

Amide-Containing Bottlebrushes via Continuous-Flow Photoiniferter Reversible Addition–Fragmentation Chain Transfer Polymerization: Micellization Behavior
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
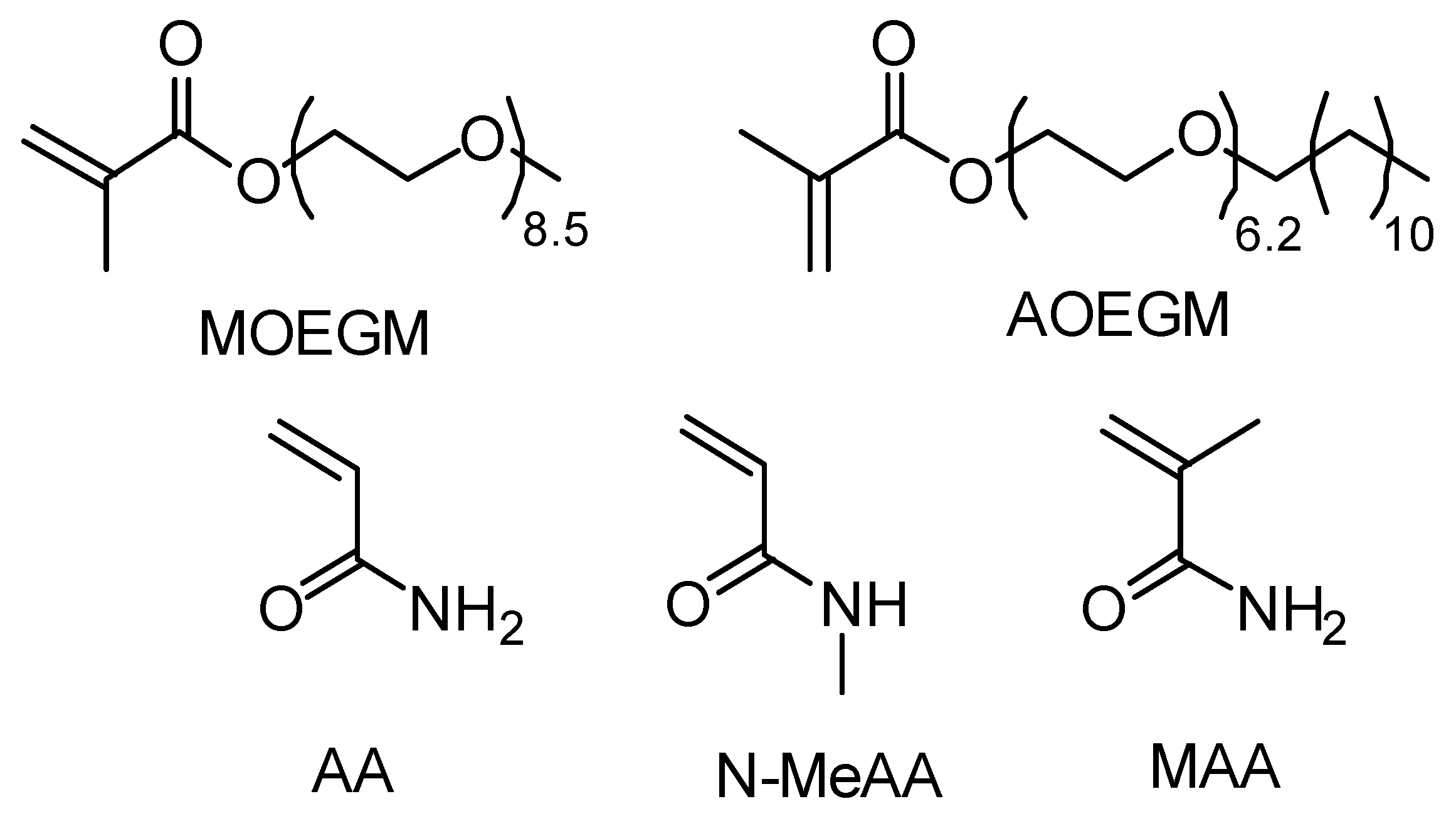
2.1. Materials
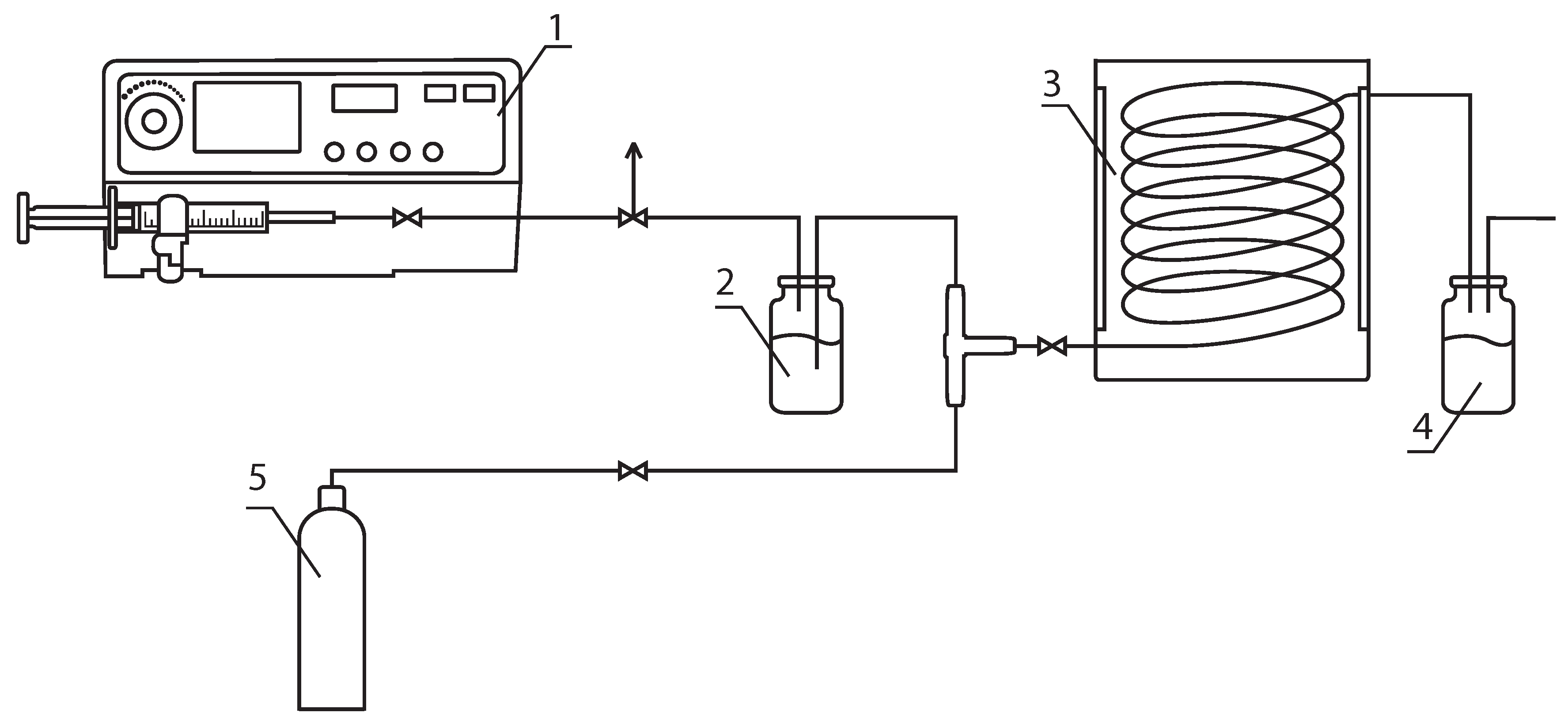
2.2. Photoiniferter RAFT Polymerization
2.3. Characterization Techniques
3. Results and Discussion
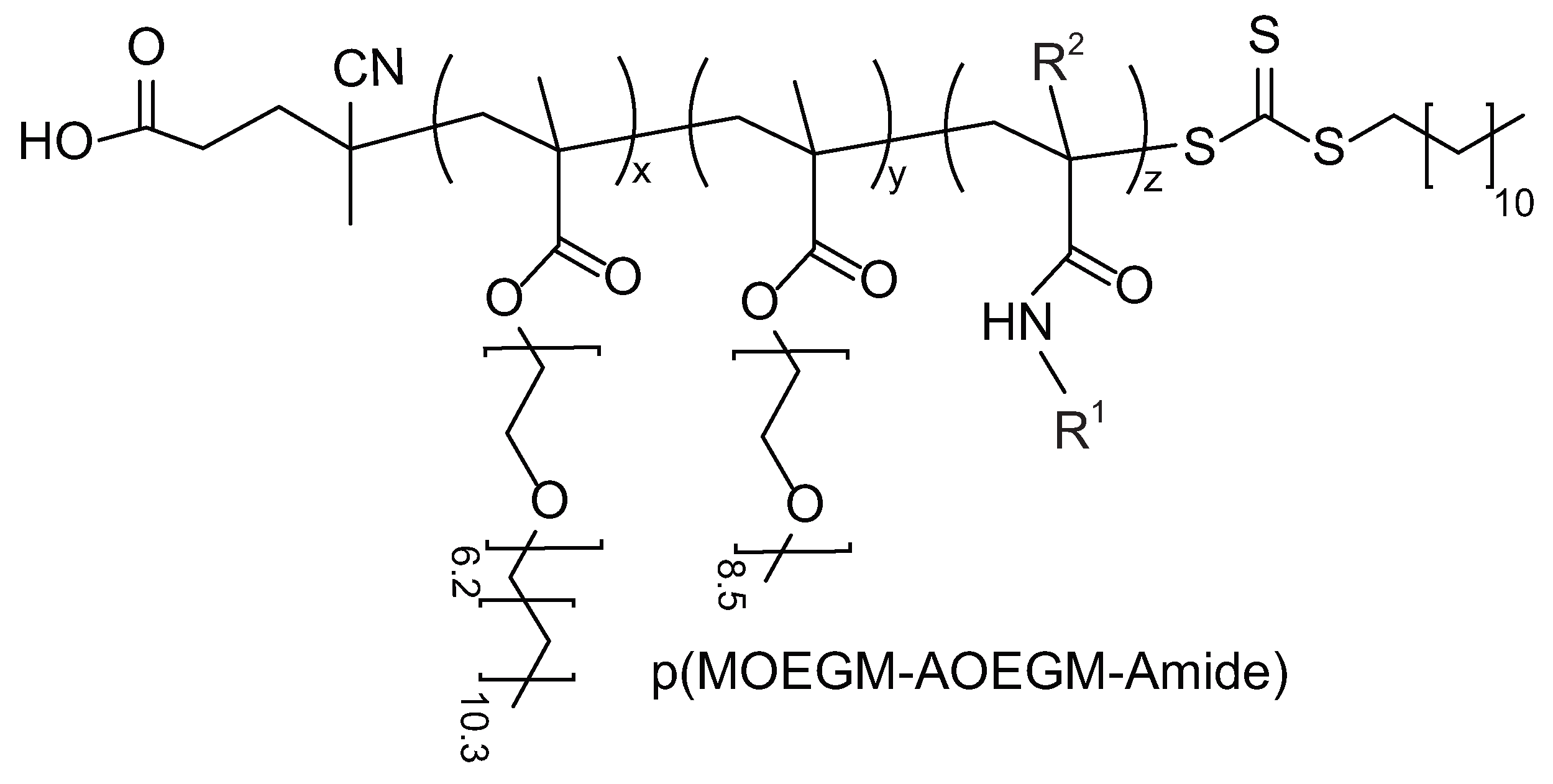
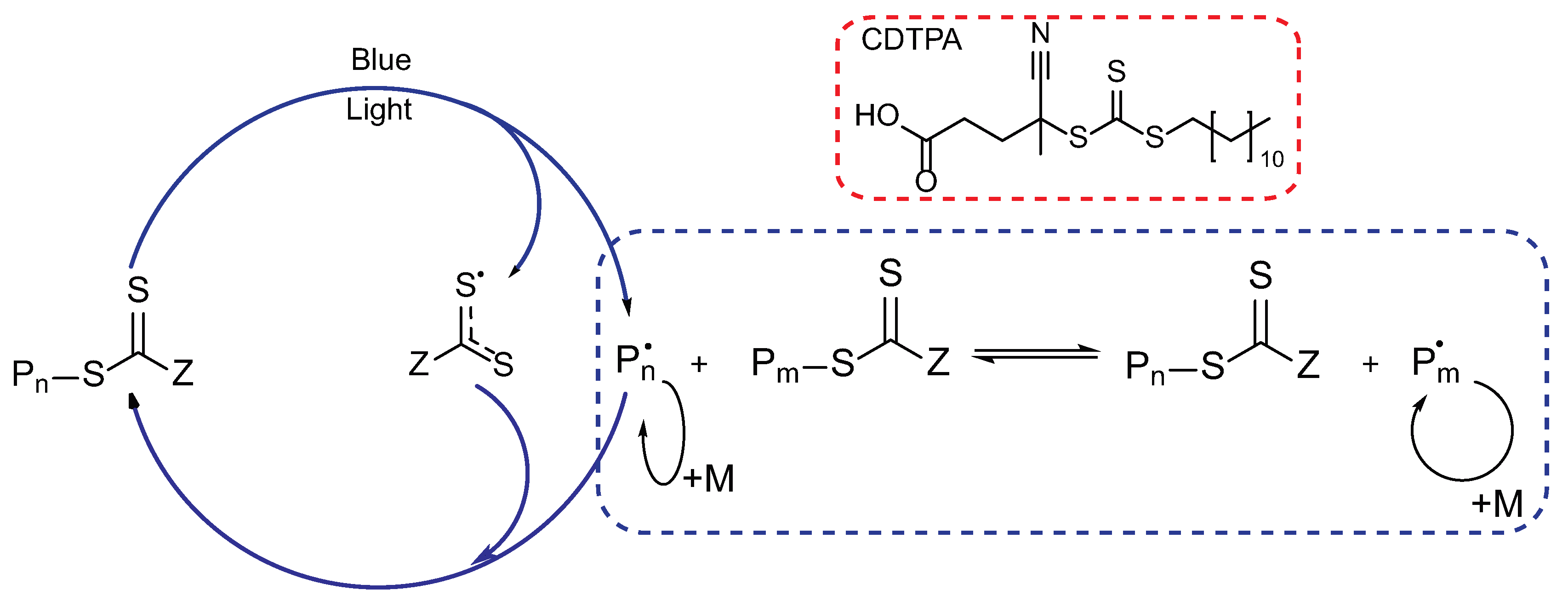
3.1. Photoiniferter RAFT Copolymerization
3.2. Thermoresponsive Properties, Hydrodynamic and Molecular Weight Characteristics of Bottlebrushes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AOEGM | alkoxy oligo(ethylene glycol) methacrylate |
| CDTPA | 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid |
| Cp | cloud point |
| DLS | dynamic light scattering |
| DSC | differential scanning calorimetry |
| LCST | lower critical solution temperature |
| LED | light-emitting diode |
| Mn,GPC | number average molecular weight determined through gel permeation chromatography |
| Mn,th | theoretical number average molecular weight |
| MOEGM | methoxy oligo(ethylene glycol) methacrylate |
| OEGMA | oligo(ethylene glycol) methacrylate |
| PET-RAFT | photo-induced electron/energy transfer RAFT |
| RAFT | reversible addition–fragmentation chain transfer |
| SLS | static light scattering |
References
- Yagci, Y.; Jockusch, S.; Turro, N.J. Photoinitiated Polymerization: Advances, Challenges, and Opportunities. Macromolecules 2010, 43, 6245–6260. [Google Scholar] [CrossRef]
- Xu, J.; Jung, K.; Atme, A.; Shanmugam, S.; Boyer, C. A Robust and Versatile Photoinduced Living Polymerization of Conjugated and Unconjugated Monomers and Its Oxygen Tolerance. JACS 2014, 136, 5508–5519. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, T.G.; Fu, Q.; Wong, E.H.H.; Dunstan, D.E.; Qiao, G.G. Visible Light Mediated Controlled Radical Polymerization in the Absence of Exogenous Radical Sources or Catalysts. Macromolecules 2015, 48, 3864–3872. [Google Scholar] [CrossRef]
