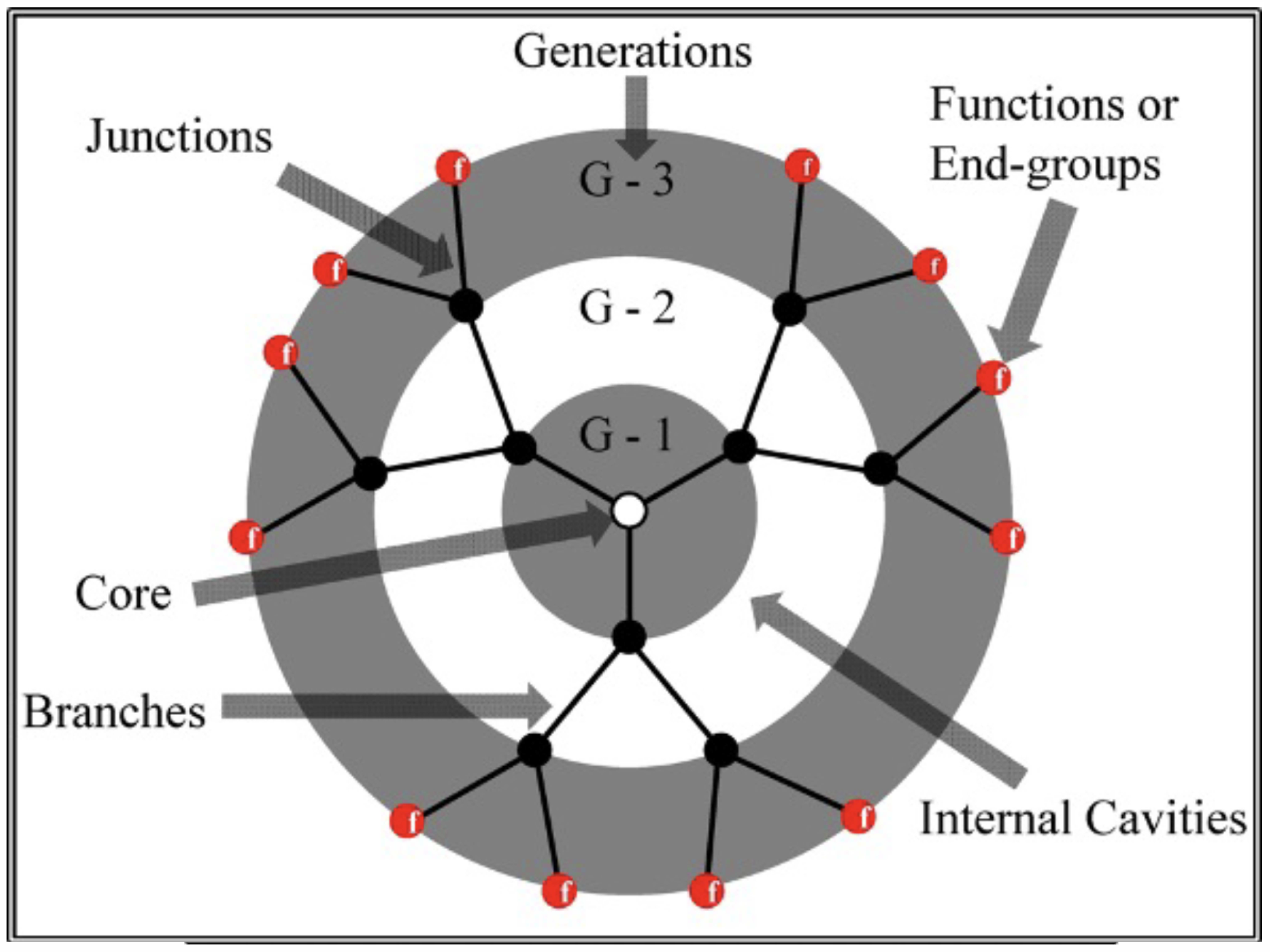
When investigating the dynamics of dendrimers using the Langevin equation, various directly measurable parameters offer useful insights into the behavior and properties of these intricate macromolecules. The comprehension of the dendrimer’s behavior in many applications, including medication delivery [
39], materials science [
40], and nanotechnology [
41], necessitates a thorough grasp of their fundamental structural and dynamical features [
42]. The measurement of intramolecular distances pertains to the determination of the spatial separation between particular spots or groups inside the dendrimer structure, hence providing insights into the local structural characteristics [
43]. Conformational flexibility refers to the dendrimer’s capacity to assume many conformations, which in turn influences its interactions with other molecules. Dynamical properties encompass various characteristics of dendrimers, one of which is the diffusion coefficient (
D). This coefficient serves as a measure of the speed at which dendrimers traverse a solvent, providing valuable insights into their mobility. The intrinsic viscosity, denoted as
, is a parameter that quantifies the resistance of a dendrimer to flow within a solvent. It provides insight into the dendrimer’s size and form characteristics. The orientational relaxation time is a measure of the duration required for dendrimers to undergo reorientation within a solvent. The concept of fractal dimensions is employed to characterize the self-similar or fractal properties exhibited by dendrimer formations. The relaxation moduli
and
characterize the elastic and viscous behaviors shown by dendrimers when subjected to external forces over varying temporal regimes.
3.1. Rheological Nature of Model Dendrimers
Rheological research is utilized to explore the influence of macromolecules on the mechanical response of materials under external stresses. The mechanical and flow properties of materials, particularly in relation to dendrimers, provide significant insights into their impact on many systems. Rheological studies in dendrimers encompass various significant elements and applications, including viscosity modification, viscoelasticity, and flow behavior analysis. The eigenvalues and normalized eigenvectors of matrices play a significant role in the computation of dynamic features of dendrimers, as discussed previously. Intrinsic viscosity is dictated by the eigenvalues, whereas the translational diffusion coefficient is affected by both the eigenvalues and the normalized eigenvectors of the matrix product of the hydrodynamic interaction matrix and the connection matrix, which is denoted as
. In contrast to star or linear polymers, dendrimers demonstrate distinctive characteristics with regard to their intrinsic viscosity. Rather than exhibiting a continuous increase throughout generations, the inherent viscosity of dendrimers achieves its maximum value at a specific generation, which is referred to as the peak or maximum. This behavior has also been documented in empirical research [
7,
16,
44] where they provided a qualitative explanation for the observed behavior of dendrimers. The primary factor contributing to this behavior is from the relationship between the mass or molecular weight of dendrimers and their production.
The mass or molecular weight of dendrimers exhibits an exponential increase as their creation progresses. Nevertheless, the pace at which their volume increases is not proportional; rather, it adheres to a cubic power-law pattern. This implies that until a specific generation is reached, the rate at which the volume of the dendrimer grows exceeds the rate at which its molecular weight increases. As a result, there is a positive correlation between the growth of the dendrimer and the intrinsic viscosity , indicating that as the generation of the dendrimer grows, so does its intrinsic viscosity. After reaching the maximum, there is a further reduction in the inherent viscosity values . The precise generation at which this maximum is observed is contingent upon the dendrimer’s topology, which refers to its specific structural arrangement. The manifestation of this behavior may vary between various generations of dendrimers owing to their distinct shapes and sizes. As previously stated, a typical trend in the behavior of intrinsic viscosity has been identified for various dendrimer models.
