
Stability Studies of Amorphous Ibrutinib Prepared Using the Quench-Cooling Method and Its Dispersions with Soluplus®


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Amorphous Ibrutinib and Formulation with Soluplus
2.3. Physical Stability Studies
2.4. Differential Scanning Calorimetry (DSC)
2.5. Thermogravimetric Analysis (TGA)
2.6. Powder X-ray Diffraction Analysis (XRPD)
2.7. Fourier Transform Infrared Spectroscopy (FTIR)
2.8. Scanning Electron Microscopy (SEM)
3. Results and Discussion
3.1. Thermal Analysis
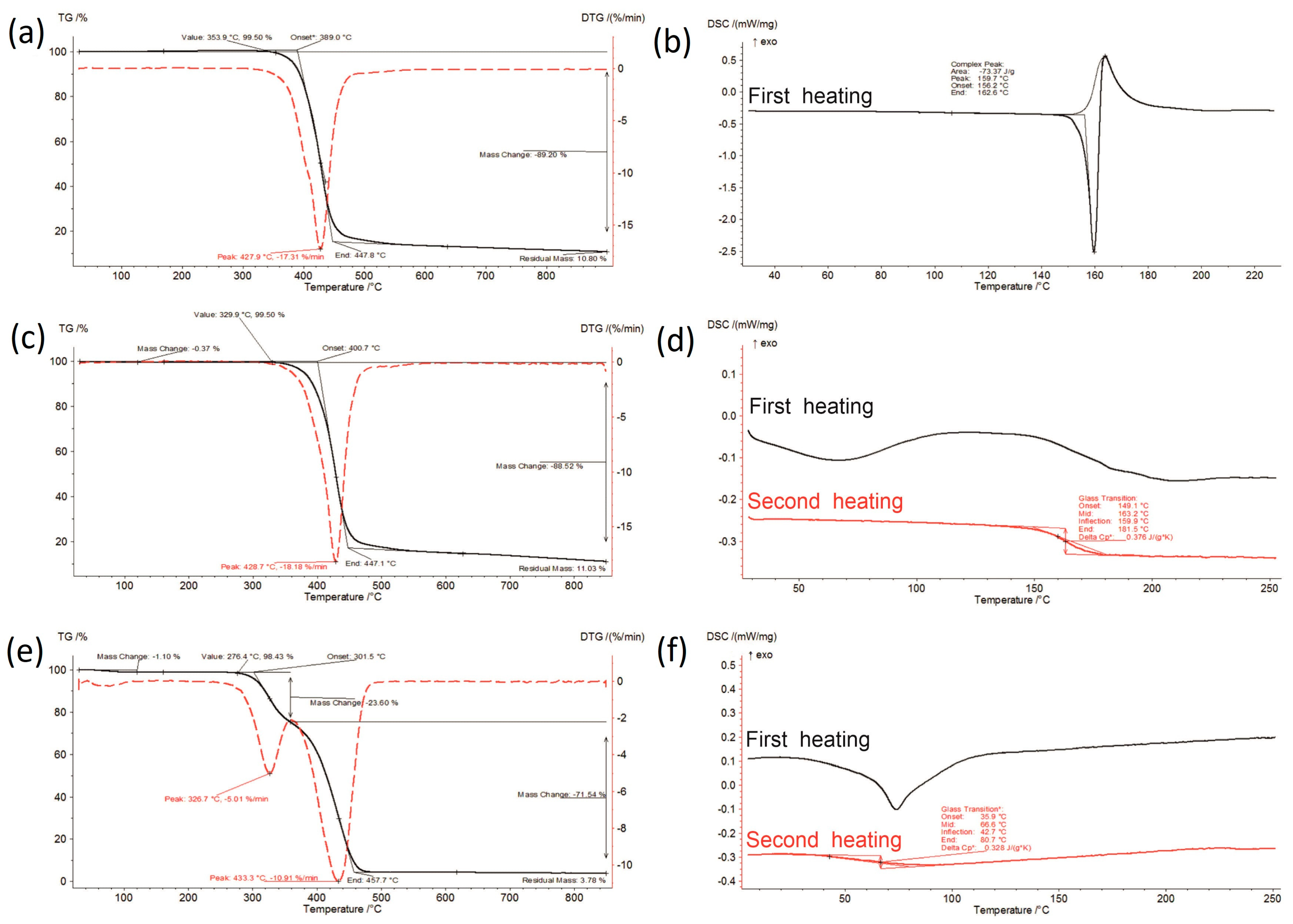


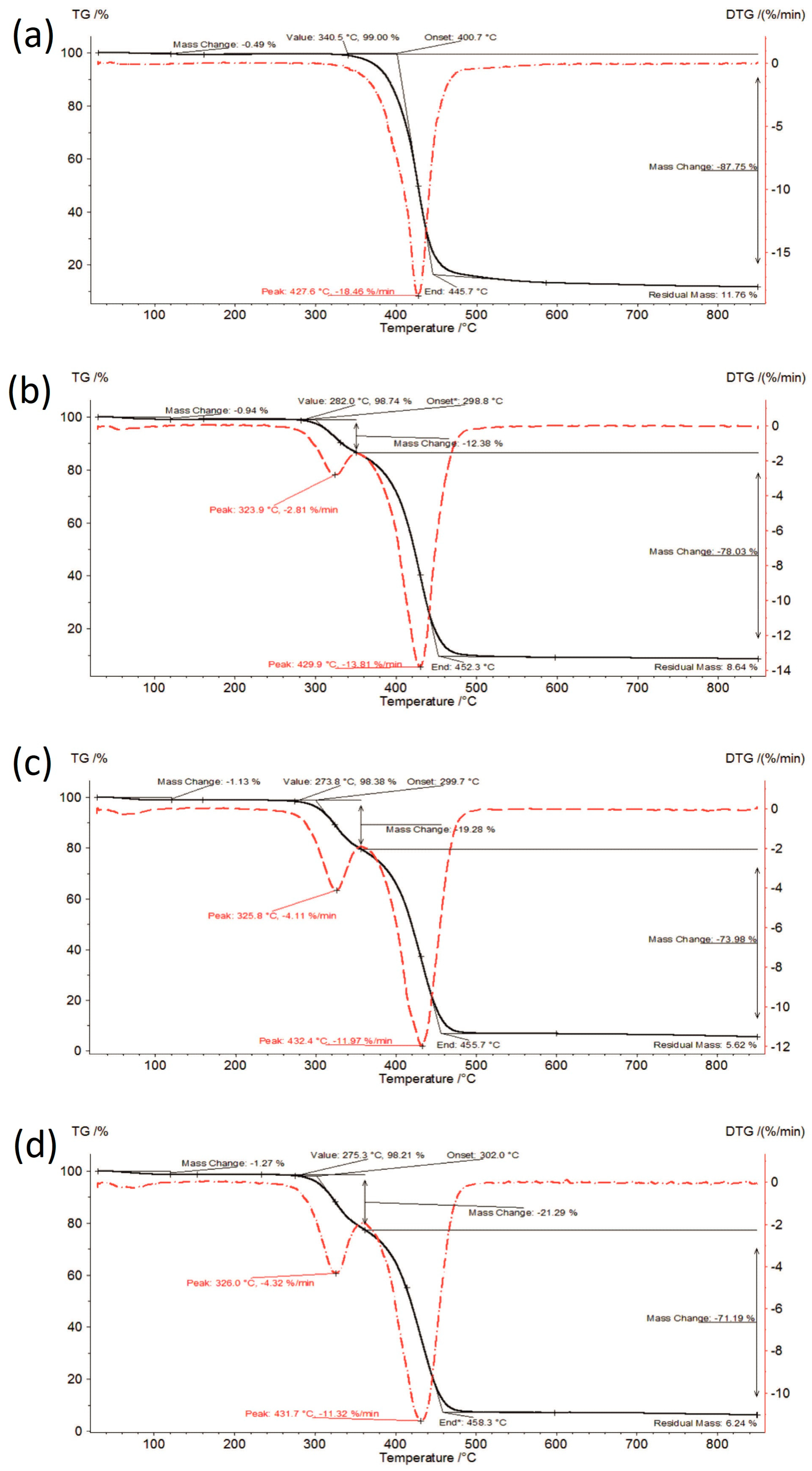

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Moisture Content [%] | I Stage of Decomposition | II Stage of Decomposition | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Temp. Range [°C] | Mass Change [%] | Tm [°C] | Rate of Mass Loss [% min−1] | Temp. Range [°C] | Mass Change [%] | Tm [°C] | Rate of Mass Loss [% min−1] | ||
| IBR raw | 0.00 | 353.9–447.8 | 89.20 | 427.9 | 17.31 | - | - | - | - |
| IBR_0 | 0.37 | 329.9–447.1 | 88.52 | 428.7 | 18.18 | - | - | - | - |
| IBR_acc | 0.41 | 345.0–446.9 | 88.27 | 428.3 | 17.97 | - | - | - | - |
| IBR_long | 0.49 | 340.5–445.7 | 87.75 | 427.6 | 18.46 | - | - | - | - |
| IBR:SOL 1:1_0 | 0.66 | 292.6–353.2 | 11.47 | 324.2 | 2.55 | 353.2–451.7 | 79.78 | 430.3 | 14.55 |
| IBR:SOL 1:1_acc | 0.97 | 289.2–348.4 | 12.19 | 325.7 | 2.81 | 348.4–451.4 | 79.48 | 429.8 | 13.57 |
| IBR:SOL 1:1_long | 0.94 | 282.0–350.2 | 12.38 | 323.9 | 2.81 | 350.2–452.3 | 78.03 | 429.9 | 13.81 |
| IBR:SOL 3:7_0 | 0.92 | 284.3–356.0 | 16.90 | 325.8 | 3.65 | 356.0–454.5 | 76.53 | 430.8 | 12.79 |