- Corrigan, N.; Xu, J.; Boyer, C. A Photoinitiation System for Conventional and Controlled Radical Polymerization at Visible and NIR Wavelengths. Macromolecules 2016, 49, 3274–3285. [Google Scholar] [CrossRef]
- Arrington, K.J.; Matson, J.B. Assembly of a visible light photoreactor: An inexpensive tool for bottlebrush polymer synthesis via photoiniferter polymerization. Polym. Chem. 2017, 8, 7452–7456. [Google Scholar] [CrossRef]
- Garcia-Soto, M.J.; Haupt, K.; Gonzato, C. Synthesis of molecularly imprinted polymers by photo-iniferter polymerization under visible light. Polym. Chem. 2017, 8, 4830–4834. [Google Scholar] [CrossRef]
- Shanmugam, S.; Cuthbert, J.; Kowalewski, T.; Boyer, C.; Matyjaszewski, K. Catalyst-Free Selective Photoactivation of RAFT Polymerization: A Facile Route for Preparation of Comblike and Bottlebrush Polymers. Macromolecules 2018, 51, 7776–7784. [Google Scholar] [CrossRef]
- Lamb, J.R.; Qin, K.P.; Johnson, J.A. Visible-light-mediated, additive-free, and open-to-air controlled radical polymerization of acrylates and acrylamides. Polym. Chem. 2019, 10, 1585–1590. [Google Scholar] [CrossRef]
- Nomeir, B.; Fabre, O.; Ferji, K. Effect of Tertiary Amines on the Photoinduced Electron Transfer-Reversible Addition–Fragmentation Chain Transfer (PET-RAFT) Polymerization. Macromolecules 2019, 52, 6898–6903. [Google Scholar] [CrossRef]
- Yang, Q.; Ladmiral, V.; Améduri, B. PhotoRAFT Polymerization of Vinylidene Fluoride Using a Household White LED as Light Source at Room Temperature. ChemPhotoChem 2019, 3, 1095–1099. [Google Scholar] [CrossRef]