Mansfield and Klushin [
45] studied the hydrodynamic radii of poly(amido amide) (PAMAM) starburst dendrimers using intrinsic viscosity measurements. It compares experimental PAMAM radii with theoretical models, particularly the Lescanec–Muthukumar configurational model [
46]. The study discovers that hydrodynamic radii rapidly grow with increasing generation number, indicating a structure with stretched tiers and a hollow core. Lescanec–Muthukumar’s [
46] simulations indicate a more folded structure with higher density toward the core. Using Zimm and Fixman’s intrinsic viscosity formulas [
47], computer simulations were utilized to calculate the hydrodynamic radii (
), radius of gyration (
), and radius
. The study found that
increases faster with each generation than
or
, indicating considerable densification as generations increase. The computed radii of Lescanec–Muthukumar [
46] starbursts with stiff spacers were consistent with experimental PAMAM radii. So, the primary takeaway is that measuring the hydrodynamic radius using inherent viscosity might be misleading if taken too literally. Mourey et al. [
44] studied the intrinsic viscosity of polyether dendrimers utilizing size exclusion chromatography (SEC) and molecular weight-sensitive detection. They identified dendrimers from the 0–4th generation with narrow chromatogram distributions and negligible widening. With increasing generation, the intrinsic viscosity reached a maximum, whilst the refractive index increased to a minimum. Hydrodynamic radii grew almost linearly with generation. These findings coincided with the Lescanec–Muthukumar model [
46], showing a decreasing density profile from the center outward. The study revealed polyether dendrimer’s distinct behavior when compared to linear polymers, demonstrating a clear maximum in intrinsic viscosity and predictable variations in density distribution during generation. Ganazzoli et al. [
36] used self-consistent free-energy minimization to study the structural characteristics and intrinsic viscosity of dendrimers under excluded-volume conditions. This approach gives information about both local conformation and overall molecular dynamics. The study derived the radius of gyration (
) and viscometric radius (
) from intrinsic viscosity. The results revealed that intramolecular swelling is localized in the core with dendrons expanding outward and little inter-dendron mixing. Universal variables for characteristic radii resulted in a master curve for any generation or solvent quality. Molecular expansion is most noticeable in the core, and intrinsic viscosity rose progressively with generation number without reaching a maximum under ideal solvent conditions. The predicted radii and inherent viscosities were consistent with experimental and simulation data, demonstrating the model’s correctness. Drew and Adolf [
48] studied the intrinsic viscosity of trifunctional dendrimers by equilibrium molecular dynamics simulations in an explicit Lennard–Jones solvent. They calculated the intrinsic viscosity of dendrimers from generations 3–7, revealing a peak at generation 5. This result is consistent with the experimental evidence and earlier stochastic simulations. For this, two methods were used: the standard Green–Kubo approach and a direct calculation method, both of which produced consistent findings. In contrast, the Flory–Fox approximation [
49] exhibited a declining tendency. The investigation confirmed that intrinsic viscosity peaks at generation five before falling, as opposed to linear polymers, which show a monotonic increase. The findings highlight the importance of structure and density distribution in determining intrinsic viscosity. Monte Carlo simulations were used to determine the intrinsic viscosity of polyamidoamine (PAMAM), polypropylene-imine (PPI), and polybenzylether (PBzE) dendrimers [
