
Effects of Ultrasonication in Water and Isopropyl Alcohol on High-Crystalline Cellulose: A Fourier Transform Infrared Spectrometry and X-ray Diffraction Investigation




Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
3. Results and Discussion
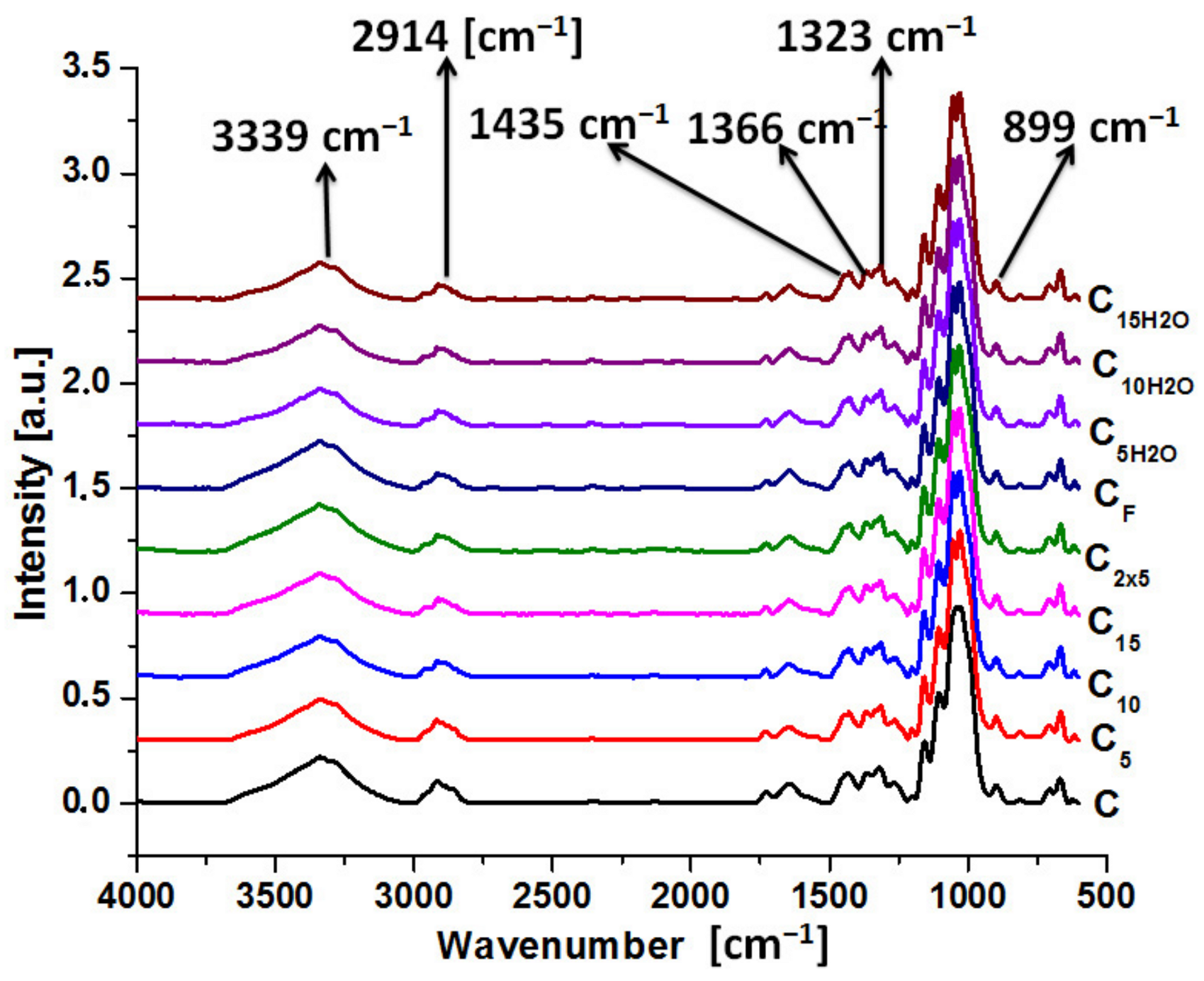
3.1. The Chemical Structure of the Irradiated Samples Established by FTIR Analysis
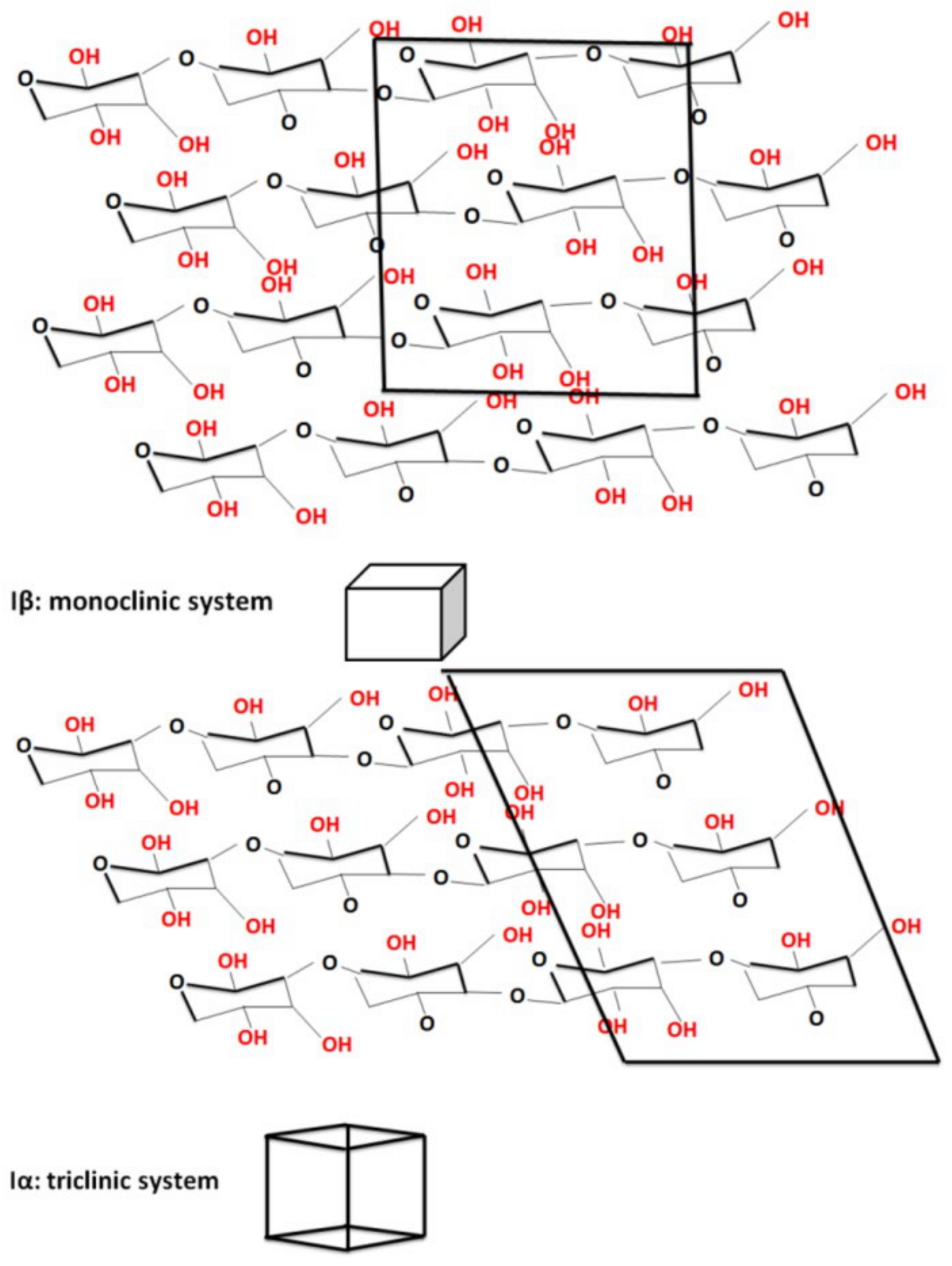
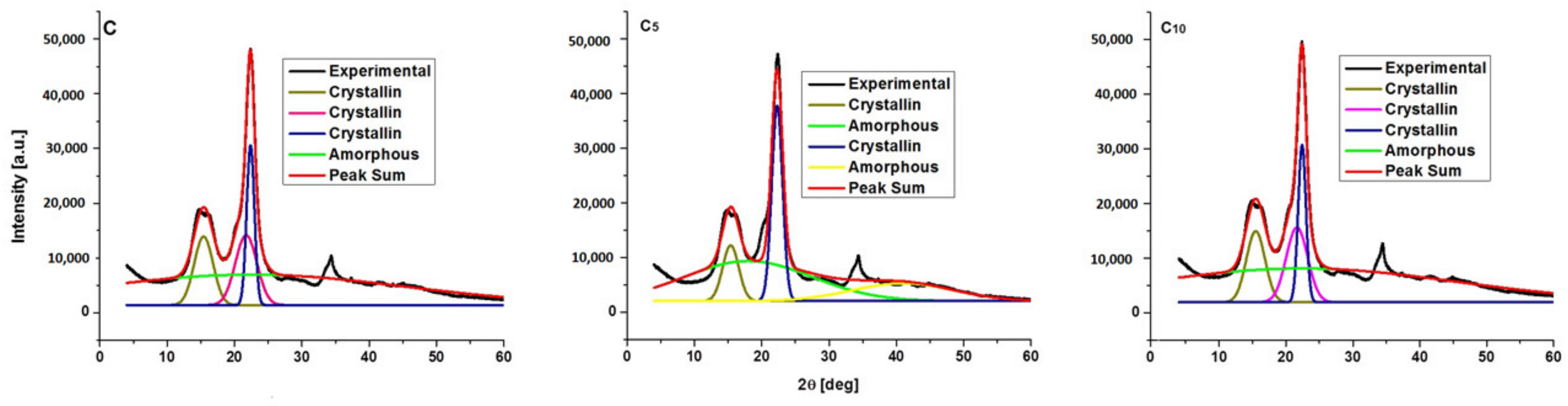
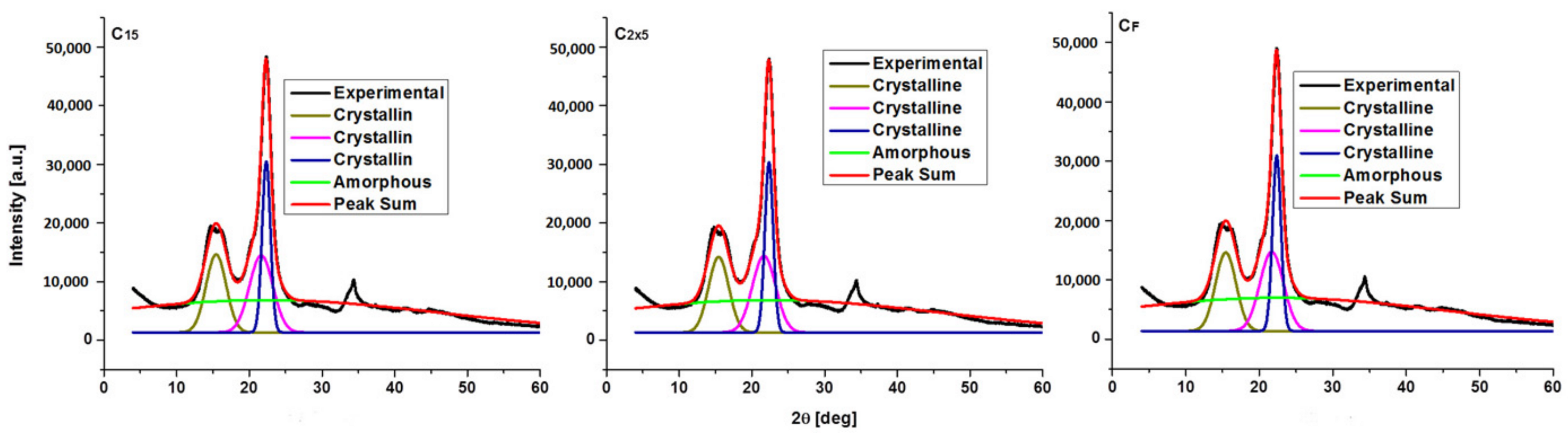
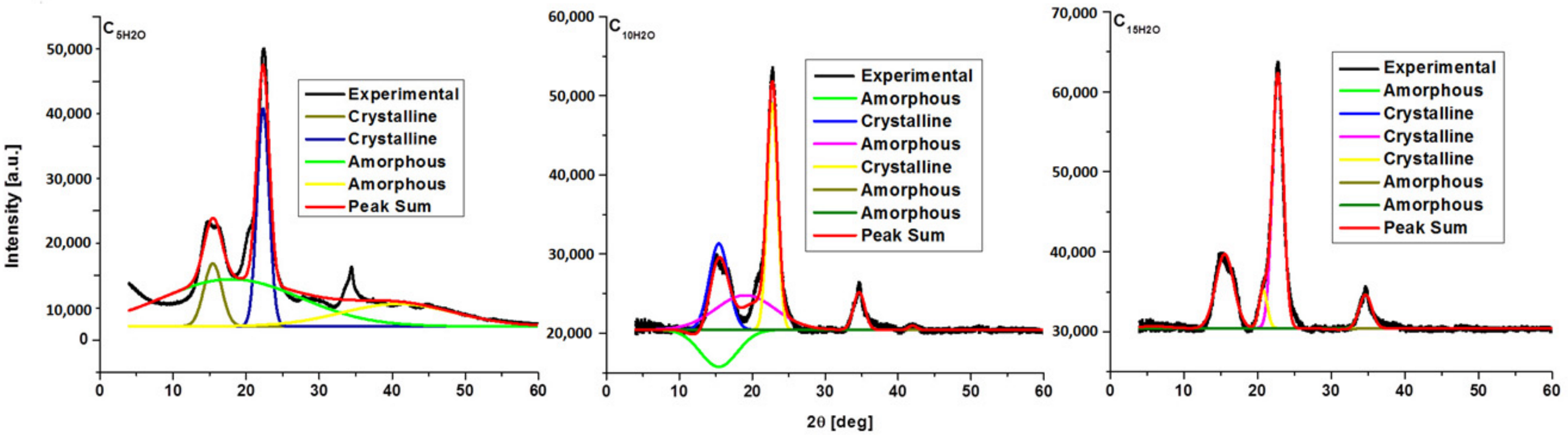
3.2. Crystalline Structure of the Ultrasonicated Sample: X-ray Diffraction Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Ungureanu, E.; Fortună, M.E.; Țopa, D.C.; Lobiuc, A.; Ungureanu, O.C.; Jităreanu, D.C. Design of the functional polymer systems to optimize the filler retention in obtaining cellulosic substrates with improved properties. Materials 2023, 16, 1904. [Google Scholar] [CrossRef]
- Li, Y.; Guan, X.; Chaffey, P.K.; Ruan, Y.; Ma, B.; Shang, S.; Himmel, M.E.; Beckham, G.T.; Longand, H.; Tan, Z. Carbohydrate-binding module O-mannosylation alters binding selectivity to cellulose and lignin. Chem. Sci. 2020, 11, 9262–9271. [Google Scholar] [CrossRef] [PubMed]
- Chowdury, Z.Z.; Hamid, S.B.A. Preparation and Characteriazation of Nanocrystalline Cellulose using ultrasonication combined with a microwave-assisted pretreatment process. Bioresources 2016, 11, 3397–3415. [Google Scholar]
- Rotaru, R.; Savin, M.; Tudorachi, N.; Peptu, C.; Samoila, P.; Sacarescu, L.; Harabagiu, V. Ferromagnetic iron oxide–cellulose nanocomposites prepared by ultrasonication. Polym. Chem. 2018, 9, 860–868. [Google Scholar]
- Li, Y.; Wang, J.; Liu, X.; Zhang, S. Towards a molecular understanding of cellulose dissolution in ionic liquids: Anion/cation effect, synergistic mechanism and physicochemical aspects. Chem. Sci. 2018, 9, 4027–4043. [Google Scholar] [CrossRef]