- Carmean, R.N.; Sims, M.B.; Figg, C.A.; Hurst, P.J.; Patterson, J.P.; Sumerlin, B.S. Ultrahigh Molecular Weight Hydrophobic Acrylic and Styrenic Polymers through Organic-Phase Photoiniferter-Mediated Polymerization. ACS Macro Lett. 2020, 9, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.-F.; Wang, L.-H.; You, Y.-Z. Photoinduced electron transfer-reversible addition-fragmentation chain transfer (PET-RAFT) polymerization using titanium dioxide. Macromol. Res. 2016, 24, 811–815. [Google Scholar] [CrossRef]
- Corrigan, N.; Rosli, D.; Jones, J.W.J.; Xu, J.; Boyer, C. Oxygen Tolerance in Living Radical Polymerization: Investigation of Mechanism and Implementation in Continuous Flow Polymerization. Macromolecules 2016, 49, 6779–6789. [Google Scholar] [CrossRef]
- Corrigan, N.; Trujillo, F.J.; Xu, J.; Moad, G.; Hawker, C.J.; Boyer, C. Divergent Synthesis of Graft and Branched Copolymers through Spatially Controlled Photopolymerization in Flow Reactors. Macromolecules 2021, 54, 3430–3446. [Google Scholar] [CrossRef]
- Shanmugam, S.; Xu, J.; Boyer, C. Exploiting Metalloporphyrins for Selective Living Radical Polymerization Tunable over Visible Wavelengths. JACS 2015, 137, 9174–9185. [Google Scholar] [CrossRef]
- Zaquen, N.; Azizi, W.A.A.W.; Yeow, J.; Kuchel, R.P.; Junkers, T.; Zetterlund, P.B.; Boyer, C. Alcohol-based PISA in batch and flow: Exploring the role of photoinitiators. Polym. Chem. 2019, 10, 2406–2414. [Google Scholar] [CrossRef]
- Corrigan, N.; Zhernakov, L.; Hashim, M.H.; Xu, J.; Boyer, C. Flow mediated metal-free PET-RAFT polymerisation for upscaled and consistent polymer production. React. Chem. Eng. 2019, 4, 1216–1228. [Google Scholar] [CrossRef]
- Li, M.; Zhang, Y.; Zhang, J.; Peng, M.; Yan, L.; Tang, Z.; Wu, Q. Continuous Gas–Liquid–Solid Slug Flow for Sustainable Heterogeneously Catalyzed PET-RAFT Polymerization. Ind. Eng. Chem. Res. 2021, 60, 5451–5462. [Google Scholar] [CrossRef]
- Li, R.; Kong, W.; An, Z. Controlling Radical Polymerization with Biocatalysts. Macromolecules 2023, 56, 751–761. [Google Scholar] [CrossRef]
