng, J. Toughened polypropylene random copolymer with olefin block copolymer. Mater. Des. 2016, 107, 295–301. [Google Scholar] [CrossRef]
- Cano, J.; Kunz, K. How to synthesize a constrained geometry catalyst (CGC)—A survey. J. Organomet. Chem. 2007, 692, 4411–4423. [Google Scholar] [CrossRef]
- McKnight, A.L.; Waymouth, R.M. Group 4 ansa-Cyclopentadienyl-Amido Catalysts for Olefin Polymerization. Chem. Rev. 1998, 98, 2587–2598. [Google Scholar] [CrossRef]
- Gibson, V.C.; Spitzmesser, S.K. Advances in non-metallocene olefin polymerization catalysis. Chem. Rev. 2003, 103, 283–316. [Google Scholar] [CrossRef] [PubMed]
- Britovsek, G.J.P.; Gibson, V.C.; Wass, D.F. The search for new-generation olefin polymerization catalysts: Life beyond metallocenes. Angew. Chem. Int. Ed. 1999, 38, 428–447. [Google Scholar] [CrossRef]
- Okuda, J. Rare earth metal complexes that contain linked amido-cyclopentadienyl ligands: Ansa-metallocene mimics and “constrained geometry” catalysts. Dalton Trans. 2003, 12, 2367–2378. [Google Scholar] [CrossRef]
- Okuda, J.; Musikabhumma, K.; Sinnema, P. The Kinetic Stability of Cationic Benzyl Titanium Complexes that Contain a Linked Amido-Cyclopentadienyl Ligand: The Influence of the Amido-Substituent on the Ethylene Polymerization Activity of “Constrained Geometry Catalysts”. Isr. J. Chem. 2002, 42, 383–392. [Google Scholar] [CrossRef]
- Soga, K.; Uozumi, T.; Nakamura, S.; Toneri, T.; Teranishi, T.; Sano, T.; Arai, T.; Shiono, T. Structures of polyethylene and copolymers of ethylene with 1-octene and oligoethylene produced with the Cp2ZrCl2 and [(C5Me4)SiMe2N(t-Bu)]TiCl2 catalysts. Macromol. Chem. Phys. 1996, 197, 4237–4251. [Google Scholar] [CrossRef]
- Stevens, J.C. 26. Insite TM Catalysts Structure/Activity Relationships for Olefin Polymerization. Stud. Surf. Sci. Catal. 1994, 89, 277–284. [Google Scholar]
- Klosin, J.; Kruper, W.J.; Nickias, P.N.; Roof, G.R.; De Waele, P.; Abboud, K.A. Heteroatom-substituted constrained-geometry complexes. Dramatic substituent effect on catalyst efficiency and polymer molecular weight. Organometallics 2001, 20, 2663–2665. [Google Scholar] [CrossRef]
- Klosin, J.; Fontaine, P.P.; Figueroa, R. Development of group IV molecular catalysts for high temperature ethylene-α-olefin copolymerization reactions. Accounts Chem. Res. 2015, 48, 2004–2016. [Google Scholar] [CrossRef]
- Martínez, S.; Ramos, J.; Cruz, V.L.; Martínez-Salazar, J. Isomeric effect of the Et(H4Ind)2Zr(CH3)2 catalyst on the copolymerization of ethylene and styrene: A computational study. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 4752–4761. [Google Scholar] [CrossRef]
- Exposito, M.; Martínez, S.; Ramos, J.; Cruz, V.; Lopez, M.; Muñoz-Escalona, A.; Haider, N.; Martínez-Salazar, J. Ethylene/styrene copolymerisation by homogeneous metallocene catalysts: Experimental and molecular simulations using rac-ethylenebis(tetrahydroindenyl)MCl2 [M=Ti,Zr] systems. Polymer 2004, 45, 9029–9038. [Google Scholar] [CrossRef]
- Yang, S.H.; Jo, W.H.; Noh, S.K. Density functional study of the insertion mechanism for ethylene-styrene copolymerization with constrained geometry catalysts. J. Chem. Phys. 2003, 119, 1824–1837. [Google Scholar] [CrossRef]
- Parveen, R.; Cundari, T.R.; Younker, J.M.; Rodriguez, G.; McCullough, L. DFT and QSAR studies of ethylene polymerization by zirconocene catalysts. ACS Catal. 2019, 9, 9339–9349. [Google Scholar] [CrossRef]
- Ciancaleoni, G.; Fraldi, N.; Cipullo, R.; Busico, V.; Macchioni, A.; Budzelaar, P.H.M. Structure/properties relationship for bis(phenoxyamine)zr(iv)-based olefin polymerization catalysts: A simple dft model to predict catalytic activity. Macromolecules 2012, 45, 4046–4053. [Google Scholar] [CrossRef]