- Spiridon, I.; Teaca, C.A.; Bodirlau, R. Structural changes evidenced by FTIR Spectroscopy in cellulosic materials after pre-treatment with ionic liquid and enzimatic hydrolysis. Bioresources 2010, 6, 400–413. [Google Scholar] [CrossRef]
- Peng, B.L.; Dhar, N.; Liu, H.L.; Tam, K.C. Chemistry and applications of nanocrystalline cellulose and its derivatives: A nanotechnology perspective. J. Chem. Eng. 2011, 9999, 1–16. [Google Scholar] [CrossRef]
- de Oliveira, H.F.N.; Farès, C.; Rinaldi, R. Beyond a solvent: The roles of 1-butyl-3-methylimidazolium chloride in the acid-catalysis for cellulose depolymerization. Chem. Sci. 2015, 6, 5215–5224. [Google Scholar] [CrossRef]
- Chen, W.; Yu, H.; Liu, Y.; Hai, Y.; Zhang, M.; Chen, P. Isolation and characterization of cellulose nanofibers from four plant cellulose fibers using a chemical-ultrasonic process. Cellulose 2011, 18, 433–442. [Google Scholar] [CrossRef]
- Li, X.F.; Ding, E.Y.; Li, G.K. A method of preparing spherical nano-crystal cellulose with mixed cryslalline forms of cellulose I and II. J. Polym. Sci. 2001, 19, 291–296. [Google Scholar]
- Li, W.; Yue, J.; Liu, S. Preparation of nanocrystalline cellulose via ultrasound and its reinforcement capability for poly(vinyl alcohol) composites. Ultrason. Sonochem. 2012, 19, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Frone, A.N.; Panaitescu, D.M.; Donescu, D.; Spataru, C.I.; Radovici, C.; Trusca, R.; Somoghi, R. Preparation and characterization of pva composites with celluose nanofibers obtained by ultrasonication. Bioresources 2011, 6, 487–512. [Google Scholar] [CrossRef]
- Chen, W.; Yu, H.; Liu, Y.; Chen, P.; Zhang, M.; Hai, Y. Individualization of cellulose nanofibers from wood using high-intensity ultrasonication combined with chemical pretreatments. Carbohydr. Polym. 2011, 83, 1804–1811. [Google Scholar] [CrossRef]