- Li, R.; Zhang, S.; Li, Q.; Qiao, G.G.; An, Z. An Atom-Economic Enzymatic Cascade Catalysis for High-Throughput RAFT Synthesis of Ultrahigh Molecular Weight Polymers. Angew. Chem. Int. Ed. 2022, 61, e202213396. [Google Scholar] [CrossRef]
- Li, R.; Kong, W.; An, Z. Enzyme Catalysis for Reversible Deactivation Radical Polymerization. Angew. Chem. Int. Ed. 2022, 61, e202202033. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.-B.; Liu, D.-D.; Chen, Y.; Zhang, L.; Tan, J.-B. Enzyme-assisted Photoinitiated Polymerization-induced Self-assembly in Continuous Flow Reactors with Oxygen Tolerance. Chin. J. Polym. Sci. 2021, 39, 1127–1137. [Google Scholar] [CrossRef]
- Li, R.; An, Z. Achieving Ultrahigh Molecular Weights with Diverse Architectures for Unconjugated Monomers through Oxygen-Tolerant Photoenzymatic RAFT Polymerization. Angew. Chem. Int. Ed. 2020, 59, 22258–22264. [Google Scholar] [CrossRef] [PubMed]
- Navarro, L.A.; Enciso, A.E.; Matyjaszewski, K.; Zauscher, S. Enzymatically Degassed Surface-Initiated Atom Transfer Radical Polymerization with Real-Time Monitoring. JACS 2019, 141, 3100–3109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, X.; Zhu, A.; Ma, K.; Lv, Y.; Wang, X.; An, Z. Enzyme-Initiated Reversible Addition–Fragmentation Chain Transfer Polymerization. Macromolecules 2015, 48, 7792–7802. [Google Scholar] [CrossRef]
- Hollmann, F.; Arends, I.W.C.E. Enzyme Initiated Radical Polymerizations. Polymers 2012, 4, 759–793. [Google Scholar] [CrossRef]
- Liu, W.; Li, Q.; Zhang, Y.; Liu, T.; Wang, L.; Li, H.; Hu, Y. Continuous-flow RAFT copolymerization of styrene and maleic anhydride: Acceleration of reaction and effect of polymerization conditions on reaction kinetics. J. Flow Chem. 2021, 11, 867–875. [Google Scholar] [CrossRef]
- Wang, J.; Hu, X.; Zhu, N.; Guo, K. Continuous flow photo-RAFT and light-PISA. Chem. Eng. J. 2021, 420, 127663. [Google Scholar] [CrossRef]
