
Bottlebrush Elastomers with Crystallizable Side Chains: Monolayer-like Structure of Backbones Segregated in Intercrystalline Regions



Abstract
1. Introduction
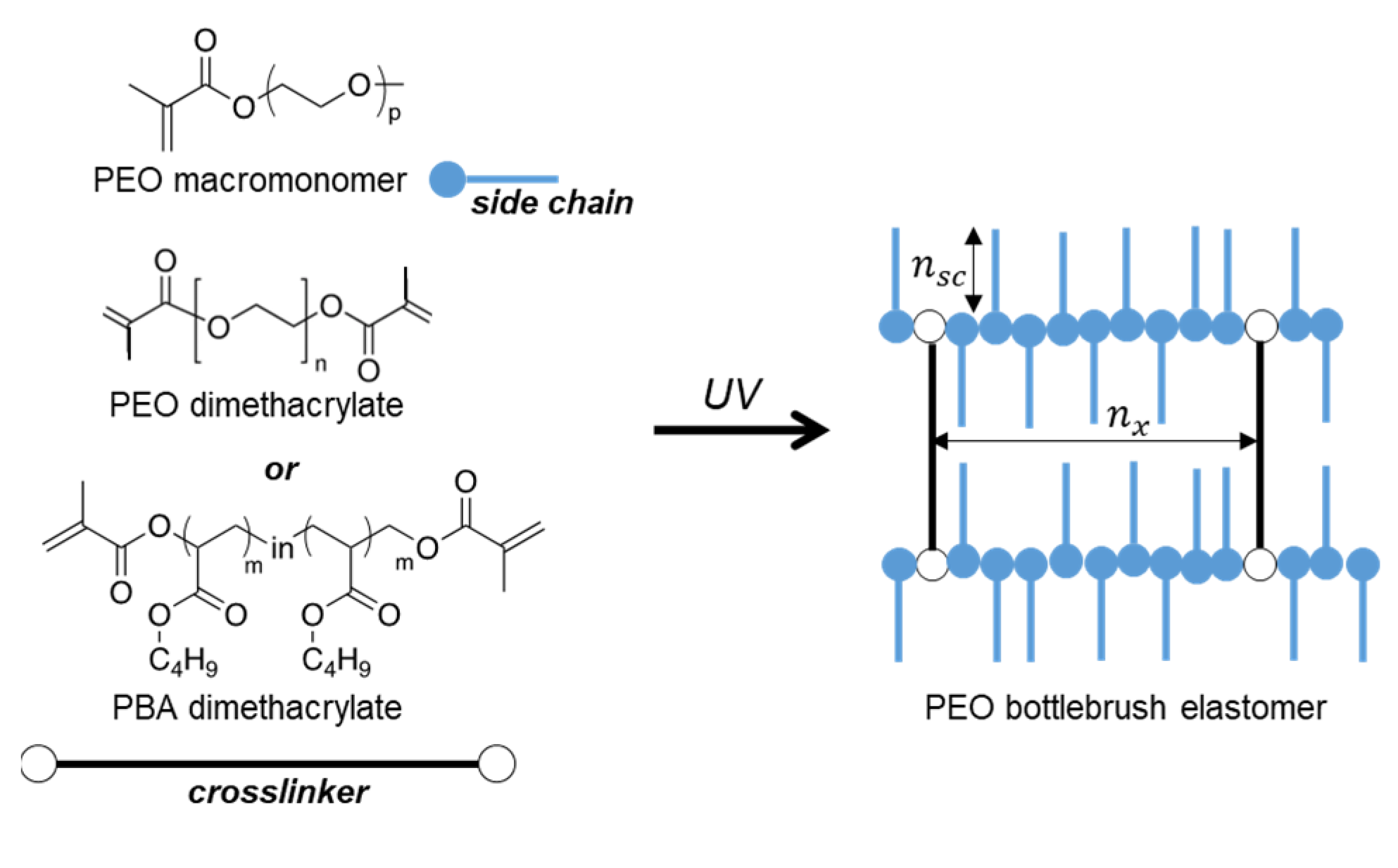
2. Materials and Methods
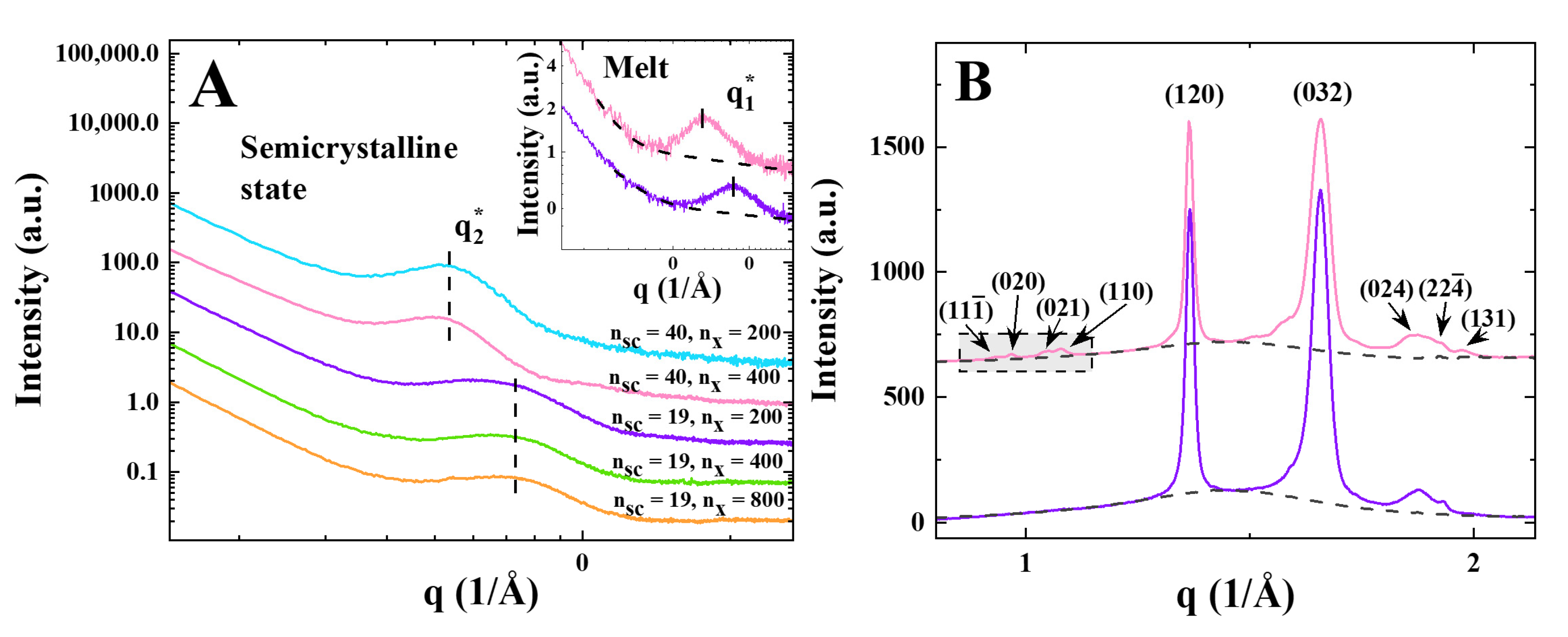
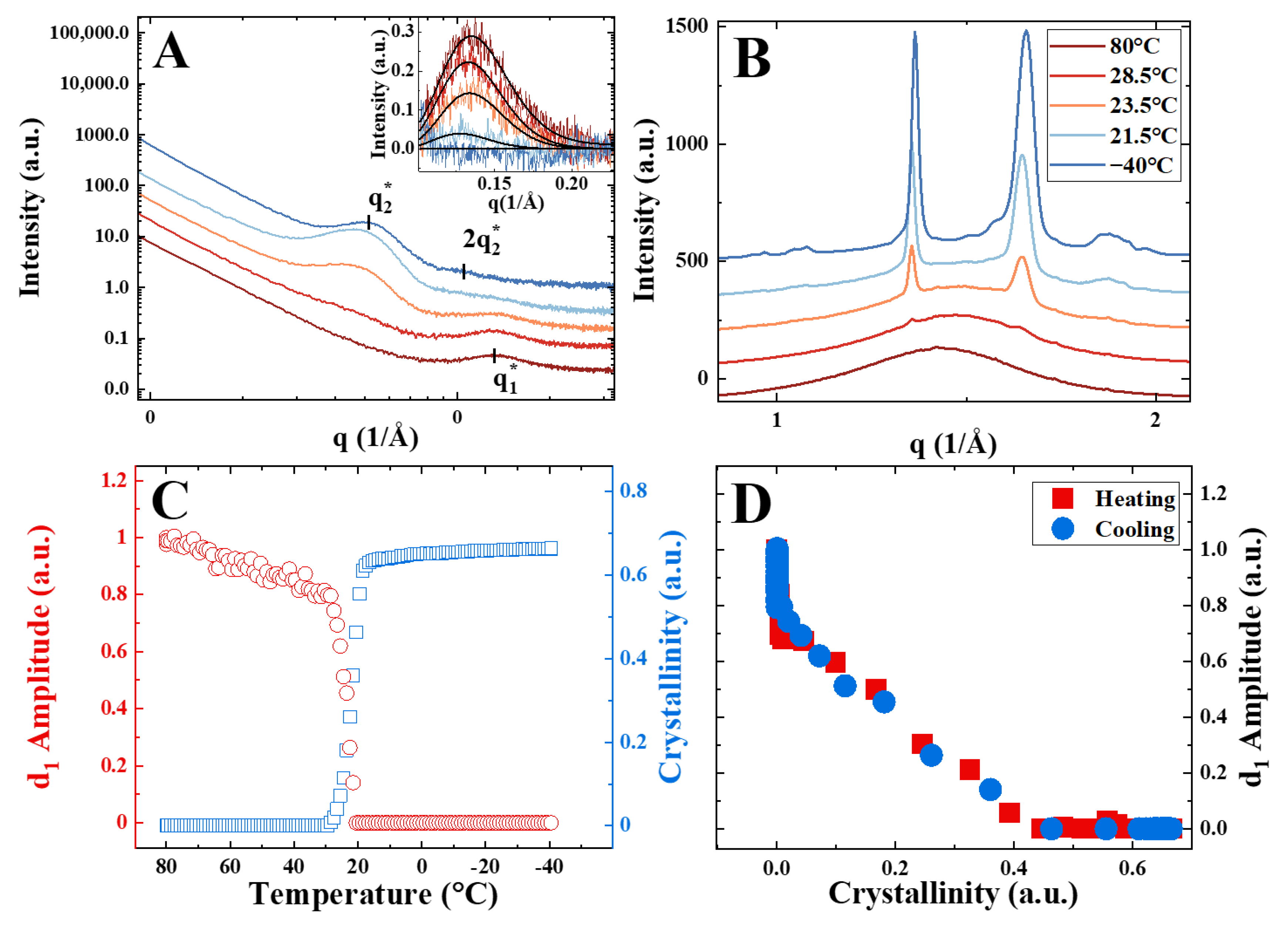
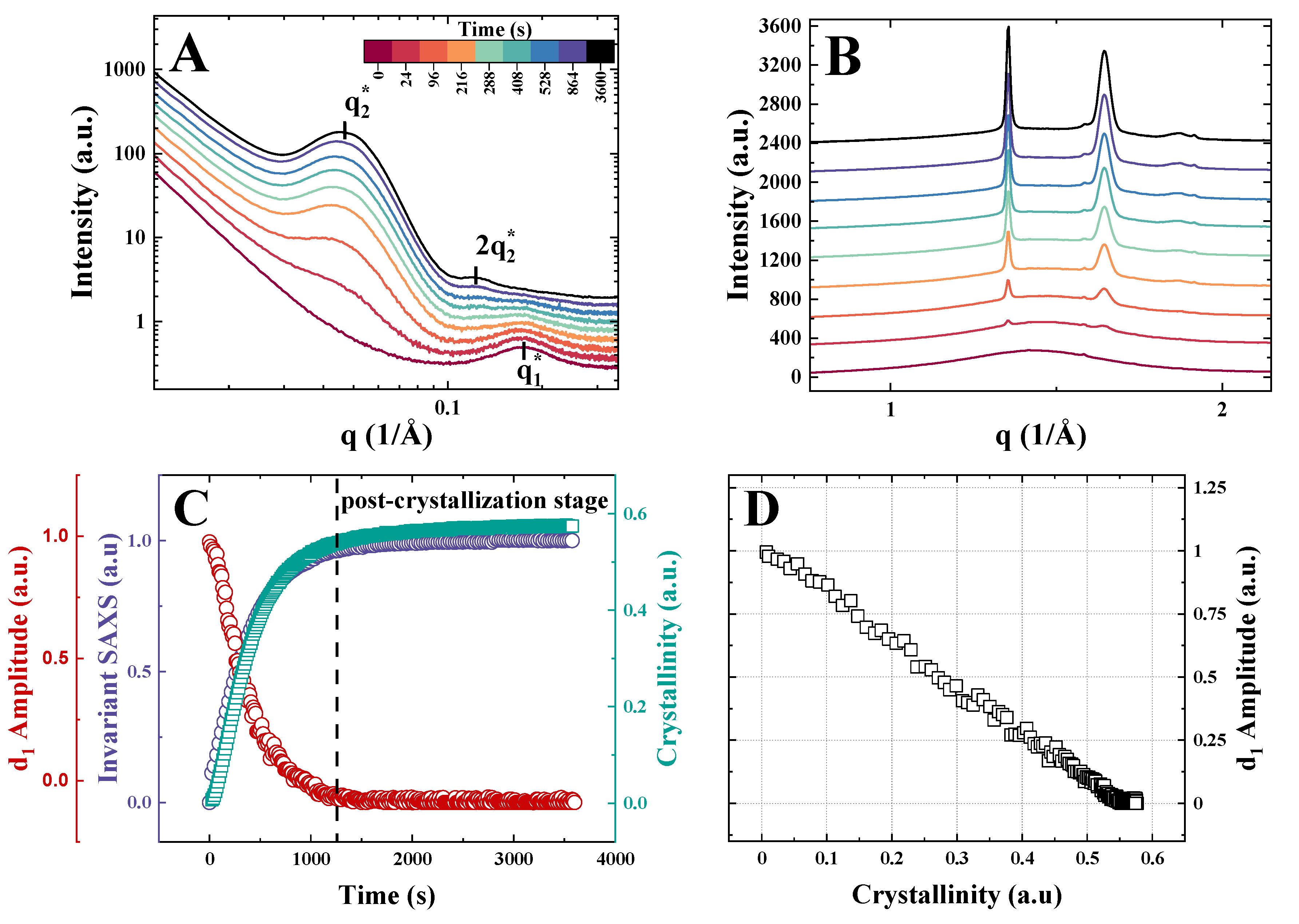
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seguela, R. Critical Review of the Molecular Topology of Semicrystalline Polymers: The Origin and Assessment of Intercrystalline Tie Molecules and Chain Entanglements. J. Polym. Sci. Part B Polym. Phys. 2005, 43, 1729–1748. [Google Scholar] [CrossRef]
- Aharoni, S.M. Increased glass transition temperature in motionally constrained semicrystalline polymers. Polym. Adv. Technol. 1998, 9, 169–201. [Google Scholar] [CrossRef]
- Ania, F.; Martinez-Salazar, J.; Baltá Calleja, F.J. Physical ageing and glass transition in amorphous polymers as revealed by microhardness. J. Mater. Sci. 1989, 24, 2934–2938. [Google Scholar] [CrossRef]
- Jonas, A.M.; Russell, T.P.; Yoon, D.Y. Synchrotron X-ray Scattering Studies of Crystallization of Poly(ether-ether-ketone) from the Glass and Structural Changes during Subsequent Heating-Cooling Processes. Macromolecules 1995, 28, 8491–8503. [Google Scholar] [CrossRef]