- Parveen, S.; Rana, S.; Ferreira, S.; Filho, A.; Fangueiro, R. Ultrasonic dispersion of micro crystalline cellulose for developing cementitious composites with excellent strength and stiffness. Ind. Crops Prod. 2018, 122, 156–165. [Google Scholar] [CrossRef]
- Girard, M.; Vidal, D.; Bertrand, F.; Tavares, J.R.; Heuzey, M.C. Ultrason. Evidence-based guidelines for the ultrasonic dispersion of cellulose nanocrystals. Ultrason. Sonochem. 2021, 71, 105378. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, A.M.; Robles, E.; Ribeiro, J.S.; Lund, R.G.; Neftali, L.V.C.; Labidi, J. Cellulose nanocrystal membranes as excipients for drug delivery systems. Materials 2016, 9, 1002. [Google Scholar] [CrossRef]
- Wong, S.; Kasapis, S.S.; Huang, D. Molecular weight and crystallinity alteration of cellulose via prolonged ultrasound fragmentation. Food Hydrocoll. 2012, 26, 365–369. [Google Scholar] [CrossRef]
- Sun, B.; Zhang, M.; Shen, J.; He, Z.; Fatehi, P.; Ni, Y. Applications of cellulose-based materials in sustained drug delivery systems. Curr. Med. Chem. 2019, 26, 2485–2501. [Google Scholar] [CrossRef]
- Rotaru, R.; Popescu, C.M.; Dascalu, A.; Timpu, D.; Asandulesa, M.; Fortuna, M.E.; Harabagiu, V. Influence of Ultrasonic Treatment and Heating/Cooling Under Electric Field on High-K Cellulose-Barium Titanate Composites. Rev. Roum. Chim. 2023, 68, 173–183. [Google Scholar] [CrossRef]
- Ciolacu, D.; Chiriac, A.I.; Pastor, F.I.J.; Kokol, V. The influence of supramolecular structure of cellulose allomorphs on the interactions with cellulose-binding domain, CBD3d from Paenibacilus barcinonensis. Bioresour. Technol. 2014, 157, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Poletto, M.; Ornaghi Júnior, H.L.; Zattera, A.J. Native Cellulose: Structure, characterization and thermal properties. Materials 2014, 7, 6105–6119. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, G.C.; Sederholm, C.H.J. Correlation of infrared stretching frequencies and hydrogen bond distances in crystals. Chem. Phys. 1956, 24, 639–641. [Google Scholar] [CrossRef]