- Yan, M.; Kang, X.; Li, S.; Xu, X.; Luo, Y.; He, S.; Chen, C. Mechanistic Studies on Nickel-Catalyzed Ethylene Polymerization: Ligand Effects and Quantitative Structure–Activity Relationship Model. Organometallics 2022, 41, 3212–3218. [Google Scholar] [CrossRef]
- Ratanasak, M.; Hasegawa, J.-Y.; Parasuk, V. Design and prediction of high potentansa-zirconocene catalyst for olefin polymerizations: Combined DFT calculations and QSPR approach. New J. Chem. 2021, 45, 8248–8257. [Google Scholar] [CrossRef]
- Zaccaria, F.; Ehm, C.; Budzelaar, P.H.M.; Busico, V. Accurate Prediction of Copolymerization Statistics in Molecular Olefin Polymerization Catalysis: The Role of Entropic, Electronic, and Steric Effects in Catalyst Comonomer Affinity. ACS Catal. 2017, 7, 1512–1519. [Google Scholar] [CrossRef]
- Maity, B.; Cao, Z.; Kumawat, J.; Gupta, V.; Cavallo, L. A Multivariate linear regression approach to predict ethene/1-olefin copolymerization statistics promoted by group 4 catalysts. ACS Catal. 2021, 11, 4061–4070. [Google Scholar] [CrossRef]
- Uborsky, D.V.; Mladentsev, D.Y.; Guzeev, B.A.; Borisov, I.S.; Vittoria, A.; Ehm, C.; Cipullo, R.; Hendriksen, C.; Friederichs, N.; Busico, V.; et al. C1-Symmetric Si-bridged (2-indenyl)(1-indenyl) ansa-metallocenes as efficient ethene/1-hexene copolymerization catalysts. Dalton Trans. 2020, 49, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lai, X.; Xu, X.; So, Y.-M.; Du, Y.; Zhang, Z.; Pan, Y. Theoretical Study on Ethylene Polymerization Catalyzed by Half-Titanocenes Bearing Different Ancillary Groups. Catalysts 2021, 11, 1392. [Google Scholar] [CrossRef]
- Lu, H.; Kang, X.; Luo, Y. Structure-based relative energy prediction model: A case study of pd(ii)-catalyzed ethylene polymerization and the electronic effect of ancillary ligands. J. Phys. Chem. B 2021, 125, 12047–12053. [Google Scholar] [CrossRef]
- Liao, Z.; Du, X.; Cheng, X.; Chen, Y.; Hong, X.; Zhou, S.; Yang, Y.; Li, W.; Wang, J.; Yang, Y. Quantitative structure-property relationship study of constrained geometry catalysts for olefin polymerization. New J. Chem. 2024, 48, 6221–6231. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- CYLview20; Legault, C.Y. Université de Sherbrooke. 2020. Available online: http://www.cylview.org (accessed on 1 June 2024).
- Falivene, L.; Cao, Z.; Petta, A.; Serra, L.; Poater, A.; Oliva, R.; Scarano, V.; Cavallo, L. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 2019, 11, 872–879. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, X.; Ren, C.; Hong, X.; Wang, J.; Yang, Y.; Liao, Z. Theoretical Study on the High Polymer Molecular Weight of Heteroatom-Substituted Constrained Geometry Catalyst. Polymers 2024, 16, 3251. https://doi.org/10.3390/polym16233251
Du X, Ren C, Hong X, Wang J, Yang Y, Liao Z. Theoretical Study on the High Polymer Molecular Weight of Heteroatom-Substituted Constrained Geometry Catalyst. Polymers. 2024; 16(23):3251. https://doi.org/10.3390/polym16233251
Chicago/Turabian StyleDu, Xinyue, Congjing Ren, Xiaodong Hong, Jingdai Wang, Yongrong Yang, and Zuwei Liao. 2024. "Theoretical Study on the High Polymer Molecular Weight of Heteroatom-Substituted Constrained Geometry Catalyst" Polymers 16, no. 23: 3251. https://doi.org/10.3390/polym16233251
APA StyleDu, X., Ren, C., Hong, X., Wang, J., Yang, Y., & Liao, Z. (2024). Theoretical Study on the High Polymer Molecular Weight of Heteroatom-Substituted Constrained Geometry Catalyst. Polymers, 16(23), 3251. https://doi.org/10.3390/polym16233251