- Safandowska, M.; Makarewicz, C.; Rozanski, A.; Idczak, R. Barrier Properties of Semicrystalline Polylactide: The Role of the Density of the Amorphous Regions. Macromolecules 2022, 55, 10077–10089. [Google Scholar] [CrossRef]
- Flory, P.J. Theoretical predictions on the configurations of polymer chains in the amorphous state. J. Macromol. Sci. Phys. 1976, 12, 1–11. [Google Scholar] [CrossRef]
- Ivanov, D.A.; Legras, R.; Jonas, A.M. The crystallization of poly(aryl-ether-ether-ketone) (PEEK). Interdependence between the evolution of amorphous and crystalline regions during isothermal cold-crystallization. Macromolecules 1999, 32, 1582–1592. [Google Scholar] [CrossRef]
- Rymaruk, M.J.; O’Brien, C.T.; György, C.; Darmau, B.; Jennings, J.; Mykhaylyk, O.O.; Armes, S.P. Small-Angle X-ray Scattering Studies of Block Copolymer Nano-Objects: Formation of Ordered Phases in Concentrated Solution during Polymerization-Induced Self-Assembly. Angew. Chem. Int. Ed. 2021, 133, 13065–13073. [Google Scholar] [CrossRef]
- Liberman, L.; Coughlin, M.L.; Weigand, S.; Bates, F.S.; Lodge, T.P. Phase Behavior of Linear-Bottlebrush Block Polymers. Macromolecules 2022, 55, 2821–2831. [Google Scholar] [CrossRef]
- Clair, C.; Lallam, A.; Rosenthal, M.; Sztucki, M.; Vatankhah-Varnosfaderani, M.; Keith, A.N.; Cong, Y.; Liang, H.; Dobrynin, A.V.; Sheiko, S.S.; et al. Strained Bottlebrushes in Super-Soft Physical Networks. ACS Macro Lett. 2019, 8, 530–534. [Google Scholar] [CrossRef]