- Segal, L.; Creely, J.J.; Martin, A.E., Jr.; Conrad, G.M. An empirical method for estimating the degree of crystallinity of native cellulose using the X-ray diffractometer. CM Tex. Res. J. 1959, 29, 786–794. [Google Scholar] [CrossRef]
- Terinte, N.; Ibbett, R.; Schuster, K.C. Overviewon native cellulose and microcrystalline cellulose i structure studied by X-ray diffraction (WAXD): Comparison between measurement techniques. Lenzinger Berichte 2011, 89, 118–131. [Google Scholar]
- Oh, S.Y.; Yoo, D.I.; Shin, Y.; Kim, H.C.; Kim, H.Y.; Chung, Y.S.; Park, W.H.; Youk, J.H. Crystalline structure analysis of cellulose treated with sodium hydroxide and carbon dioxide by means of X-ray diffraction and FTIR spectroscopy. Carbohydr. Res. 2005, 340, 2376–2391. [Google Scholar] [CrossRef] [PubMed]
- Barzic, A.I.; Soroceanu, M.; Rotaru, R.; Doroftei, F.; Asandulesa, M.; Tugui, C.; Dascalu, I.A.; Harabagiu, V. Cellulose derivative/barium titanate composites with high refractive index, conductivity and energy density. Cellulose 2022, 29, 873–878. [Google Scholar] [CrossRef]
- Markovic, S.; Mitric, M.; Starcevic, G.; Uskokovic, D. Ultrasonic de-agglomeration of barium titanate powder. Ultrason. Sonochem. 2008, 15, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Santos, H.M.; Lodeiro, C.; Capelo-Martinez, J.L. Ultrasound in Chemistry: Analytical Applications, Chapter 1.3.2.3 Dead Zones; Capelo-Martínez, J.-L., Ed.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; ISBN 978-3-527-31934-3. [Google Scholar]