50]. The study’s goal was to reproduce intrinsic viscosity over generations and better understand experimental differences. The results revealed a maximum inherent viscosity as a function of generation number, which is governed by friction radius and structural features. The models for PAMAM, PPI, and PBzE were consistent with experimental results for radius of gyration and inherent viscosity. PAMAM-EDA’s intrinsic viscosity in water peaked at generation 5. The study discovered that the frictional radius influences both the intrinsic viscosity value and the location of its maximum, stressing the need to precisely model hydrodynamic interactions. The simulation method for determining dendrimer intrinsic viscosity has been improved [
51]. The work used Monte Carlo simulations and the Fixman variational technique to produce numerous major results. First, adding a correction term for individual friction beads increased the accuracy of intrinsic viscosity estimations for PPI dendrimers in water. Furthermore, the use of realistic angle distributions improved the consistency between simulation findings and experimental data for PBzE monodendrons and tridendrons in THF. The distance distribution generated from MD simulations with explicit solvent was a better fit to experimental data for both the radius of gyration and intrinsic viscosity of PBzE dendrimers. The improved model effectively recreated the experimental intrinsic viscosity maximum for tridendrons, demonstrating the value of rich structural information in simulations.
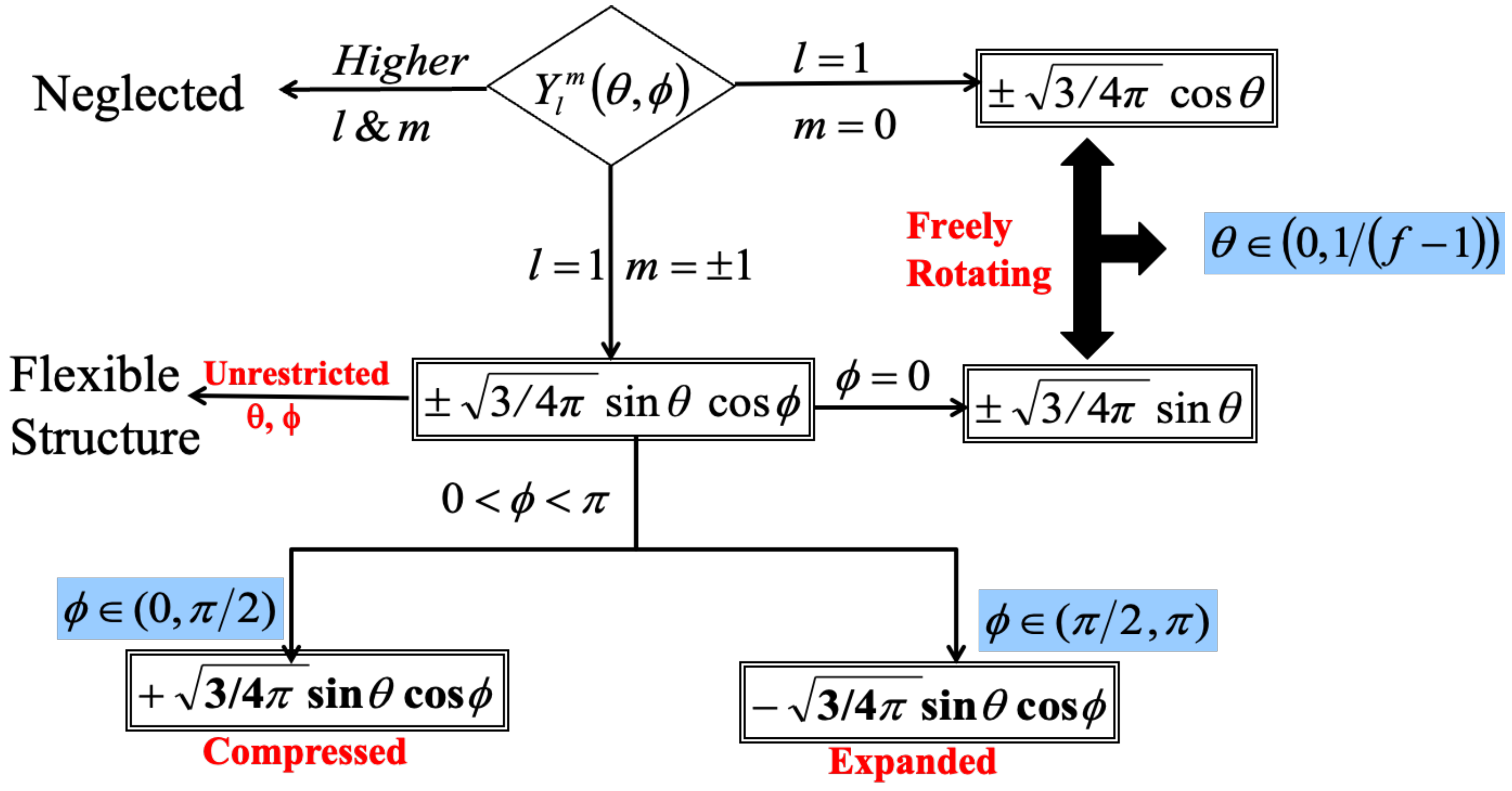
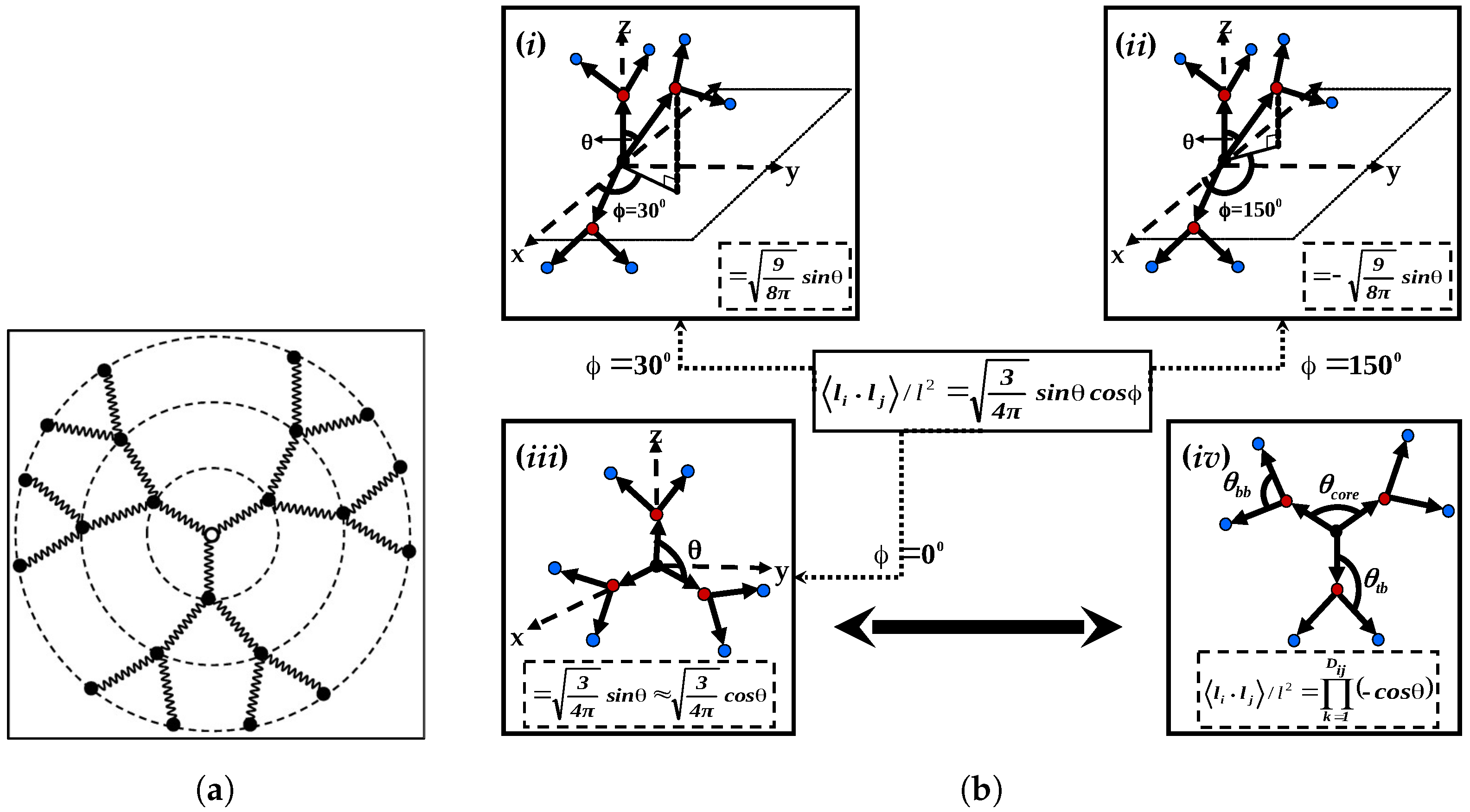
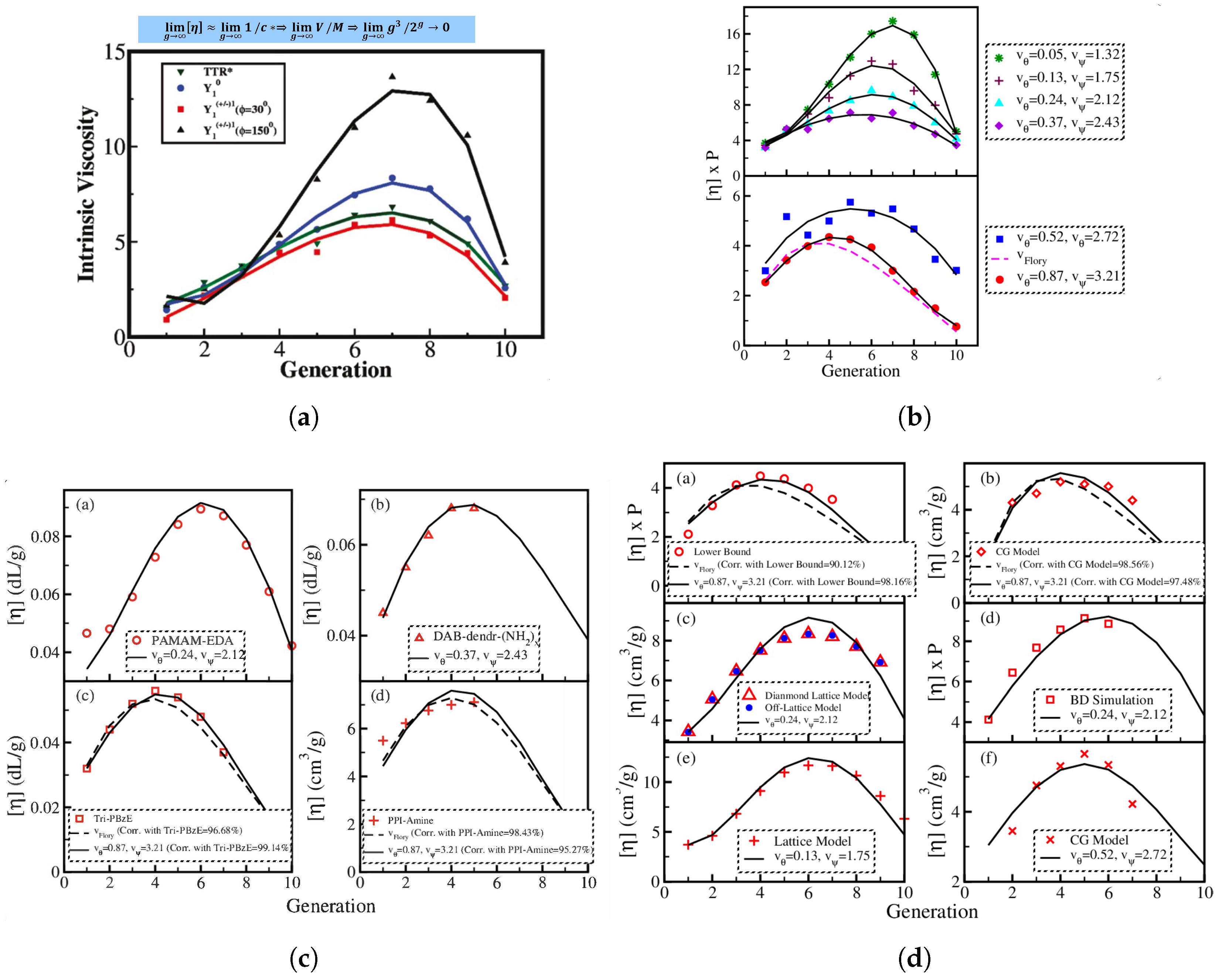
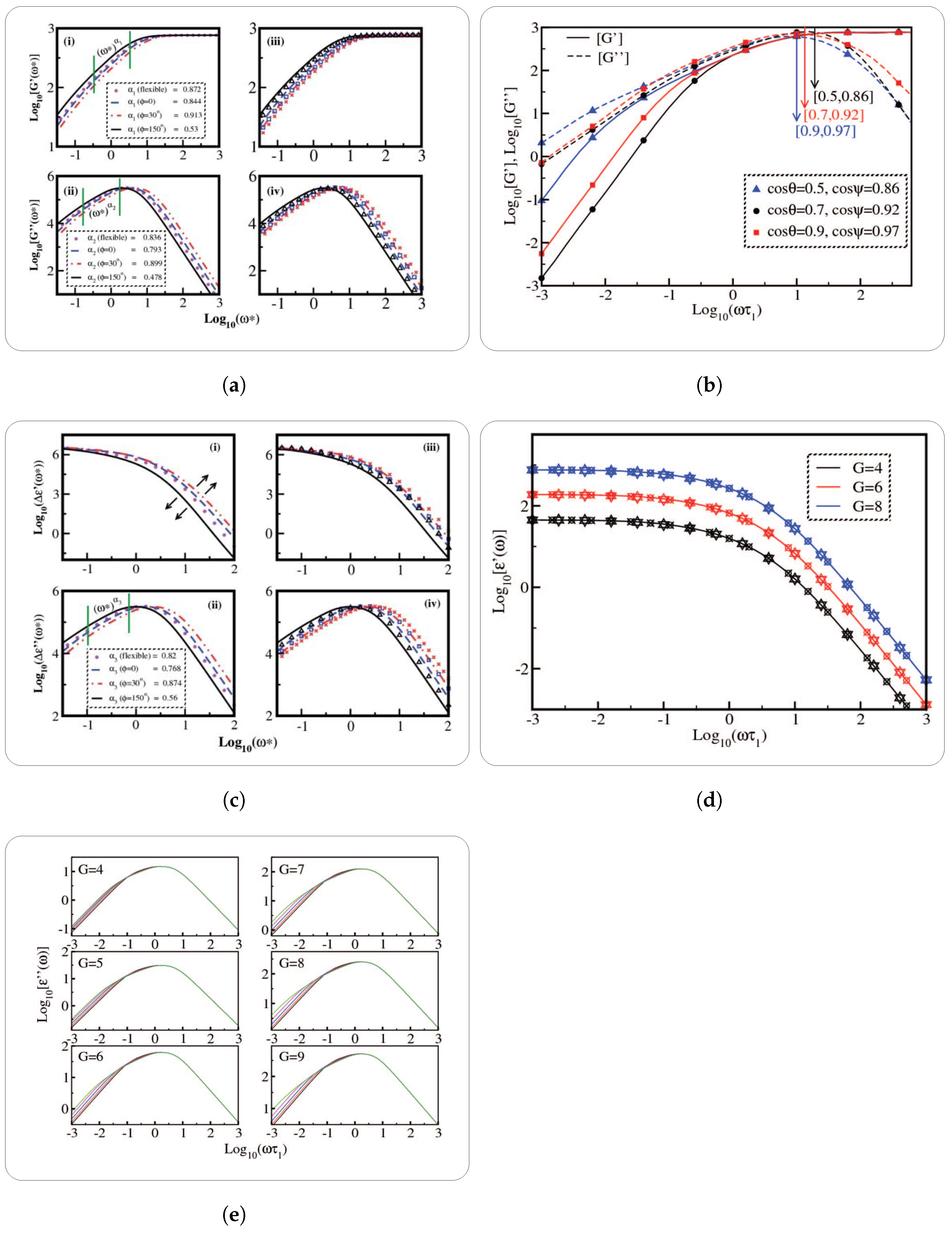
The researchers [
16] conducted a comparative analysis between their findings on the intrinsic viscosity of semiflexible dendrimers, namely those with bond orientation angles of
,
, and
. This comparison was made with La Ferla’s TTR model, which offers an alternative method for incorporating stiffness in dendrimers. A resemblance was noted in the qualitative behavior displayed in
Figure 5a. All models exhibited a distinct peak in intrinsic viscosity at a certain generation, although the precise location of these peaks differed across the different models. In the scenario when
, the intrinsic viscosity pattern exhibited by Kumar’s model [
16] bears a striking resemblance to that of La Ferla’s TTR model [
30], including a comparable range. Nevertheless, the numerical values of intrinsic viscosity
exhibited a little elevation compared to the comparable values obtained from the La Ferla model. Transitioning to the compressed semiflexible dendrimers characterized by a bond orientation angle of
, it was observed that their intrinsic viscosity exhibits comparable patterns and magnitudes to those of the TTR model [
30]. In contrast, the enlarged dendrimers with a bond orientation angle of
exhibited a wider range of intrinsic viscosity values, notably in the vicinity of the peak, while maintaining a qualitatively similar overall shape. Dendrimers exhibiting a bond orientation angle (
) of
occupy an intermediate position in relation to the expanded and compressed conformations, as evidenced by their intrinsic viscosity behavior. Additionally, it was observed that the dendrimers with a bond angle of
exhibited much shorter relaxation times compared to dendrimers with higher bond angle limitations, such as
and
. Consequently, the highest values of intrinsic viscosity maxima [
16] was observed in dendrimers that impose the most stringent bond orientation limitations, specifically at
. The aforementioned observation elucidates the complex correlation between bond orientation and the intrinsic viscosity characteristics of semiflexible dendrimers.