- Vashahi, F.; Martinez, M.; Cong, Y.; Dashtimoghadam, E.; Fahimpour, F.; Keith, A.N.; Bersenev, E.A.; Ivanov, D.A.; Zhulina, E.B.; Matyjaszewski, K.; et al. Injectable hydrogels with tissue-adaptive gelation and mechanical properties. Sci. Adv. 2022, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bersenev, E.A.; Nikitina, E.A.; Sheiko, S.S.; Ivanov, D.A. Bottlebrush Elastomers with Crystallizable Side Chains: Monitoring Configuration of Polymer Backbones in the Amorphous Regions during Crystallization. ACS Macro Lett. 2022, 11, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, M.; Faust, L.; Wilhelm, M. Comb and Bottlebrush Polymers with Superior Rheological and Mechanical Properties. Adv. Mater. 2019, 31, 1806484. [Google Scholar] [CrossRef] [PubMed]
- Mukumoto, K.; Averick, S.E.; Park, S.; Nese, A.; Mpoukouvalas, A.; Zeng, Y.; Koynov, K.; Leduc, P.R.; Matyjaszewski, K. Phototunable Supersoft Elastomers Using Coumarin Functionalized Molecular Bottlebrushes for Cell-Surface Interactions Study. Macromolecules 2014, 47, 7852–7857. [Google Scholar] [CrossRef]
- Johnson, J.A.; Lu, Y.Y.; Burts, A.O.; Xia, Y.; Durrell, A.C.; Tirrell, D.A.; Grubbs, R.H. Drug-Loaded, Bivalent-Bottle-Brush Polymers by Graft-through ROMP. Macromolecules 2010, 43, 10326–10335. [Google Scholar] [CrossRef]