- Yeo, J.; Woo, J.; Choi, S.; Kwon, K.; Lee, J.-K.; Kim, M. Comprehensive studies of continuous flow reversible addition–fragmentation chain transfer copolymerization and its application for photoimaging materials. Polym. Chem. 2022, 13, 4535–4546. [Google Scholar] [CrossRef]
- Parkinson, S.; Knox, S.T.; Bourne, R.A.; Warren, N.J. Rapid production of block copolymer nano-objects via continuous-flow ultrafast RAFT dispersion polymerisation. Polym. Chem. 2020, 11, 3465–3474. [Google Scholar] [CrossRef]
- Wenn, B.; Junkers, T. Continuous Microflow PhotoRAFT Polymerization. Macromolecules 2016, 49, 6888–6895. [Google Scholar] [CrossRef]
- Parkinson, S.; Hondow, N.S.; Conteh, J.S.; Bourne, R.A.; Warren, N.J. All-aqueous continuous-flow RAFT dispersion polymerisation for efficient preparation of diblock copolymer spheres, worms and vesicles. React. Chem. Eng. 2019, 4, 852–861. [Google Scholar] [CrossRef]
- Peng, J.; Xu, Q.; Ni, Y.; Zhang, L.; Cheng, Z.; Zhu, X. Visible light controlled aqueous RAFT continuous flow polymerization with oxygen tolerance. Polym. Chem. 2019, 10, 2064–2072. [Google Scholar] [CrossRef]
- Chen, M.; Johnson, J.A. Improving photo-controlled living radical polymerization from trithiocarbonates through the use of continuous-flow techniques. Chem. Commun. 2015, 51, 6742–6745. [Google Scholar] [CrossRef] [PubMed]
- Lauterbach, F.; Rubens, M.; Abetz, V.; Junkers, T. Ultrafast PhotoRAFT Block Copolymerization of Isoprene and Styrene Facilitated through Continuous-Flow Operation. Angew. Chem. Int. Ed. 2018, 57, 14260–14264. [Google Scholar] [CrossRef] [PubMed]
- Rubens, M.; Latsrisaeng, P.; Junkers, T. Visible light-induced iniferter polymerization of methacrylates enhanced by continuous flow. Polym. Chem. 2017, 8, 6496–6505. [Google Scholar] [CrossRef]