| IBR:SOL 3:7_acc | 0.88 | 284.1–356.2 | 18.43 | 326.2 | 4.05 | 356.2–455.7 | 74.91 | 431.4 | 12.33 |
| IBR:SOL 3:7_long | 1.13 | 273.8–356.1 | 19.28 | 325.8 | 4.11 | 356.1–455.7 | 73.98 | 432.4 | 11.97 |
| IBR:SOL 1:9_0 | 1.13 | 269.6–357.9 | 22.68 | 326.3 | 4.75 | 357.9–454.4 | 73.67 | 432.8 | 11.29 |
| IBR:SOL 1:9_acc | 0.93 | 279.2–361.5 | 22.16 | 326.5 | 4.63 | 361.5–458.0 | 71.65 | 434.9 | 11.59 |
| IBR:SOL 1:9_long | 1.27 | 275.3–361.7 | 21.29 | 326.0 | 4.32 | 361.7–458.3 | 71.19 | 431.7 | 11.32 |
| SOL_0 | 1.10 | 276.4–358.5 | 23.60 | 326.7 | 5.01 | 358.5–457.7 | 71.54 | 433.3 | 10.91 |
| SOL_acc | 1.21 | 278.2–360.0 | 23.70 | 327.2 | 5.08 | 360.0–458.1 | 70.81 | 435.4 | 10.89 |
| SOL_long | 1.37 | 271.7–360.2 | 24.02 | 326.3 | 5.03 | 360.2–457.5 | 69.47 | 433.5 | 10.58 |
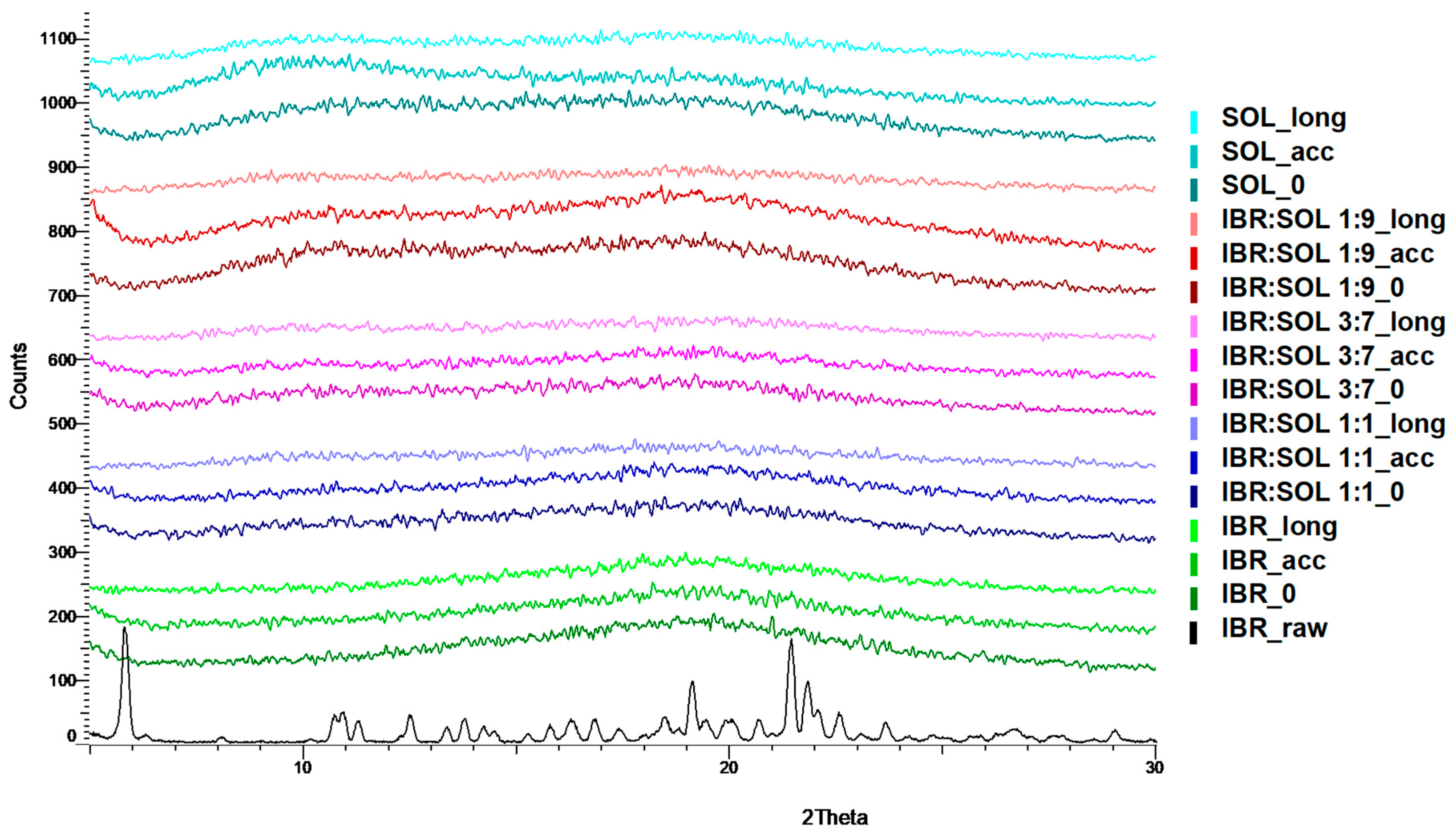
3.2. Powder X-ray Diffraction Analysis (XRPD)
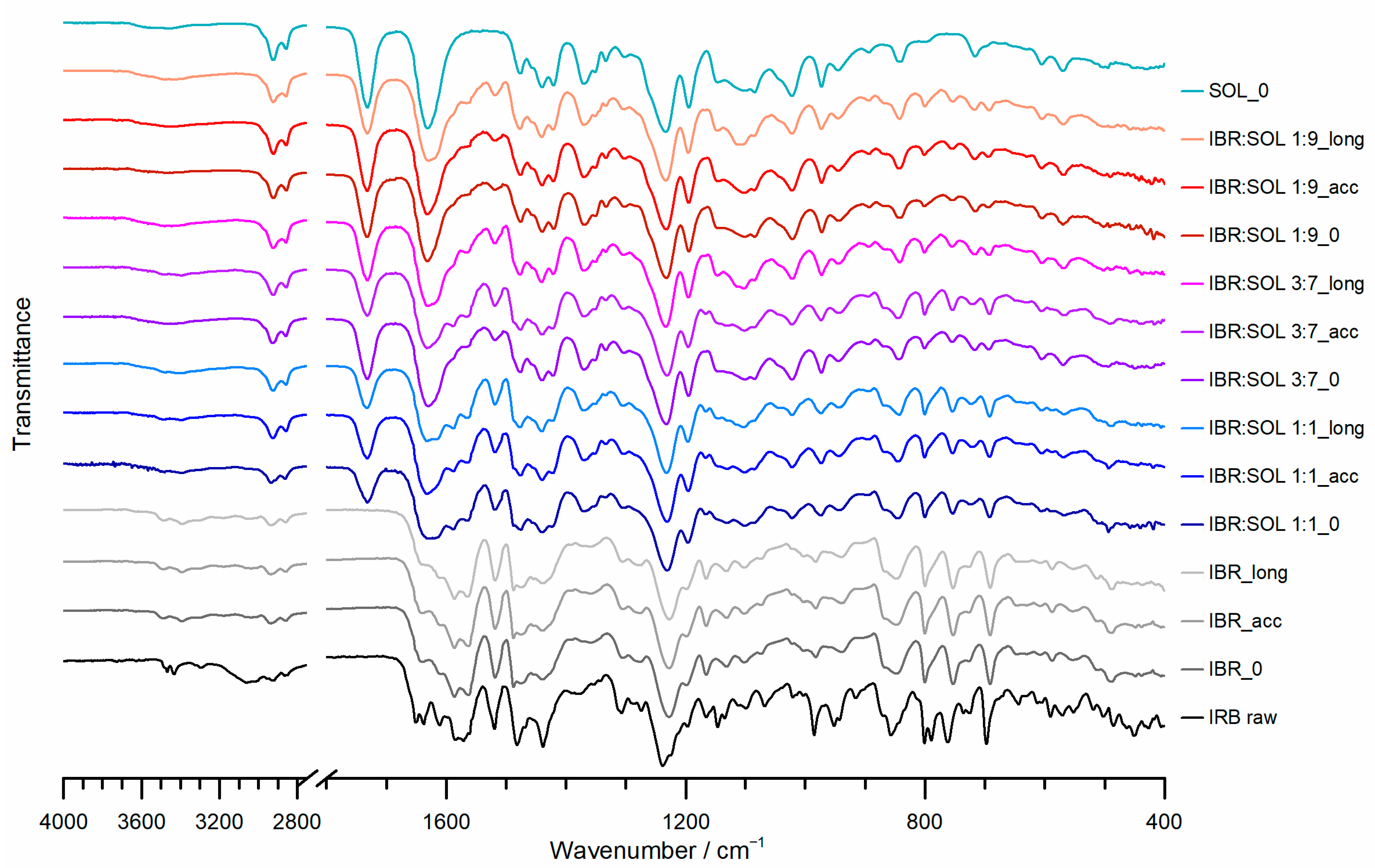
3.3. Fourier Transform Infrared Spectroscopy (FTIR)
3.4. Scanning Electron Microscopy (SEM)
3.5. Physical Stability Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Yu, L. Amorphous pharmaceutical solids: Preparation, characterization and stabilization. Adv. Drug Deliv. Rev. 2001, 48, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Karmwar, P.; Graeser, K.; Gordon, K.C.; Strachan, C.J.; Rades, T. Investigation of properties and recrystallisation behaviour of amorphous indomethacin samples prepared by different methods. Int. J. Pharm. 2011, 417, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Savolainen, M.; Heinz, A.; Strachan, C.; Gordon, K.C.; Yliruusi, J.; Rades, T.; Sandler, N. Screening for Differences in the Amorphous State of Indomethacin Using Multivariate Visualization. Eur. J. Pharm. Sci. 