Lu et al. [
53] introduced a new theory for the intrinsic viscosity of polymers with various architectures, from linear chains to dendrimers, based on a partially permeable sphere model. This model incorporates two phenomenological functions,
and
, to describe hydrodynamic interactions. The theory combines Debye’s [
54] and Einstein’s theories [
55], resulting in a simple expression for intrinsic viscosity that avoids complex multi-body interaction calculations. An intrinsic viscosity expression accounting for both Debye and Einstein contributions was obtained. The calculated intrinsic viscosities for linear and star polystyrene in both
and good solvents closely match experimental data, typically deviating by less than 4%. The theory also predicted that the Mark–Houwink–Sakurada (MHS) exponent for hyperbranched polymers is less than 0.4 and decreases with molecular weight. For dendrimers, it accurately predicts the non-monotonic behavior and the peak in intrinsic viscosity. Thus, the findings highlighted that polymer architecture significantly influences intrinsic viscosity with different behaviors observed for various structures. Furthermore, the authors in the referenced work [
16] conducted intrinsic viscosity estimates by manipulating orientation angles while taking into account hydrodynamic interactions as well in their absence and the excluded volume interactions. In subsequent years, an examination was conducted on the rheological properties of model dendrimers in order to observe the distinct peaks in intrinsic viscosity as a function of generation growth. This analysis incorporated the consideration of excluded volume interactions, utilizing the Rouse–Zimm theory as the theoretical framework. Rai et al. [
52] conducted a study on the dynamics, and their investigation focused on the relationship between the intrinsic viscosity of dendrimers and their excluded volume parameters, namely
and
, as well as their generation. This was compared to dendrimers with the mean-field excluded volume, where the value of
was determined using Flory’s theory [
56]. The variable
displayed a comparable pattern in the intrinsic viscosity of dendrimers. The numerical value roughly corresponds to the result obtained by the geometric technique for the given parameters
. This alignment was in accordance with the geometric methodology employed for the computation of excluded volumes, as outlined within the framework of Flory’s theory [
56]. The observation was clear that dendrimers exhibiting different excluded volume parameters displayed the same qualitative pattern, which was characterized by a distinct maximum at specified stages of growth. The observed tendency was in opposition to the previously suggested dendrimer model under conditions of favorable solvent [
36], where they conducted a study on conformational properties. Nevertheless, these findings are consistent with facts derived from a range of experimental, modeling [
27,
57], and theoretical investigations [
16]. The intrinsic viscosity is influenced by the precise values of the excluded volume parameters. Dendrimers characterized by lower values of
and
demonstrated elevated inherent viscosity across all generations with the exception of the initial generation. A significant peak in intrinsic viscosity was seen at the sixth generation when the excluded volume parameters were set to low values (
and
. As the strength of excluded volume interactions decreases, the location of the maximum changes toward higher generations. The displacement of the typical maximum has been shown in previous theoretical [
16], computational [
7], and experimental [
7] investigations as well.
Figure 5 depicts graphically the comparisons between different analytical models (
Figure 5a,b), an analytical model with experimental results (
Figure 5c), and an analytical model with simulation results (
Figure 5d).
Interestingly, in contrast to the common misconception that dendrimers have a dense core and open periphery, a careful explanation [
58] between the molecular weight and volume of the dendrimer for estimating intrinsic viscosity was discovered and displayed the current picture of dendrimers as a transition from a non-compact sphere to a dense space-filling hyperbolic topology [
58]. The exceptional maximum in the generation dependent intrinsic viscosity was examined [
58]. A cross-over from Euclidean to non-Euclidean dimensions marks the beginning of this structural transition, which was marked by the position of this maximum. The topology of dendrimers is characterized by this structural transition. In three dimensions, the real accessible volume is not a continuous cubic power-law but a discontinuous function of generation. This suggests that the terminal groups are dispersed throughout the core and periphery of dendrimers, whereas the core is dense. Due to their enormous density and densely packed peripheries, huge dendrimers demonstrate a fractal nature with fractal dimensions larger than 3 (refer to the highlighted equation in
Figure 5). As a result, it was discovered that dendrimers have compact cores and dispersed terminal groups in both the core and periphery. Large dendrimers therefore display a fractal nature with a fractal dimension greater than 3, as they are extremely dense entities with crowded peripheries. A maximum in the
plot is produced by such a shift in shape, which is followed by a negative slope. The value of
, which here refers to the exponent factor from the Mark–Houwink–Kuhn–Sakurada (MHKS) equation, changed from being positive for lower generations to being negative after a specific value of
corresponding to a threshold generation. Therefore, as dendrimers develop through generations, the location of the peak in intrinsic viscosity denotes the beginning of a structural change. This shift is accompanied by an internal reorganization of the dendrimer’s component monomers, which results in a change in the dendrimer’s overall shape.
The viscoelastic qualities are a subset of rheological properties that focus on the materials’ combined viscous and elastic behavior. Relaxation moduli, specifically the storage modulus
and the loss modulus
, are key factors in understanding dendrimer viscoelasticity. The storage modulus
measures the elastic energy stored in a material, indicating its stiffness and capacity to return to its original shape after deformation. In contrast, the loss modulus
measures the viscous energy dissipated as heat, indicating the material’s damping and flow characteristics. These moduli are especially important in polymer research because they help characterize the mechanical properties and relaxation dynamics of complicated structures like dendrimers. These moduli are measured using rheological techniques, which provide a full understanding of the material’s relaxation spectrum across a range of frequencies. Theoretical models, such as the extended Gaussian structure model, are used to understand how molecular architecture and topology affect polymer mechanical characteristics and relaxation behavior.
Table 1 highlights studies undertaken to determine various relaxation moduli, such as the storage modulus, mechanical relaxation modulus, and shear stress relaxation modulus, including their time and frequency dependency.
3.2. Conformational Behavior of Model Dendrimers
Conformational transitions in dendrimers pertain to alterations in the spatial configuration and morphology of dendrimer molecules in reaction to external stimuli or environmental circumstances. The comprehension of conformational transitions has significant importance in the customization of dendrimer characteristics to suit certain applications. The transitions observed in the dendrimers can be characterized by changes in their flexibility, compactness, and overall form. The integration of theoretical and computational inquiries with experimental examination of macromolecules facilitates the comprehension of the relationship between their molecular structure and conformational attributes in a solvent environment. Scattering experiments [
64,
65] and computer simulations [
66,
67,
68,
69] play a crucial role in the examination of the morphology and internal composition of dendrimers across different domains. The determination of the static structural factor
of dendrimers in a solution is carried out in scattering experiments. Valuable information, such as the pair correlation function and the segment density profile, can be derived from the provided data by employing techniques such as inverse Fourier transform [
57,
67,
68] and square root deconvolution [
64,
65]. In contrast, computer simulations provide a means to directly assess the radial density profile and pair correlaetion function, enabling the subsequent determination of
using Fourier transformation. So, both experimental and simulation methodologies offer valuable insights into important parameters such as the radius of gyration
and hydrodynamic radius
by directly analyzing the pair correlation function. Moreover, the shape factor, denoted as
, which is the ratio of
to
, is a significant parameter that provides insights into the morphology, dimensions, and organization of dendrimers and other particle systems.