- Chen, K.; Han, W.; Hu, X.; Liu, Y.; Hu, Y.; Zhao, S.; Zhu, N.; Fang, Z.; Guo, K. Microreactor-based chemo-enzymatic ROP-ROMP platform for continuous flow synthesis of bottlebrush polymers. Chem. Eng. J. 2022, 437, 135284. [Google Scholar] [CrossRef]
- Xiang, L.; Qiu, M.; Shang, M.; Su, Y. Continuous synthesis of star polymers with RAFT polymerization in cascade microreactor systems. Polymer 2021, 222, 123669. [Google Scholar] [CrossRef]
- Zaquen, N.; Kadir, A.M.N.B.P.H.A.; Iasa, A.; Corrigan, N.; Junkers, T.; Zetterlund, P.B.; Boyer, C. Rapid Oxygen Tolerant Aqueous RAFT Photopolymerization in Continuous Flow Reactors. Macromolecules 2019, 52, 1609–1619. [Google Scholar] [CrossRef]
- Rubens, M.; Vrijsen, J.H.; Laun, J.; Junkers, T. Precise Polymer Synthesis by Autonomous Self-Optimizing Flow Reactors. Angew. Chem. Int. Ed. 2019, 58, 3183–3187. [Google Scholar] [CrossRef]
- Harmon, M.E.; Tang, M.; Frank, C.W. A microfluidic actuator based on thermoresponsive hydrogels. Polymer 2003, 44, 4547–4556. [Google Scholar] [CrossRef]
- Schmaljohann, D. Thermo- and pH-responsive polymers in drug delivery. Adv. Drug Deliv. Rev. 2006, 58, 1655–1670. [Google Scholar] [CrossRef] [PubMed]
- Crespy, D.; Rossi, R.M. Temperature-responsive polymers with LCST in the physiological range and their applications in textiles. Polym. Int. 2007, 56, 1461–1468. [Google Scholar] [CrossRef]
- Teotia, A.K.; Sami, H.; Kumar, A. 1—Thermo-responsive polymers: Structure and design of smart materials. In Switchable and Responsive Surfaces and Materials for Biomedical Applications; Zhang, Z., Ed.; Woodhead Publishing: Oxford, UK, 2015; pp. 3–43. [Google Scholar]
- Kim, Y.-J.; Matsunaga, Y.T. Thermo-responsive polymers and their application as smart biomaterials. J. Mater. Chem. B 2017, 5, 4307–4321. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.-F.; Akdemir, Ö.; Hoth, A. Point by Point Comparison of Two Thermosensitive Polymers Exhibiting a Similar LCST: Is the Age of Poly(NIPAM) Over? JACS 2006, 128, 13046–13047. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Wu, F.; Wang, E. Thermosensitive Behavior of Poly(ethylene Glycol)-Based Block Copolymer (PEG-b-PADMO) Controlled via Self-Assembled Microstructure. J. Phys. Chem. B 2011, 115, 5913–5922. [Google Scholar] [CrossRef]
- Xu, Y.; Xie, J.; Chen, L.; Yuan, C.; Pan, Y.; Cheng, L.; Luo, W.; Zeng, B.; Dai, L. A novel hybrid random copolymer poly(MAPOSS-co-NIPAM-co-OEGMA-co-2VP): Synthesis, characterization, self-assembly behaviors and multiple responsive properties. Macromol. Res. 2013, 21, 1338–1348. [Google Scholar] [CrossRef]
- Roy, D.; Brooks, W.L.A.; Sumerlin, B.S. New directions in thermoresponsive polymers. Chem. Soc. Rev. 2013, 42, 7214–7243. [Google Scholar] [CrossRef]
- Lutz, J.-F. Polymerization of oligo(ethylene glycol) (meth)acrylates: Toward new generations of smart biocompatible materials. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 3459–3470. [Google Scholar] [CrossRef]
- Lutz, J.-F.; Hoth, A.; Schade, K. Design of Oligo(ethylene glycol)-Based Thermoresponsive Polymers: An Optimization Study. Des. Monomers Polym. 2009, 12, 343–353. [Google Scholar] [CrossRef]
- Becer, C.R.; Hahn, S.; Fijten, M.W.M.; Thijs, H.M.L.; Hoogenboom, R.; Schubert, U.S. Libraries of methacrylic acid and oligo(ethylene glycol) methacrylate copolymers with LCST behavior. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 7138–7147. [Google Scholar] [CrossRef]
- Fournier, D.; Hoogenboom, R.; Thijs, H.M.L.; Paulus, R.M.; Schubert, U.S. Tunable pH- and Temperature-Sensitive Copolymer Libraries by Reversible Addition−Fragmentation Chain Transfer Copolymerizations of Methacrylates. Macromolecules 2007, 40, 915–920. [Google Scholar] [CrossRef]
- Hoogenboom, R.; Becer, C.R.; Hahn, S.; Fournier, D.J.R.; Schubert, U.S. Thermo- and pH-responsive copolymers based on oligoethyleneglycol methacrylates. Polym. Prepr. 2007, 48, 161–162. [Google Scholar]