2007, 30, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Graeser, K.A.; Strachan, C.J.; Patterson, J.E.; Gordon, K.C.; Rades, T. Physicochemical Properties and Stability of Two Differently Prepared Amorphous Forms of Simvastatin. Cryst. Growth Des. 2008, 8, 128–135. [Google Scholar] [CrossRef]
- Feng, T.; Pinal, R.; Carvajal, M.T. Process induced disorder in crystalline materials: Differentiating defective crystals from the amorphous form of griseofulvin. J. Pharm. Sci. 2008, 97, 3207–3221. [Google Scholar] [CrossRef] [PubMed]
- Wojnarowska, Z.; Grzybowska, K.; Adrjanowicz, K.; Kaminski, K.; Paluch, M.; Hawelek, L.; Wrzalik, R.; Dulski, M.; Sawicki, W.; Mazgalski, J.; et al. Study of the Amorphous Glibenclamide Drug: Analysis of the Molecular Dynamics of Quenched and Cryomilled Material. Mol. Pharm. 2010, 7, 1692–1707. [Google Scholar] [CrossRef] [PubMed]
- Baghel, S.; Cathcart, H.; Redington, W.; O’Reilly, N.J. An investigation into the crystallization tendency/kinetics of amorphous active pharmaceutical ingredients: A case study with dipyridamole and cinnarizine. Eur. J. Pharm. Biopharm. 2016, 104, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Trasi, N.S.; Byrn, S.R. Mechanically induced amorphization of drugs: A study of the thermal behavior of cryomilled compounds. AAPS PharmSciTech 2012, 13, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Chikhalia, V.; Forbes, R.T.; Storey, R.A.; Ticehurst, M. The effect of crystal morphology and mill type on milling induced crystal disorder. Eur. J. Pharm. Sci. 2006, 27, 19–26. [Google Scholar] [CrossRef]