The radius of gyration
is an important metric to calculate first when researching dendrimer conformations since it offers a basic assessment of the dendrimer’s overall size and spatial extent. Meanwhile, features such as the shape factor, asphericity, and branching density provide extensive information on the dendrimer’s structure. This fundamental understanding of the dendrimer’s size and density distribution is required for comparing different generations, studying solvent interactions, and determining physical attributes. Determining
initially offers the framework for a thorough examination of more complex structural characteristics.
Table 2 below summarizes major discoveries on
for several types of dendrimers.
A metric that depicts how the complexity of a fractal pattern increases with scale is referred to as the fractal dimension, which is denoted by the symbol . In many cases, it results in non-integer values that encapsulate the pattern’s intricacy and space-filling features. It does this by quantifying how the detail or density of a fractal changes as the observation scale changes. A more critical description of natural and irregular shapes is provided by the fractal dimension, in contrast to the usual dimensions, which are integers (for example, 1D for lines and 2D for planes).
The relationship between the radius of gyration
and the fractal dimension
is
Here, the aggregate mass is denoted by
M, the radius of gyration is denoted by
, the radius of the single constituent particle (monomer) is denoted by
a, and the fractal distance is denoted by
. Although some general trends have been reported for the fractal dimension
of dendrimers,
Table 3 shows that lower fractal dimensions, around 1.67, lead to more open and less compact structures. A magnitude between 2.4 and 2.8 is a moderate level of compactness, and fractal dimensions closer to 3.0 are associated with highly compact, space-filling dendrimers. We discuss in the later half of this section that the fractal dimension (
) of semiflexible dendrimers depends on their conformation and bond orientation angle (
). For compressed conformations (
between 0 and
),
is nearly constant at about 4, indicating compact structures. For extended conformations (
between
and
),
decreases as
increases, ranging from 2.00 to 1.63, suggesting less space-filling structures. Higher generation dendrimers exhibit fractal dimensions greater than 4 due to their highly branching topology. Thus, the findings highlight the role of
in determining dendrimer conformations and interactions. Some authors [
52] claimed that a Euclidean to non-Euclidean conformation transition possibly causes
> 3.
Burchard [
76] and Hearst [
77] have conducted studies on static phenomena and sedimentation, respectively. Kleppinger et al. [
78] used small angle X-ray scattering (SAXS) to experimentally analyze highly diluted solutions of poly(benzyl ether) dendrimers, and the results provide important insights into the structural properties of individual molecules. Ramzi et al. [
79] aimed to investigate the structural characteristics and intermolecular interactions of amine-terminated poly(propylene imine) dendrimers in solution using small-angle neutron scattering (SANS). The study examines how dendrimer concentration and acidity influence these interactions, showing that the intermolecular interactions between dendrimers increase with concentration, leading to a pronounced interference peak. Meanwhile, the investigation [
80] regarding the structural characteristics of poly(propyleneimine) dendrimers up to the fifth generation employing SANS, viscosimetry, and molecular dynamics simulations to determine the dimensions and density distributions in various solvents concluded that PPI dendrimers exhibit a compact, space-filling structure with a fractal dimensionality of approximately 3. Ballauff [
81] analyzed the structure of a fifth-generation dendrimer in solution using SANS. The analysis effectively elucidates the radial scattering length density and highlights the strong structural fluctuations within the dendrimer. Other studies featured the use of SANS to understand how counterions and the protonation of amino groups affect the dendrimer’s molecular conformation and interactions [
82], understanding how dendrimer generation affects counterion association and molecular conformation under varying acidity conditions [
83] and how molecular protonation and dendrimer concentration affect the dendrimer’s radial density profile and overall conformation using SANS [
84].
The study of scattering patterns at lower concentrations primarily reveals form factor contributions from individual molecules. In this context, form factors refer to the scattering profiles attributable to the shape and size of single particles without considering interactions between them. As the concentration of particles increases, a notable decrease in forward scattering intensity is observed, which indicates the onset of significant intermolecular interactions. These interactions disrupt the scattering pattern predicted solely by form factors and need to be considered in the analysis. In dilute solutions, the Guinier approximation is effective for determining the radius of gyration (
), a measure of the overall size of the particles, up to a certain
q-value (where
q is the scattering vector). The Guinier plot, which is a plot of
versus
, where
is the scattering intensity, remains linear at low
q-values, allowing for the accurate determination of
. However, at higher concentrations, the Guinier approximation fails due to the increased influence of intermolecular interactions, leading to deviations from linearity in the Guinier plot. To understand how particle size changes with concentration, the radius of gyration (
) was determined. This measure provides insight into the spatial dimensions of the molecules in solution and how they are affected by concentration. The Kratky plot, which plots
versus
q, is useful for analyzing the shape of the particles. In this study, the Kratky plot for dendrimer complexes displayed a maximum, which is indicative of a transition to a more compact, globular shape for the dissolved molecules. This finding was pivotal in identifying the structural characteristics of dendrimer molecules, as a peak in the Kratky plot typically signifies a globular structure. In comparison, Topp et al. [
85] conducted an extensive study using SANS and X-ray scattering to examine changes in dendrimer size in concentrated solutions. Their work revealed a distinctive peak in the Kratky plot, which is characteristic of particles with a relatively high internal segment density. Using SANS, Topp et al. [
85] systematically examined the average dimensions of poly(amido amine) (PAMAM) dendrimers under various conditions, including temperature fluctuations, mixture compositions of good solvents and non-solvents, and different solvent environments. Their findings indicated that the size and shape of PAMAM dendrimers are highly sensitive to the surrounding environment, providing detailed insights into their structural behavior in solution. Similarly, Chen et al. [
86] investigated the behavior of dendrimers in coatings using small-angle scattering techniques. Their study focused on understanding how dendrimers interact within a coating matrix and how these interactions affect the overall properties of the coating. They observed that dendrimers tend to form compact structures at higher concentrations, which is similar to the findings of the Kratky plot analysis in the present study. Their work underscored the importance of considering concentration effects and intermolecular interactions when analyzing the structural properties of dendrimers in various applications.
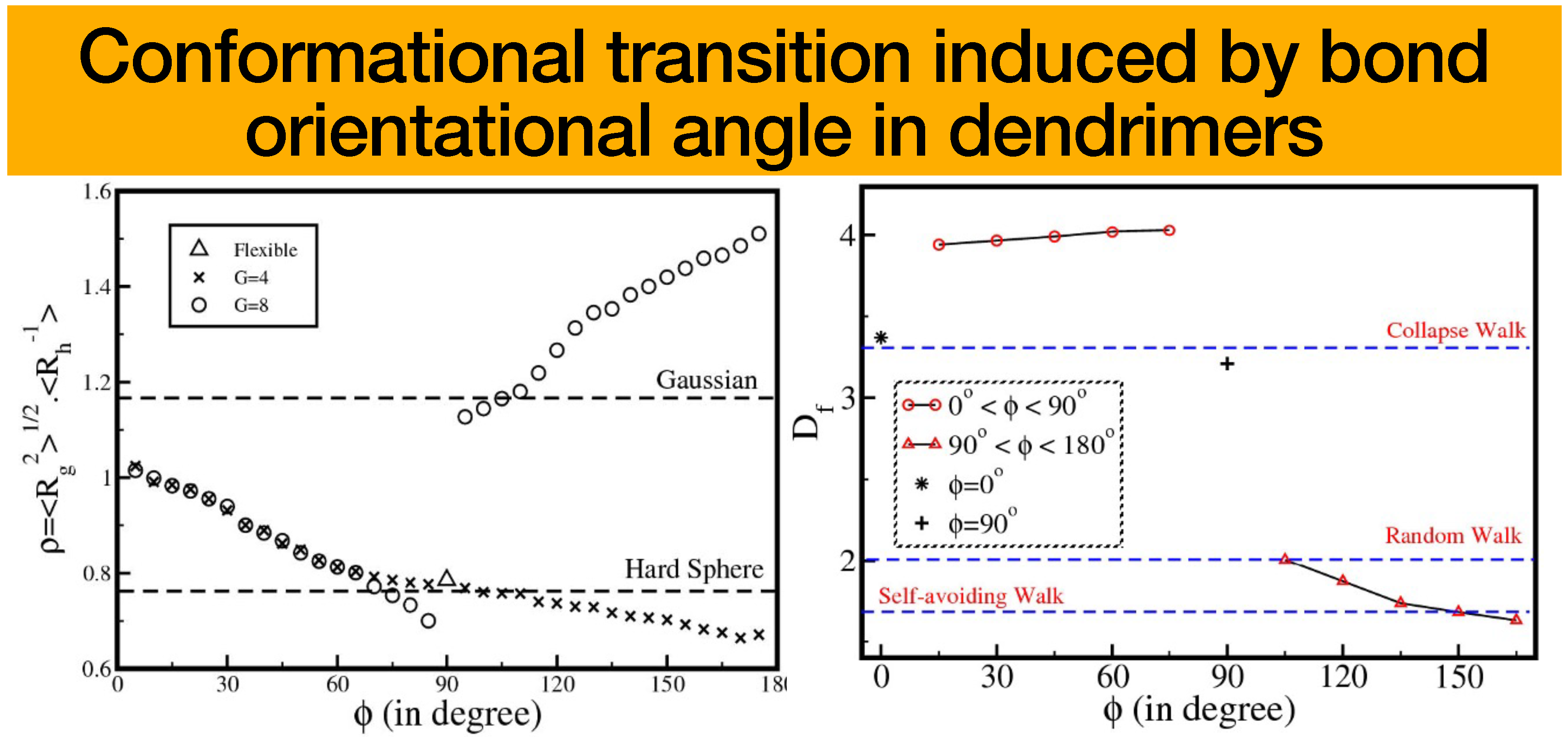
In the study conducted by Kumar and Biswas [
18], they examined the impact of several environmental factors on the conformational characteristics and morphology of dendrimers. The researchers employed various methodologies, including scattering experiments and computer simulations, in order to obtain insights on the behavior of dendrimers. The presented study investigated the relationship between the shape factor, denoted as
, and the bond orientation angle, referred to as
, in the context of semiflexible dendrimers at generations 4 and 8. It is worth mentioning that in compressed conformations where the angle
is less than
, the parameter
exhibited a decreasing trend as the angle
grows regardless of whether it pertains to a lower or higher generation. In contrast, it has been shown that for extended conformations where the bond orientation angle
is more than
, and for higher-generation semiflexible dendrimers with a generation value of 8
, there is an increase in the shape factor,
, as the bond orientation angle
increases. In the case of lower generation expanded conformations, there is a drop in the value of
as the value of
grows. These observations were consistent with experimental results, which demonstrate that the quality of the solvent has an impact on the values of
. Moreover, the findings indicate that when the variable
grows, the shape factor tends to approximate the limit of a hard sphere for compressed conformations (where
), and it shifts toward more open structures for extended conformations (where
), as illustrated in
Figure 6 (left).