- SriBala, G.; Chennuru, R.; Mahapatra, S.; Vinu, R. Effect of alkaline ultrasonic pretreatment on crystalline morphology and enzymatic hydrolysis of cellulose. Cellulose 2016, 23, 1725–1740. [Google Scholar] [CrossRef]
- Sumari, S.; Roesyadi, A.; Sumarno, S. Effects of ultrasound on the morphology, particle size, crystallinity, and crystallite size of cellulose. Sci. Study Res. Chem. Chem. Eng. 2013, 14, 229–239. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Time [min] | Amplitude [%] | Liquid | Vessel |
|---|---|---|---|---|
| C | - | - | - | - |
| C5 | 5 | 50 | C3H7OH | Berzelius |
| C10 | 10 | 50 | C3H7OH | Berzelius |
| C15 | 15 | 50 | C3H7OH | Berzelius |
| C2x5 | 5 | 100 | C3H7OH | Berzelius |
| CF | 5 | 50 | C3H7OH | Round-bottomed flask |
| C5H2O | 5 | 50 | H2O | Berzelius |
| C10H2O | 10 | 50 | H2O | Berzelius |
| C15H2O | 15 | 50 | H2O | Berzelius |
| WC [cm−1] | Type of Vibration | Wsamples [cm−1] | Sample | ΔW [cm−1] |
|---|---|---|---|---|
| 3339 | hydroxyl groups | 3336 | C5 | 3 |
| 3341 | CF, C5H2O | −2 | ||
| 3352 | C10H2O | −13 | ||
| 3435 | C15H2O | −96 | ||
| 2914 | νCH | 2918 | C5 | −4 |
| 2916 | C10 | −2 | ||
| 2962 | C10 | −48 | ||
| 2897 | C10 | 17 | ||
| 2962 | C15 | −48 | ||
| 2851 | C15 | 63 | ||
| 2916 | C2x5 | −2 | ||
| 2955 | C2x5 | −41 | ||
| 2899 | C2x5 | 15 | ||
| 2962 | CF | −48 | ||
| 2901 | CF | −13 | ||
| 2962 | C5H2O | −48 | ||
| 2897 | C5H2O | 17 | ||
| 2849 | C5H2O | 65 | ||
| 2903 | C10H2O | 11 | ||
| 2959 | C15H2O | −45 | ||
| 2905 | C15H2O | 9 | ||
| 1435 | δCH2 | 1429 | C5, C10, C15, C2x5, C5H2O | 6 |
| 1427 | CF | 8 | ||
| 1433 | C10H2O | 2 | ||
| 1437 | C15H2O | −2 | ||
| 1366 | δCH, νCOO | 1367 | C5, C10, C15, C2x5, CF, C5H2O | −1 |
| 1371 | C10H2O | −5 | ||
| 1375 | C15H2O | −9 | ||
| 1323 | CH2 | 1315 | C5, C10, C15, C2x5, CF, C5H2O | 8 |
| 1319 | C10H2O, C15H2O | 4 | ||
| 1157 | νCO, δOH | 1159 | C5, C10, C15, C2x5, CF, C5H2O, C10H2O, C15H2O | |