- Simões, M.F.; Nogueira, B.A.; Tabanez, A.M.; Fausto, R.; Pinto, R.M.A.; Simões, S. Enhanced solid-state stability of amorphous ibrutinib formulations prepared by hot-melt extrusion. Int. J. Pharm. 2020, 579, 119156. [Google Scholar] [CrossRef]
- Karolewicz, B.; Górniak, A.; Marciniak, D.M.; Mucha, I. Molecular mobility and stability studies of amorphous imatinib mesylate. Pharmaceutics 2019, 11, 304. [Google Scholar] [CrossRef] [PubMed]
- Mucha, I.; Baranowski, P.; Owczarek, A.; Gajda, M.; Pluta, J.; Górniak, A.; Karolewicz, B. Thermal stability and decompositions kinetics under non-isothermal conditions of imatinib mesylate α form. J. Pharm. Biomed. Anal. 2016, 129, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Schmitt, E.A.; Law, D.; Brackemeyer, P.J.; Zhang, G.G.Z. Assessing Physical Stability Risk Using the Amorphous Classification System (ACS) Based on Simple Thermal Analysis. Mol. Pharm. 2019, 16, 2742–2754. [Google Scholar] [CrossRef] [PubMed]
- Vora, C.; Patadia, R.; Mittal, K.; Mashru, R. Preparation and characterization of dipyridamole solid dispersions for stabilization of supersaturation: Effect of precipitation inhibitors type and molecular weight. Pharm. Dev. Technol. 2016, 21, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Caron, V.; Hu, Y.; Tajber, L.; Erxleben, A.; Corrigan, O.I.; McArdle, P.; Healy, A.M. Amorphous solid dispersions of sulfonamide/Soluplus® and sulfonamide/PVP prepared by ball milling. AAPS PharmSciTech 2013, 14, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Lin, H.L.; Chi, Y.T.; Huang, Y.T.; Kao, C.Y.; Hsieh, W.H. Thermoanalytical and Fourier transform infrared spectral curve-fitting techniques used to investigate the amorphous indomethacin formation and its physical stability in Indomethacin-Soluplus® solid dispersions. Int. J. Pharm. 