- Maw, M.; Dashtimoghadam, E.; Keith, A.N.; Morgan, B.J.; Tanas, A.K.; Nikitina, E.A.; Ivanov, D.A.; Vatankhah-Varnosfaderani, M.; Dobrynin, A.V.; Sheiko, S.S. Sticky architecture: Encoding pressure sensitive adhesion in polymer network. ACS Cent. Sci. 2023, 9, 197–205. [Google Scholar] [CrossRef]
- Obhi, N.K.; Jarrett-Wilkins, C.N.; Hicks, G.E.J.; Seferos, D.S. Self-Assembly of Poly(3-Hexylthiophene) Bottlebrush Polymers into End-On-End Linear Fnikiiber Morphologies. Macromolecules 2020, 53, 8592–8599. [Google Scholar] [CrossRef]
- Verduzco, R.; Li, X.; Pesek, S.L.; Stein, G.E. Structure, Function, Self-Assembly, and Applications of Bottlebrush Copolymers. Chem. Soc. Rev. 2015, 44, 2405–2420. [Google Scholar] [CrossRef]
- Runge, M.B.; Bowden, N.B. Synthesis of High Molecular Weight Comb Block Copolymers and Their Assembly into Ordered Morphologies in the Solid State. J. Am. Chem. Soc. 2007, 129, 10551–10560. [Google Scholar] [CrossRef]
- Chremos, A.; Theodorakis, P.E. Morphologies of Bottle-Brush Block Copolymers. ACS Macro Lett. 2014, 3, 1096–1100. [Google Scholar] [CrossRef]
- Zhang, D.; Dashtimoghadam, E.; Fahimipour, F.; Hu, X.; Li, Q.; Bersenev, E.A.; Ivanov, D.A.; Vatankhah-Varnoosfaderani, M.; Sheiko, S.S. Tissue-Adaptive Materials with Independently Regulated Modulus and Transition Temperature. Adv. Mater. 2020, 2005314, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Tadokoro, H. Structural Study of Polyethers, (-(CH2)m-O-)n. Crystal Structure of Poly(ethylene oxide). Macromolecules 1973, 6, 672–675. [Google Scholar] [CrossRef]