| 1055, 1030 | νCO | 1059, 1032 | C10H2O | −4, −2 |
| 1059, 1038 | C15H2O | −4, −8 | ||
| 899 | δCH2 | 895 | C10H2O, C15H2O | 4 |
| Sample | LOI | TCI | HBI |
|---|---|---|---|
| C | 1.68 | 1.39 | 1.29 |
| C5 | 1.22 | 1.45 | 1.19 |
| C10 | 1.45 | 1.72 | 1.40 |
| C15 | 1.50 | 1.89 | 1.39 |
| C2x5 | 1.46 | 1.75 | 1.37 |
| CF | 1.64 | 1.93 | 1.36 |
| C5H2O | 1.48 | 1.94 | 1.23 |
| C10H2O | 2.05 | 0.76 | 2.12 |
| C15H2O | 1.29 | 0.56 | 2.92 |
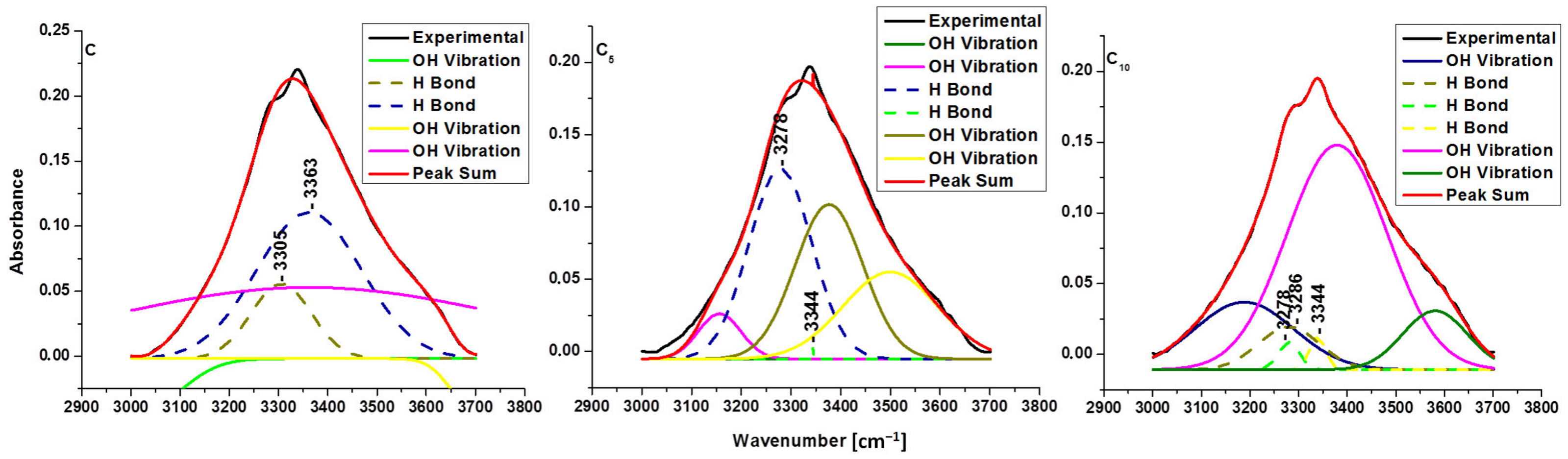
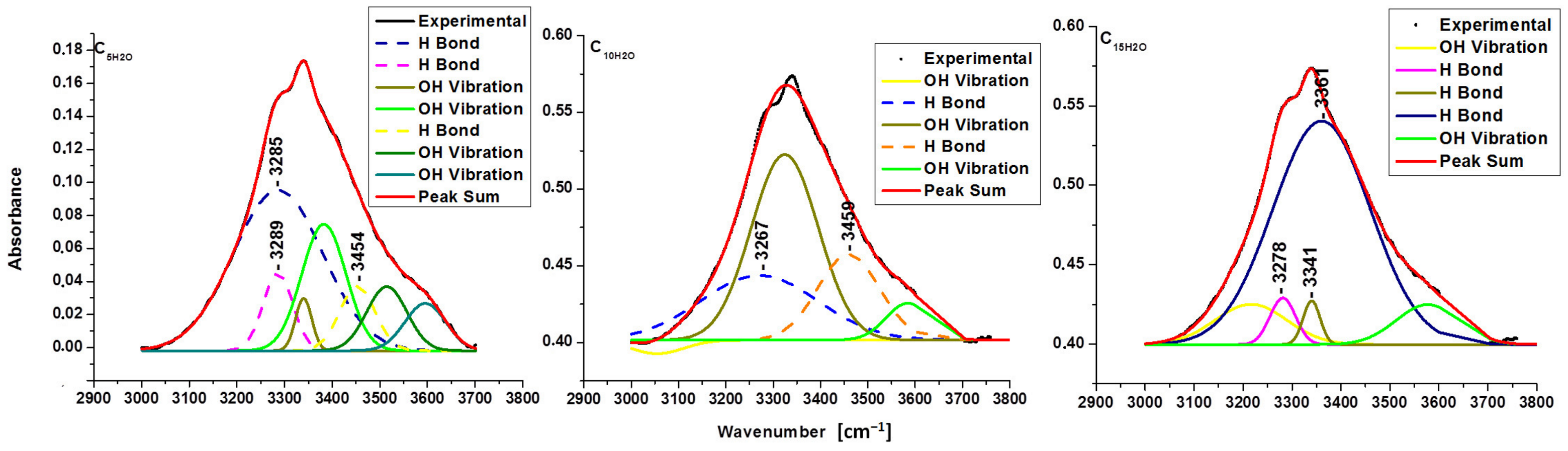
| Sample | Type of Hydrogen Bond | EH [kJ] | R [Å] |
|---|---|---|---|
| C | intermolecular O(6)H-O(3) | E3305: 23.43 | R3305: 2.773 |
| intramolecular O(3)H-O(5) | E3363: 18.49 | R3363: 2.786 | |
| C5 | intermolecular O(6)H-O(3) | E3278: 25.78 | R3278: 2.767 |
| intramolecular O(3)H-O(5) | E3344: 20.09 | R3344: 2.782 | |
| C10 | intermolecular O(6)H-O(3) | E3278: 25.78 | R3278: 2.767 |
| intermolecular O(6)H-O(3) | E3286: 25.08 | R3286: 2.769 | |
| intramolecular O(3)H-O(5) | E3344: 20.09 | R3344: 2.782 | |
| C15 | intermolecular O(6)H-O(3) | E3251: 28.17 | R3251: 2.761 |
| intermolecular O(6)H-O(3) | E3286: 25.08 | R3286: 2.769 | |
| C2x5 | intermolecular O(6)H-O(3) | E3234: 29.70 | R3234: 2.757 |
| intermolecular O(6)H-O(3) | E3276: 25.96 | R3276: 2.766 | |
| CF | intermolecular O(6)H-O(3) | E3269: 26.57 | R3269: 2.765 |
| intermolecular O(6)H-O(3) | E3280: 25.60 | R3280: 2.767 | |
| intramolecular O(3)H-O(5) | E3342: 20.26 | R3342: 2.781 | |
| C5H2O | intermolecular O(6)H-O(3) | E3285: 25.17 | R3285: 2.768 |
| intermolecular O(6)H-O(3) | E3289: 24.82 | R3289: 2.769 | |
| intramolecular O(2)H-O(6) | E3454: 11.09 | R3454: 2.807 | |
| C10H2O | intermolecular O(6)H-O(3) | E3267: 26.75 | R3267: 2.764 |
| intramolecular O(2)H-O(6) | E3459: 10.70 | R3459: 2.808 | |
| C15H2O | intermolecular O(6)H-O(3) | E3278: 25.78 | R3278: 2.798 |