2015, 496, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.Q.; Lai, H.W.; Zhang, Y.; Feng, B.; Xiao, X.; Zhang, H.M.; Li, Z.Q.; Qi, X.R. On the inherent properties of Soluplus and its application in ibuprofen solid dispersions generated by microwave-quench cooling technology. Pharm. Dev. Technol. 2018, 23, 573–586. [Google Scholar] [CrossRef]
- Chen, Z.; Zhai, J.; Liu, X.; Mao, S.; Zhang, L.; Rohani, S.; Lu, J. Solubility measurement and correlation of the form A of ibrutynib in organic solvents from 278.15 to 323.15 K. J. Chem. Thermodyn. 2016, 103, 342–348. [Google Scholar] [CrossRef]
- Zvoníček, V.; Skořepová, E.; Dušek, M.; Babor, M.; Žvátora, P.; Šoóš, M. First crystal structures of pharmaceutical ibrutinib: Systematic solvate screening and characterization. Cryst. Growth Des. 2017, 17, 3116–3127. [Google Scholar] [CrossRef]
- Surana, R.; Pyne, A.; Suryanarayanan, R. Effect of Preparation Method on Physical Properties of Amorphous Trehalose. Pharm. Res. 2004, 21, 1167–1176. [Google Scholar] [CrossRef]
- Graeser, K.A.; Patterson, J.E.; Zeitler, J.A.; Gordon, K.C.; Rades, T. Correlating Thermodynamic and Kinetic Parameters with Amorphous Stability. Eur. J. Pharm. Sci. 2009, 37, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, X.; Zhu, Y.; Yin, T.; Gou, J.; Wang, Y.; He, H.; Zhang, Y.; Tang, X. Characterization of nimodipine amorphous nanopowder prepared by quenching cooling combined with wet milling and spray drying. Int. J. Pharm. 2022, 628, 122332. [Google Scholar] [CrossRef]