- Pielichowski, K.; Kinga, F. Differential scanning calorimetry studies on poly (ethylene glycol) with different molecular weights for thermal energy storage materials. Polym. Adv. Technol. 2002, 13, 690–696. [Google Scholar] [CrossRef]
- Buckley, C.P.; Kovacs, A.J. Melting behaviour of low molecular weight poly (ethylene-oxide) fractions. Colloid Polym. Sci. 1976, 254, 695–715. [Google Scholar] [CrossRef]
- Thelen, J.L.; Chen, X.C.; Inceoglu, S.; Balsara, N.P. Influence of Miscibility on Poly(ethylene oxide) Crystallization from Disordered Melts of Block Copolymers with Lithium and Magnesium Counterions. Macromolecules 2017, 50, 4827–4839. [Google Scholar] [CrossRef]
- Dong, X.-H.; Van Horn, R.; Chen, Z.; Ni, B.; Yu, X.; Wurm, A.; Schick, C.; Lotz, B.; Zhang, W.-B.; Cheng, S.Z.D. Exactly Defined Half-Stemmed Polymer Lamellar Crystals with Precisely Controlled Defects’ Locations. Phys. Chem. Lett. 2013, 4, 2356–2360. [Google Scholar] [CrossRef]
- Pulst, M.; Samiullah, M.H.; Baumeister, U.; Prehm, M.; Balko, J.; Thurn-Albrecht, T.; Busse, K.; Golitsyn, Y.; Reichert, D.; Kressler, J. Crystallization of Poly(ethylene oxide) with a Well-Defined Point Defect in the Middle of the Polymer Chain. Macromolecules 2016, 49, 6609–6620. [Google Scholar] [CrossRef]