In addition, the investigation also examined the potential influence of factors such as the bond direction angle , generation G, and functionality f on the conformational transition’s dependence on . It was observed that the parameter exhibits a linear increase with respect to the variable across all three conformations. However, this relationship does not suggest any notable structural transition. This observation implies that for a given value of , the configurations of dendrimers do not undergo significant alterations as a result of variations in . The thermodynamic validation of the substantial conformational transition in dendrimers, which is dependent on variations in the shape factor and the bond orientation angle , was achieved through an examination of the configurational free energy. Subsequently, it became apparent that the configurational free energy exhibits an upward trend as the angle grows in dendrimers possessing a bond orientation angle of , but it remains rather stable for dendrimers with bond orientation angles of and . The variation of free energy with respect to is continuous for a certain value of , where is situated between the compressed state at and the expanded state at .
In order to advance their research regarding semiflexibility [
18], their work closes the gap between earlier models and dendrimers with excluded volume interactions, offering insightful knowledge into the structural adaptability of semiflexible dendrimers by successfully identifying the fractal dimension of semiflexible dendrimers [
19]. There are three main scaling regimes that are typical of polymers with various levels of branch flexibility at a given generation, and the fractal dimension ranges widely. The study showed that either adjusting the hierarchy of possible conformations at a given generation or reducing the number of generations during growth can be used to precisely control the internal structure of dendrimers. Any macromolecule’s fractal dimension
can be calculated using the scaling equation between the number of monomers
and the radius of gyration
. Experimental and theoretical studies show discrepancies in the reported values of
. For instance, Mallamace et al. experimentally found that
for higher-generation dendrimers, while molecular dynamics simulations reported
. This article reconciles these differences by tuning the bond orientation angle
within the optimized Rouse–Zimm model framework. They determined the fractal dimension of flexible dendrimers using observed scattering patterns. The discussion centering on the effects of bond orientation angles on higher generation dendrimer fractal dimensions was held by the group. They pointed out that in earlier experimental and theoretical studies, there have been inconsistencies in the reported values of fractal dimensions. The fractal dimension is nearly constant at about 4 for compressed conformations of semiflexible dendrimers (
is between 0 and
). This is consistent with the results of simulations of dendrimers of higher generations. In contrast, the fractal dimensions for extended conformations (where
is between
and
) fall as
increases and can range between 2.00 and 1.63. As
rises in the extended conformation regime, semiflexible dendrimers become less space-filling structures, with values that are similar to those of the random walk and self-avoiding walk models of linear polymers. Overall, they emphasized the importance of
in capturing the physical characteristics of various dendrimer conformations and their resemblance to interactions between polymers and solvents; see
Figure 6 (right). They also pointed out that compressed conformations of lower generation dendrimers display behaviors ranging from a random walk to extremely compact globular forms. These compressed conformations surpass the proportions of a compact collapsed walk as generations progress and grow much denser. Particularly at higher generations, the highly branching topology of dendrimers produces a pseudo-fractal structure with a fractal dimension greater than 4, which denotes a very uneven surface. The results of this work demonstrate that the parameter
may be used to predict the degree of polymer–solvent and polymer–polymer interactions precisely, enabling the investigation of a variety of dendrimer conformations.
3.3. Relaxation Dynamics of Model Dendrimer
Dendrimer’s relaxation moduli act as windows into their environment, allowing us to observe how these intricate molecules move and interact. They provide information on how rigid, flexible, or semiflexible dendrimers respond to stress as well as how they manage changes over time. Semiflexibility has a noticeable impact on the normal modes as seen in some relaxation moduli examples, such as
and
. The phenomenon of dielectric relaxation is a fundamental observation that occurs in dendrimers and other materials when they are exposed to an external electric field [
87]. In the study of dendrimers, a comprehensive comprehension of dielectric relaxation is necessary for the purpose of characterizing their electrical characteristics and dynamic behavior. The dielectric constant of dendrimers is characterized by two components: the real component, denoted as
, and the imaginary component, which is denoted as
. The real component,
, is a critical parameter that quantifies the ability of dendrimers to undergo polarization and store energy in the form of electric dipoles. Similarly, the imaginary component,
, holds equal significance in dendrimers. The measurement pertains to the efficacy of dendrimers in dissipating energy, primarily in the form of heat, due to molecular reorientations, electronic transitions, or other relaxing processes taking place inside the dendritic structure. A greater value of
indicates a more significant level of energy dissipation. Both
and
exhibit frequency dependence, and their changes with respect to frequency provide valuable information. The utilization of frequency dependency enables the comprehension of the dispersion of relaxation durations inside dendrimers.
The investigation of dielectric susceptibility components in the research unveiled fascinating patterns within semiflexible dendrimers across different bond orientation angles and generations [
17]. For lower frequencies, the magnitudes of these components were equal across all bond orientations and generations. However, they showed a rise that was dependent on frequency and proportional to the inverse square of the normalized frequency,
. In the region of high frequency, the decay of these entities followed an inverse relationship with respect to
, which was subsequent to the attainment of local maxima. This pattern was particularly evident for generations exceeding four unless the entities were in expanded conformations. In the latter scenario, the decay occurred at a faster rate, which was characterized by a power law exponent of about −1.3. The displacement of distinctive peaks moved toward higher frequencies as the generation increases, resulting in an expansion of the susceptibility’s dynamic range (
Figure 7a). It is worth mentioning that the peak values observed in the semiflexible dendrimer with an angle of
coincided with those of
. Conversely, for
, the peak values were observed at lower frequencies in comparison to the models with
. Thus, the aforementioned results offered significant contributions to the understanding of the dynamic characteristics of semiflexible dendrimers across different circumstances, elucidating their relaxation properties and frequency-dependent reactions.
Moreover, the behavior of the real portion of the dielectric susceptibility, designated as
, in the context of dendrimers with variable excluded volume parameters and dendrimer generations was covered by the authors in another study [
38]. For dendrimers with various properties, they displayed their findings through a double logarithmic plot of
against normalized frequency
. Surprisingly, their findings showed that the magnitude of
remained constant despite changes in the excluded volume parameters, indicating that excluded volume interactions between adjacent non-bonded monomers within the dendrimer structure do not affect the dielectric susceptibility (
Figure 7b). Furthermore, they observed that the dynamical range of
values widens with increased dendrimer generations, indicating a larger frequency dependency. In contrast to earlier theoretical predictions,
displayed a power-law decrease in the high-frequency band with an exponent of roughly
[
88]. The authors hypothesized that this discrepancy might be caused by how eigenvalues and eigenvectors interact throughout the mathematical operations. Intriguingly, they discovered that the expanded conformations of semiflexible dendrimers had power-law exponents that mimic those of dendrimers with excluded volume interactions, indicating that the latter accurately simulates the effect of excluded volume interactions in dendrimers. So, the complex dielectric behavior of dendrimers and how they respond to various structural features were clarified by these studies. For the imaginary part, they found that for all generations and excluded volume parameters, the numerical values of
increased proportionally with normalized frequency in the low-frequency region, following a power law of about
. It is interesting to note that the magnitudes in this location showed a substantial relationship with the parameters that were excluded from the volume calculations with magnitudes falling as the parameters were increased. The behavior of
in the high-frequency zone, however, was constant for all excluded volume factors with all curves overlapping. The values of
in this high-frequency regime declined with normalized frequency as
with local maxima in the intermediate frequency range occurring before this for all generations. This behavior in the low- and high-frequency regimes remarkably resembled expanded semiflexible dendrimer conformations. Additionally, the authors observed a cross-over in
with different excluded volume parameters, showing that numerical values are higher for larger excluded volume parameters in the intermediate regime (
Figure 7c).