- Zi, P.; Zhang, C.; Ju, C.; Su, Z.; Bao, Y.; Gao, J.; Sun, J.; Lu, J.; Zhang, C. Solubility and bioavailability enhancement study of lopinavir solid dispersion matrixed with a polymeric surfactant-Soluplus. Eur. J. Pharm. Sci. 2019, 134, 233–245. [Google Scholar] [CrossRef]
- Linn, M.; Collnot, E.M.; Djuric, D.; Hempel, K.; Fabian, E.; Kolter, K.; Lehr, C.M. Soluplus® as an effective absorption enhancer of poorly soluble drugs in vitro and in vivo. Eur. J. Pharm. Sci. 2012, 45, 336–433. [Google Scholar] [CrossRef]
- Punčochová, K.; Vukosavljevic, B.; Hanuš, J.; Beránek, J.; Windbergs, M.; Štěpánek, F. Non-invasive insight into the release mechanisms of a poorly soluble drug from amorphous solid dispersions by confocal Raman microscopy. Eur. J. Pharm. Biopharm. 2016, 101, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Xu, T.; Huang, W.; Fan, B.; Sheng, X. Stability and bioavailability enhancement of telmisartan ternary solid dispersions: The synergistic effect of polymers and drug-polymer(s) interactions. AAPS PharmSciTech 2019, 20, 143. [Google Scholar] [CrossRef] [PubMed]
- Purro, N.; Smyth, M.S.; Goldman, E.; Wirth, D.D. Crystalline Forms of a Bruton’s Tyrosine Kinase Inhibitor. U.S. Patent 2017/0100404 A1, 13 April 2017. [Google Scholar]
- Shi, X.; Fan, B.; Zhou, X.; Chen, Q.; Shen, S.; Xing, X.; Deng, Y. Preparation and characterization of Ibrutinib amorphous solid dispersions: A discussion of interaction force. J. Pharm. Innov. 2022, 17, 1074–1083. [Google Scholar] [CrossRef]
- Zvoníček, V.; Skořepová, E.; Dušek, M.; Žvátora, P.; Šoóš, M. Ibrutinib polymorphs: Crystallographic study. Cryst. Growth Des. 2018, 18, 1315–1326. [Google Scholar] [CrossRef]
- Patnaik, S.; Chunduri, L.A.; Akilesh, M.S.; Bhagavatham, S.S.; Kamisetti, V. Enhanced dissolution characteristics of piroxicam–Soluplus® nanosuspensions. J. Exp. Nanosci. 2016, 11, 916–929. [Google Scholar] [CrossRef]