- Qi, H.; Liu, X.; Henn, D.M.; Mei, S.; Staub, M.C.; Zhao, B.; Li, C.Y. Breaking Translational Symmetry via Polymer Chain Overcrowding in Molecular Bottlebrush Crystallization. Nat. Commun. 2020, 11, 2152. [Google Scholar] [CrossRef]
- Fritzsching, K.J.; Mao, K.; Schmidt-Rohr, K. Avoidance of Density Anomalies as a Structural Principle for Semicrystalline Polymers: The Importance of Chain Ends and Chain Tilt. Macromolecules 2017, 50, 1521–1540. [Google Scholar] [CrossRef]
- Rosenthal, M.; Burghammer, M.; Bar, G.; Samulski, E.T.; Ivanov, D.A. Switching Chirality of Hybrid Left–Right Crystalline Helicoids Built of Achiral Polymer Chains: When Right to Left Becomes Left to Right. Macromolecules 2014, 47, 8295–8304. [Google Scholar] [CrossRef]
- Zardalidis, G.; Mars, J.; Allgaier, J.; Mezger, M.; Richter, D.; Floudas, G. Influence of chain topology on polymer crystallization: Poly(ethylene oxide) (PEO) rings vs. linear chains. Soft Matter 2016, 12, 8124–8134. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.sigmaaldrich.com/specification-sheets/426/514/447951-BULK_______ALDRICH__.pdf (accessed on 15 January 2024).
- Kieffer, J.; Karkoulis, D. PyFAI, a versatile library for azimuthal regrouping. J. Phys. Conf. Ser. 2013, 20, 425. [Google Scholar] [CrossRef]