| intramolecular O(3)H-O(5) | E3341: 20.34 | R3341: 2.781 | |
| intramolecular O(3)H-O(5) | E3361: 18.66 | R3361: 2.786 |
| Sample | JCPDS Card No | Space Group | Crystal System | Lattice Parameters [Å] | Lattice Volume [Å] | Crystallite Size [Å] | ||
|---|---|---|---|---|---|---|---|---|
| a | b | c | ||||||
| C | 4114994 | 4:P1121 | Monoclinic (C-unique) | 7.82 | 8.03 | 10.35 | 647.9 | 41 |
| C5 | 4114383 | 1:P1 | Triclinic | 10.45 | 6.56 | 6.03 | 331.6 | 49 |
| C10 | 4114383 | 1:P1 | Triclinic | 10.43 | 6.46 | 5.93 | 332.2 | 46 |
| C15 | 4114383 | 1:P1 | Triclinic | 10.26 | 6.49 | 5.85 | 324.6 | 56 |
| C2x5 | 4114383 | 1:P1 | Triclinic | 10.40 | 6.61 | 5.91 | 317.4 | 27 |
| CF | 4114383 | 1:P1 | Triclinic | 10.32 | 6.53 | 5.97 | 342.1 | 63 |
| C5H2O | 4114994 | 4:P1121 | Monoclinic (C-unique) | 8.02 | 8.37 | 10.40 | 695.3 | 39 |
| C10H2O | 4114994 | 4:P1121 | Monoclinic (C-unique) | 7.96 | 8.25 | 10.38 | 680.6 | 54 |
| C15H2O | 4114994 | 4:P1121 | Monoclinic (C-unique) | 7.98 | 8.15 | 10.43 | 676.3 | 36 |
| Samples | CrI [%] |
|---|---|
| C | 67.6 |
| C5 | 74.2 |
| C10 | 74.6 |
| C15 | 77.8 |
| C2x5 | 77.4 |
| CF | 77.6 |
| C5H2O | 66.9 |
| C10H2O | 51.6 |
| C15H2O | 50.4 |
| Sample | Lenghts of Fiber [μm] | Treatment | Time of Treatment [min] | CI [%] before Treatment | CI [%] after Treatment | References |
|---|---|---|---|---|---|---|
| cellulose nano-crystals | 0.105 | ultrasound assisted acid hydrolysis | 35 | 55.3–63.8-72.4 | 88.3 | [3] |
| cellulose microfibers | 1 | ultrasonication in isopropyl alchool | 5–15 | 67.6 | 72.9–77.6 | [this work] |
| cellulose microfibers | 1 | ultrasonication in Milli-Q water | 5–15 | 67.6 | 50–4–66.9 | [this work] |
| cellulose microfibers | 20 | ultrasonication in Milli-Q water | 15 | 42.5 | 42.4 | [4] |
| cellulose microfibers with anorganic nanoparticle | 20 | ultrasonication in Milli-Q water | 15 | 42.5 | 9.6 | [4] |