- Shi, X.; Song, S.; Ding, Z.; Fan, B.; Xu, T.; Huang, W. Improving the Solubility and Dissolution of Ibrutinib by Preparing Solvates. J. Pharm. Innov. 2020, 15, 569–580. [Google Scholar] [CrossRef]
- Shamma, R.N.; Basha, M. Soluplus®: A novel polymeric solubilizer for optimization of Carvedilol solid dispersions: Formulation design and effect of method of preparation. Powder Technol. 2013, 237, 406–414. [Google Scholar] [CrossRef]
- Ohtake, S.; Shalaev, E. Effect of water on the chemical stability of amorphous pharmaceuticals: I. Small molecules. J. Pharm. Sci. 2013, 102, 1139–1154. [Google Scholar] [CrossRef] [PubMed]
- Aso, Y.; Yoshioka, S.; Kojima, S. Relationship Between Water Mobility, Measured as Nuclear Magnetic Relaxation Time, and the Crystallization Rate of Amorphous Nifedipine in the Presence of Some Pharmaceutical Excipients. Chem. Pharm. Bull. 1996, 44, 1065–1067. [Google Scholar] [CrossRef]
- Kilburn, D.; Townrow, S.; Meunier, V.; Richardson, R.; Alam, A.; Ubbink, J. Organization and Mobility of Water in Amorphous and Crystalline Trehalose. Nat. Mater. 2006, 5, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Heljo, V.P.; Nordberg, A.; Tenho, M.; Virtanen, T.; Jouppila, K.; Salonen, J.; Maunu, S.L.; Juppo, A.M. The effect of water plasticization on the molecular mobility and crystallization tendency of amorphous disaccharides. Pharm. Res. 2012, 29, 2684–2697. [Google Scholar] [CrossRef] [PubMed]
- Makower, B.; Dye, W.B. Sugar Crystallization, Equilibrium Moisture Content and Crystallization of Amorphous Sucrose and Glucose. J. Agric. Food Chem. 1956, 4, 72–77. [Google Scholar] [CrossRef]