- Glatter, O.; Kratky, O. Small Angle X-ray Scattering; Academic Press Inc. Ltd.: London, UK, 1982. [Google Scholar]
- Stribeck, N.; Ruland, W. Determination of the interface distribution function of lamellar two-phase systems. J. Appl. Crystallogr. 1978, 11, 535. [Google Scholar] [CrossRef]
- Ivanov, D.A.; Bar, G.; Dosière, M.; Koch, M.H.J. A Novel View on Crystallization and Melting of Semirigid Chain Polymers: The Case of Poly(trimethylene terephthalate). Macromolecules 2008, 41, 9224. [Google Scholar] [CrossRef]
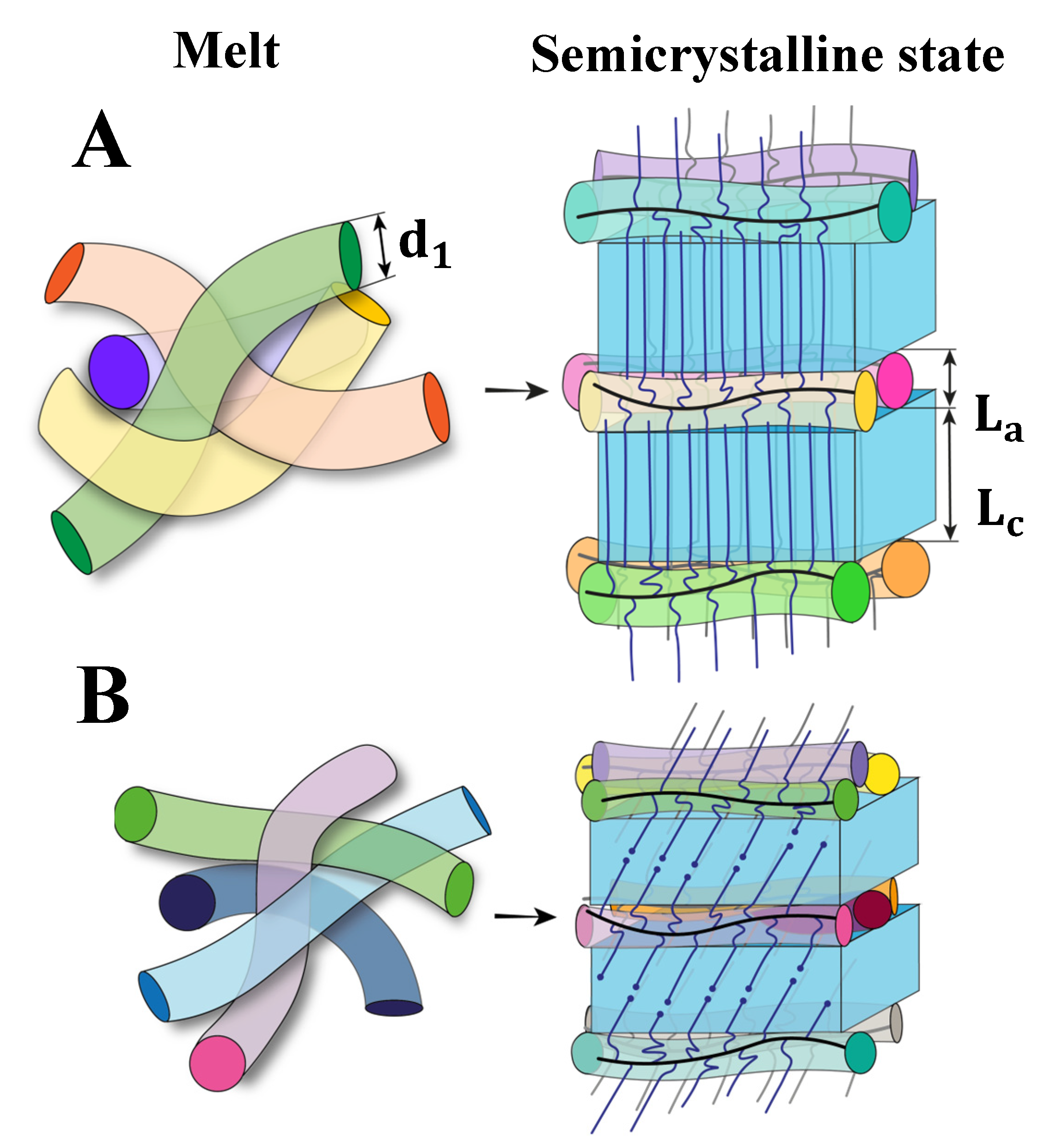

{kind=link}
{kind=link}
{kind=link}
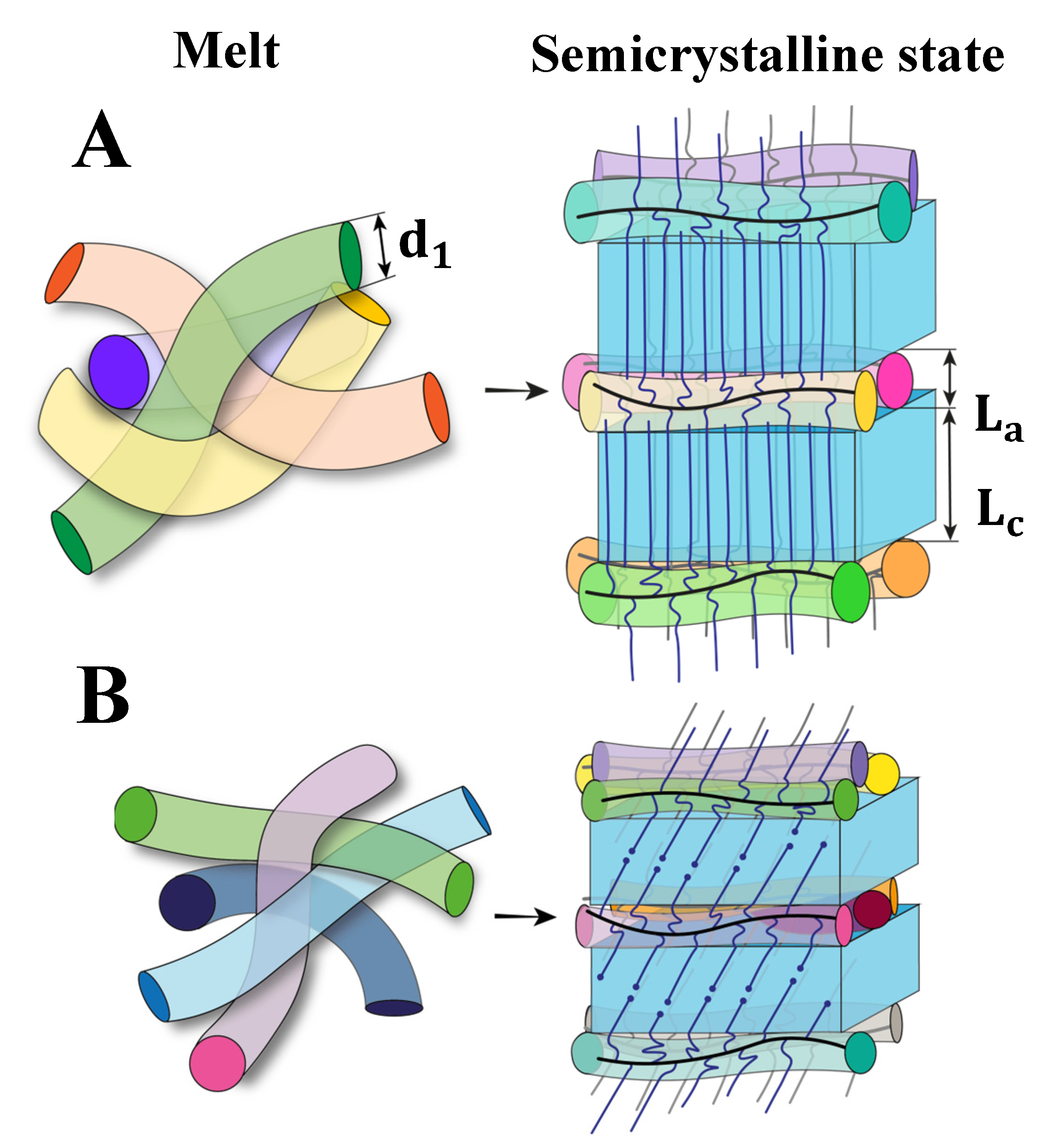{kind=link}
{kind=link}
| Sample Name | nsc 1 | nx 2 | Cross-Linker | ΔHm 3 J/g | Tm 4 °C | Tc 4 °C |
|---|---|---|---|---|---|---|
| PGX_950_200 | 19 | 200 | PEO | 79.6 | 35.2 | 9.3 |
| PGX_950_400 | 19 | 400 | PEO | 81.1 | 35.9 | 8.5 |
| PGX_950_800 | 19 | 800 | PEO | 85.2 | 38.4 | 6.9 |
| PGX_2k_200 | 40 | 200 | PEO | 80.2 | 43.3 | 13.7 |
| PGX_2k_400 | 40 | 400 | PEO | 87.3 | 43.4 | 14.9 |
| PBX_950_150 | 19 | 150 | PBA | 69.1 | 30.1 | −21.8 |
| PBX_950_300 | 19 | 300 | PBA | 72.8 | 34.6 | −20.9 |
| Sample Name | d1 1 nm | Lp 2 nm | La 2 nm | Lc 2 nm | 3 | 4 |
|---|---|---|---|---|---|---|
| PGX_950_200 | 3.6 | 8.0 | 2.6 | 5.4 | 67.5 | 52.6 |
| PGX_950_400 | 3.6 | 8.0 | 2.3 | 5.7 | 71.3 | 53.9 |
| PGX_950_800 | 3.6 | 7.9 | 2.1 | 5.8 | 73.4 | 52.2 |