| cellulose microfibers | 20 | ultrasonication in sodium hydroxide solution (1–10%) | 240 | 77 | 73.8 | [29] |
| cellulose microfibers | 50 | ultrasonication in sodium hydroxide solution | 240 | 79.8 | 68.2 | [29] |
| cellulose microfibers | 100 | ultrasonication in sodium hydroxide solution | 240 | 78.8 | 67.7 | [29] |
| cellulose microfibers | 180 | ultrasonication in sodium hydroxide solution | 240 | 76.5 | 71.7 | [29] |
| cellulose microfibers | 180 | ultrasonication in sodium hydroxide solution | 120 | 76.5 | 71.65 | [29] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rotaru, R.; Fortună, M.E.; Ungureanu, E.; Brezuleanu, C.O. Effects of Ultrasonication in Water and Isopropyl Alcohol on High-Crystalline Cellulose: A Fourier Transform Infrared Spectrometry and X-ray Diffraction Investigation. Polymers 2024, 16, 2363. https://doi.org/10.3390/polym16162363
Rotaru R, Fortună ME, Ungureanu E, Brezuleanu CO. Effects of Ultrasonication in Water and Isopropyl Alcohol on High-Crystalline Cellulose: A Fourier Transform Infrared Spectrometry and X-ray Diffraction Investigation. Polymers. 2024; 16(16):2363. https://doi.org/10.3390/polym16162363
Chicago/Turabian StyleRotaru, Răzvan, Maria E. Fortună, Elena Ungureanu, and Carmen O. Brezuleanu. 2024. "Effects of Ultrasonication in Water and Isopropyl Alcohol on High-Crystalline Cellulose: A Fourier Transform Infrared Spectrometry and X-ray Diffraction Investigation" Polymers 16, no. 16: 2363. https://doi.org/10.3390/polym16162363
APA StyleRotaru, R., Fortună, M. E., Ungureanu, E., & Brezuleanu, C. O. (2024). Effects of Ultrasonication in Water and Isopropyl Alcohol on High-Crystalline Cellulose: A Fourier Transform Infrared Spectrometry and X-ray Diffraction Investigation. Polymers, 16(16), 2363. https://doi.org/10.3390/polym16162363