- Kedward, C.J.; MacNaughtan, W.; Mitchell, J.R. Crystallization Kinetics of Amorphous Lactose as a Function of Moisture Content Using Isothermal Differential Scanning Calorimetry. J. Food Sci. 2000, 65, 324–328. [Google Scholar] [CrossRef]
- Konno, H.; Taylor, L.S. Ability of different polymers to inhibit the crystallization of amorphous felodipine in the presence of moisture. Pharm. Res. 2008, 25, 969–978. [Google Scholar] [CrossRef]
- Morrow, E.A.; Terban, M.W.; Thomas, L.C.; Gray, D.L.; Bowman, M.J.; Billinge, S.L.; Schmidt, S.J. Effect of amorphization method on the physicochemical properties of amorphous sucrose. J. Food Eng. 2018, 243, 125–141. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mucha, I.; Karolewicz, B.; Górniak, A. Stability Studies of Amorphous Ibrutinib Prepared Using the Quench-Cooling Method and Its Dispersions with Soluplus®. Polymers 2024, 16, 1961. https://doi.org/10.3390/polym16141961
Mucha I, Karolewicz B, Górniak A. Stability Studies of Amorphous Ibrutinib Prepared Using the Quench-Cooling Method and Its Dispersions with Soluplus®. Polymers. 2024; 16(14):1961. https://doi.org/10.3390/polym16141961
Chicago/Turabian StyleMucha, Igor, Bożena Karolewicz, and Agata Górniak. 2024. "Stability Studies of Amorphous Ibrutinib Prepared Using the Quench-Cooling Method and Its Dispersions with Soluplus®" Polymers 16, no. 14: 1961. https://doi.org/10.3390/polym16141961
APA StyleMucha, I., Karolewicz, B., & Górniak, A. (2024). Stability Studies of Amorphous Ibrutinib Prepared Using the Quench-Cooling Method and Its Dispersions with Soluplus®. Polymers, 16(14), 1961. https://doi.org/10.3390/polym16141961