Dendrimers also display orientational relaxation dynamics, which is different from dielectric relaxation. The ability of dendrimers or segments to change their orientation or alignment in response to environmental factors is directly related to this phenomenon. It entails reorganization in order to achieve a more thermodynamically advantageous orientation. Researchers frequently use NMR analytical theories, experimental approaches [
89], and computer simulations [
90,
91] to analyze orientational relaxation dynamics. These studies offer insightful information about the processes, mechanisms, and variables affecting dendrimer reorientation. The orientational relaxation dynamics of semiflexible dendrimers was carried out inside the optimized Rouse–Zimm formalism in a novel theoretical framework created by Kumar and Biswas [
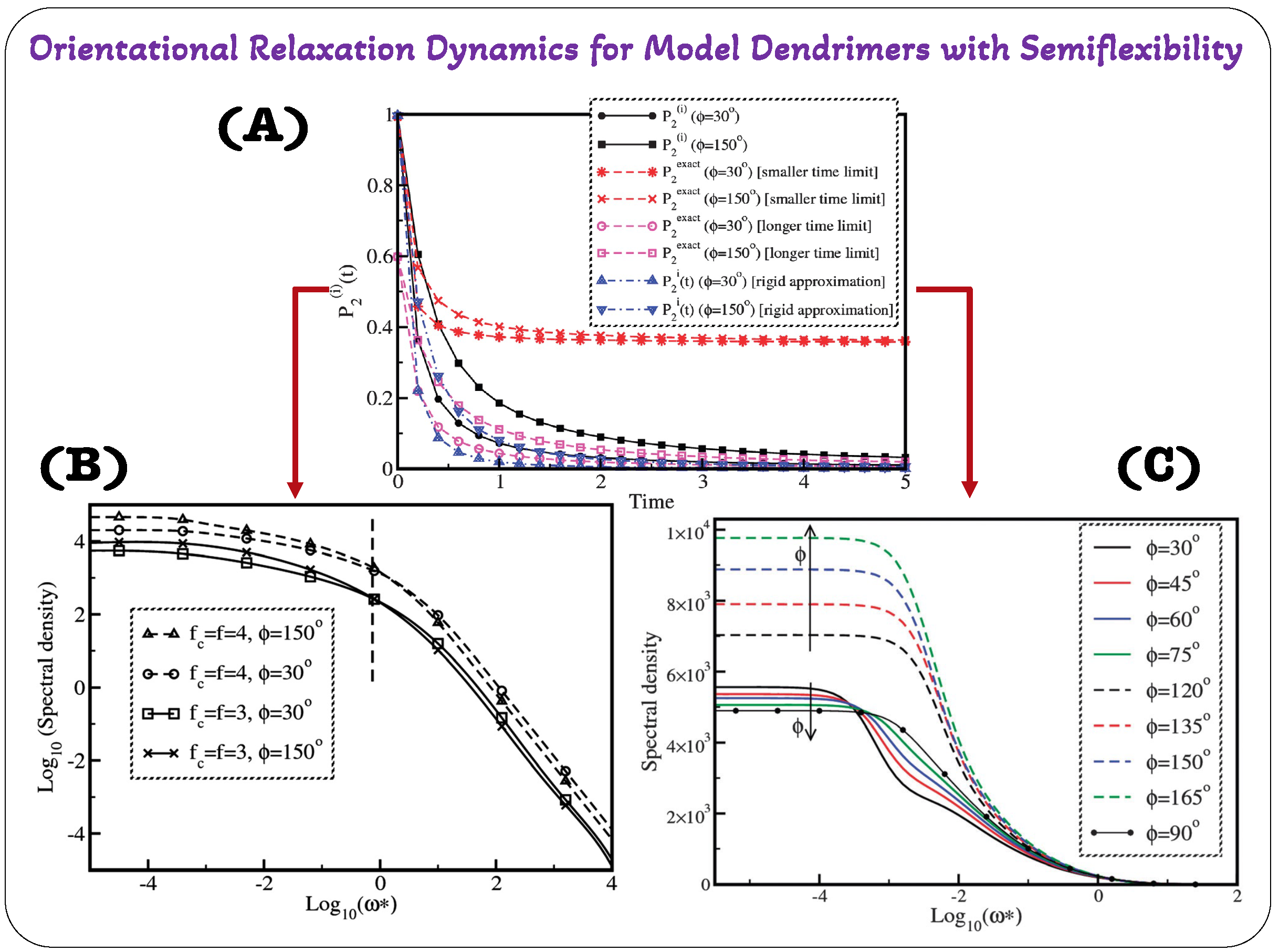
28]. By using a graph theory approach to provide precise limits on the direction and orientation of the individual bond vectors within the dendrimer structure; the concept of semiflexibility was introduced in this analytical model. The preaveraged Oseen tensor was also used to represent hydrodynamic interactions, which are crucial to the behavior of the dendrimer. The analysis of two crucial parameters—the temporal autocorrelation function, designated as
, and the second-order orientational autocorrelation function, denoted as
, coupled with its cosine Fourier transform, spectral density
, were the main emphasis of the study. Two important variables were examined in relation to these functions: the branch-point functionality and the level of semiflexibility. The innovative way that Kumar and Biswas calculated
set their methodology apart from earlier research by Perico and Guenza [
92,
93]. (For a comprehensive understanding of the formalism used in this study, readers are referred to the referenced article [
28].) Their research addressed an issue where the equation for
did not adequately distinguish between flexible and semiflexible dendrimers.
Figure 8A illustrates the contrast between these two analytical models through orientational autocorrelation function
[
28,
92].
The article [
28] also discusses the evaluation of the local orientational mobility of dendrimers through the utilization of the spectral density function
, which is obtained by performing a cosine Fourier transform of
. The authors introduced an approximation technique for computing these functions at both short and long time scales. Furthermore, they compared the accuracy of these approximations with exact expressions for specific scenarios, as documented in the works of Perico and Guenza [
92,
93].
Observations were made regarding the behavior of the second rank orientational autocorrelation function,
(
Figure 8A), for compressed and expanded conformations of semiflexible dendrimers at both small and large times. In this context,
showed distinct patterns for these two conformation types. The curves of
gradually approached zero as time extends to infinity, indicating that at large times, the orientation within the dendrimers becomes increasingly random, suggesting isotropic behavior. At small times, however, both compressed and expanded conformations exhibited similar behavior, rapidly relaxing from an initial value of 1. Interestingly, at large times,
relaxes from a finite value; specifically, it approaches 0.6 for both compressed and expanded conformations of semiflexible dendrimers. Upon closer examination, it becomes evident that the values of
obtained from the theories of Kumar [
19] and Perico [
92] align with the behavior of
[
19]. Particularly,
also diminishes to zero at extended times for both compressed and expanded conformations of semiflexible dendrimers, signifying randomness in an isotropic system. This reduction to zero happens instantaneously for all times except at
, where
equals 1, indicating that
values obtained from the theories of Kumar [
19] and Perico [
92] share qualitative similarity with the behavior of
. Examining the numerical values of
for compressed and expanded conformations of semiflexible dendrimers, it became evident that at small times, these values closely resemble those derived from equation obtained from the works of [
92,
93]. However, for all other time intervals, the magnitudes of
fall between the values of
obtained from the equations in [
28,
92] for both conformations of semiflexible dendrimers. Notably, the numerical magnitudes of
calculated from Equation [
92] consistently surpass those computed within the rigid approximation [
28,
92] across the entire time range for both compressed and expanded conformations of semiflexible dendrimers. This discrepancy in numerical magnitudes of
was particularly pronounced in the intermediate time zone, where
values derived from the graphical theory approach [
28] significantly exceeded those obtained from the rigid approximation for both compressed and expanded conformations of semiflexible dendrimers. Equations (
11) and (
12) are provided for readers to better comprehend and correlate the expressions for
and
.