| PGX_2k_200 | 4.9 | 11.0 | 3.5 | 7.5 | 68.2 | 67.0 |
| PGX_2k_400 | 4.8 | 11.5 | 3.6 | 7.9 | 68.7 | 66.5 |
| PBX_950_150 5 | 3.7 | 8.0 | 2.7 | 5.3 | 66.3 | 56.6 |
| PBX_950_300 5 | 3.7 | 7.2 | 2.3 | 4.8 | 67.6 | 56.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikitina, E.A.; Dashtimoghadam, E.; Sheiko, S.S.; Ivanov, D.A. Bottlebrush Elastomers with Crystallizable Side Chains: Monolayer-like Structure of Backbones Segregated in Intercrystalline Regions. Polymers 2024, 16, 296. https://doi.org/10.3390/polym16020296
Nikitina EA, Dashtimoghadam E, Sheiko SS, Ivanov DA. Bottlebrush Elastomers with Crystallizable Side Chains: Monolayer-like Structure of Backbones Segregated in Intercrystalline Regions. Polymers. 2024; 16(2):296. https://doi.org/10.3390/polym16020296
Chicago/Turabian StyleNikitina, Evgeniia A., Erfan Dashtimoghadam, Sergei S. Sheiko, and Dimitri A. Ivanov. 2024. "Bottlebrush Elastomers with Crystallizable Side Chains: Monolayer-like Structure of Backbones Segregated in Intercrystalline Regions" Polymers 16, no. 2: 296. https://doi.org/10.3390/polym16020296
APA StyleNikitina, E. A., Dashtimoghadam, E., Sheiko, S. S., & Ivanov, D. A. (2024). Bottlebrush Elastomers with Crystallizable Side Chains: Monolayer-like Structure of Backbones Segregated in Intercrystalline Regions. Polymers, 16(2), 296. https://doi.org/10.3390/polym16020296