The spectral density, denoted as
, derived from the cosine Fourier transformation of the second-order orientation autocorrelation function represented as
, was also derived in the studies of Kumar [
28]. As depicted in
Figure 8B, they have presented a double logarithmic plot showcasing the normalized spectral density, labeled as
, in relation to the reduced frequency,
. The investigation centered on two distinct configurations of semiflexible dendrimers, specifically those with
, encompassing the compressed
and expanded
forms. Additionally, they considered variations in branch-point functionalities, which were denoted as
. Within the low-frequency spectrum, they observed an intriguing pattern wherein the magnitude of the spectral density remains constant across all dendrimer types regardless of frequency. However, it becomes apparent that the magnitude of
exhibits a pronounced dependence on both the degree of semiflexibility and the branch-point functionality. To clarify, dendrimers characterized by higher branch-point functionality and increased semiflexibility demonstrate numerically higher values of
. In simpler terms, those with
and
display the highest
values, while their counterparts with
and
exhibit the lowest values. Transitioning into the high-frequency range reveals a distinct behavior. The spectral density decreased with frequency, following a decay pattern of
, which was a phenomenon observed for both the compressed and expanded configurations of semiflexible dendrimers featuring branch-point functionalities of
and 4. Notably, an interesting occurrence takes place in the intermediate frequency spectrum, which was marked by the dotted line in
Figure 8B. In this range, the spectral density curves for compressed and expanded dendrimers diverge noticeably. Crucially, within this intermediate spectrum, the spectral density values were higher for compressed dendrimers than for their expanded counterparts, owing to the observed cross-over effect. This implies that in shaping the spectral density’s behavior in semiflexible dendrimers, the degree of semiflexibility primarily governs the outcome, while changes in branch-point functionality exert a relatively minor influence.
Figure 8C presents the frequency-dependent behavior of the spectral density in
dendrimers with branch-point functionality
. Regardless of
f, the spectral density showed a consistent pattern across the entire frequency range. Notably, a cross-over effect was observed in the intermediate frequency region—specifically in the compressed dendrimer conformations
compared to the expanded ones
. The characteristic area beneath the spectral density curve strongly correlated with the degree of semiflexibility, which is noteworthy as it is independent of the correlation time. In the expanded conformation zone, the characteristic area is maximized and increases with higher
values. In contrast, for compressed dendrimers, this area decreases with
, which is accompanied by a cross-over in the spectral density in the intermediate frequency region. However, the relative change in the characteristic area was more pronounced in expanded conformations. Lastly, flexible dendrimers
exhibited the smallest characteristic area compared to both compressed and expanded conformations.
3.4. Application toward Randomly Hyperbranched Geometries
Randomly hyperbranched structures are a class of polymers characterized by a random and highly branched architecture, which is distinct from the regular branching patterns of dendrimers. They exhibit a high degree of branching, allowing for the incorporation of diverse monomers and leading to amorphous, compact structures with unique physical and chemical properties. They may contain defects and dead ends in their branching structure [
94]. These defects result from the more irregular and random nature of the branching process. As a consequence, randomly hyperbranched polymers exhibit a less structured, more complex architecture, making them distinct from other cascade molecules. Due to their peculiar topology-dependent behavior, hyperbranched polymers, which are characterized by their random branching patterns, exhibit equilibrium and relaxation properties that are noticeably different from those of typical dendrimers [
30,
37,
95,
96]. These analytical models, which take into account elements like semiflexibility and excluded volume interactions, were extended to examine the behavior of more complex branched macromolecules known as hyperbranched structures, in addition to research concentrating on the dynamic behavior of dendrimers. Furthermore, research involving novel systems proved the validity of these characteristics. Therefore, it was necessary to classify these macromolecules in order to determine which properties are most affected by the structural disorder and which ones are mostly untouched both theoretically and experimentally.
When analyzing the scaling behavior of viscoelastic relaxation moduli with respect to the dimensionless frequency,
, a clear contrast between randomly hyperbranched polymers and dendrimers becomes obvious. In this environment, dendrimers display a distinctive non-scaling behavior in contrast to randomly branching polymers. In an application of further study, the authors [
20] sought to examine the structural and dynamic characteristics of semiflexible randomly hyperbranched polymers within the context of an improved Rouse–Zimm formalism. By placing restrictions on the bond vector’s direction and orientations along with utilizing the preaveraged Oseen tensor to represent hydrodynamic interactions, they added semiflexibility to the model. Understanding how the bond orientation angle
relates to the frequency-dependent relaxation moduli, specifically the storage modulus
and the loss modulus
[
20], they looked into this relationship for a certain branching topology and a particular number of growth generations. The shape factor
, configurational free energy
, static structure factor
, and scattering intensity
were among the structural properties that the authors looked at in order to characterize the molecular conformation of these polymers (
Figure 9 (left)).
Examining the intermediate frequency range of the mechanical moduli for both compressed and expanded conformations revealed the peculiar properties of semiflexible randomly hyperbranched polymers. The bond vector’s orientation in this situation brought back the typical power-law scaling behavior at intermediate frequencies. In contrast to prior research [
31], where such power-law scaling was absent in semiflexible randomly branched polymers due to their lower disorder, this behavior was akin to the modeling of hyperbranched polymers as Vicsek fractals. Furthermore, it was discovered that both the quantity of monomers and the level of semiflexibility, as determined by the parameter
, affected the extent of the power-law regime in the intermediate frequency range. Additionally, hyperbranched polymers had a noticeable structural change when the parameter
was increased. The expanded conformation’s compactness declined as
rose, whereas the compressed conformation’s compactness stayed largely unaltered. This structural shift was corroborated by the fractal dimensions estimated using Porod’s scaling equation, which changed from
to
for the compressed
and extended
conformations of semiflexible randomly hyperbranched polymers, respectively. In polymers with complicated topologies, the excluded volume interactions were successfully accounted for by this approach [
20].
Semiflexibility and excluded volume interactions are very important factors to take into account in the analytical models. We now look at the results of how excluded volume [
97] affected the rheology and transport dynamics of randomly hyperbranched polymers after discussing semiflexibility in the previous section. Through the frequency-dependent mechanical moduli
and
, rheological properties were evaluated [
97]. Meanwhile, the inherent viscosity and self-diffusion coefficient are frequently used to describe the transport dynamics. The study found that collective modes with slower relaxation rates were primarily responsible for mechanical moduli, and that these modes grew stronger as the excluded volume interactions reduced. Higher relaxation rate local modes were unaffected. The mechanical moduli’s intermediate frequency range clearly reflected the internal structure of randomly hyperbranched polymers, which displayed a distinctive power-law behavior that suggested their fractal origin. The excluded volume interactions had a significant impact on the middle frequency. Here, as the excluded volume strength decreased or the quantity of polymer shells increased, the length of the power-law region grew. The strength of the excluded volume interactions was the only factor that determined the precise values of the power-law exponents. Importantly, when the number of monomers crossed a crucial threshold, the randomly hyperbranched polymers exhibited a more broad power-law region than dendrimers.
The intrinsic viscosity of randomly hyperbranched polymers was examined in relation to the number of shells and various excluded volume parameters,
and
. These results were compared to dendrimers and star polymers with equivalent monomer counts under the influence of excluded volume interactions (
Figure 9 (right)). The plot revealed that as the number of shells increases, the gap between intrinsic viscosity curves of randomly hyperbranched polymers widens for different excluded volume strengths. Notably, the impact of excluded volume interactions is more pronounced in higher shell layers due to an increased number of non-bonded interactions. The intrinsic viscosity’s magnitude grows as excluded volume interactions weaken with maximum and minimum values occurring at specific excluded volume parameters [
] and [
,
], respectively. This demonstrates an inverse relationship between intrinsic viscosity and the strength of excluded volume interactions. Furthermore, the trend of intrinsic viscosity variation with the number of shells falls between that of star polymers and dendrimers with the same monomer count. Intrinsic viscosity increases linearly, similar to star polymers, for very low excluded volume parameters. However, this trend becames predominantly nonlinear with higher excluded volume parameters, plateauing at advanced shell growth. Beyond a certain shell count, intrinsic viscosity reaches a plateau, which slightly dips with further increases in excluded volume interaction parameters,
, corresponding to
and
. This results in a characteristic broad maximum in intrinsic viscosity at intermediate shells, which is akin to dendrimers for stronger excluded volume interactions. Additionally, the existence of a compact, space-filling conformation beyond a critical shell count is noted especially for randomly hyperbranched polymers with larger excluded volume interaction parameters. Conversely, those with smaller excluded volume interaction parameters exhibit voids and resemble linear or star polymers. These findings align with earlier theoretical and experimental studies. Thus, the numerical magnitudes of intrinsic viscosity for randomly hyperbranched polymers fall between those of dendrimers and star polymers, indicating that randomly hyperbranched polymers are relatively more compact than star polymers but less compact than dendrimers with the same monomer count and identical excluded volume interactions. Importantly, this behavior can be fine-tuned by adjusting the strength of excluded volume interactions, which can be achieved experimentally by manipulating solvent conditions. Among the extensive category of hyperbranched polymers, Vicsek fractals hold particular theoretical significance, especially in the company of highly symmetrical entities like dendrimers, which are beyond the scope of this article [
98].







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}