- Liu, M.; Leroux, J.-C.; Gauthier, M.A. Conformation–function relationships for the comb-shaped polymer pOEGMA. Prog. Polym. Sci. 2015, 48, 111–121. [Google Scholar] [CrossRef]
- Zhang, X.; Dai, Y. Recent development of brush polymers via polymerization of poly(ethylene glycol)-based macromonomers. Polym. Chem. 2019, 10, 2212–2222. [Google Scholar] [CrossRef]
- Badi, N. Non-linear PEG-based thermoresponsive polymer systems. Prog. Polym. Sci. 2017, 66, 54–79. [Google Scholar] [CrossRef]
- Vancoillie, G.; Frank, D.; Hoogenboom, R. Thermoresponsive poly(oligo ethylene glycol acrylates). Prog. Polym. Sci. 2014, 39, 1074–1095. [Google Scholar] [CrossRef]
- Terashima, T.; Sugita, T.; Fukae, K.; Sawamoto, M. Synthesis and Single-Chain Folding of Amphiphilic Random Copolymers in Water. Macromolecules 2014, 47, 589–600. [Google Scholar] [CrossRef]
- Terashima, T.; Sugita, T.; Sawamoto, M. Single-chain crosslinked star polymers via intramolecular crosslinking of self-folding amphiphilic copolymers in water. Polym. J. 2015, 47, 667. [Google Scholar] [CrossRef]
- Sivokhin, A.; Orekhov, D.; Kazantsev, O.; Sivokhina, O.; Orekhov, S.; Kamorin, D.; Otopkova, K.; Smirnov, M.; Karpov, R. Random and Diblock Thermoresponsive Oligo(ethylene glycol)-Based Copolymers Synthesized via Photo-Induced RAFT Polymerization. Polymers 2022, 14, 137. [Google Scholar] [CrossRef]
- Sivokhin, A.Р.; Orekhov, D.V.; Kazantsev, O.A.; Gubanova, O.V.; Kamorin, D.M.; Zarubina, I.S.; Bolshakova, E.A.; Zaitsev, S.D. Amphiphilic thermoresponsive copolymer bottlebrushes: Synthesis, characterization, and study of their self-assembly into flower-like micelles. Polym. J. 2021, 53, 655–665. [Google Scholar] [CrossRef]
- Orekhov, D.V.; Kazantsev, O.A.; Orekhov, S.V.; Sivokhin, A.P.; Kamorin, D.M.; Simagin, A.S.; Savinova, M.V.; Bolshakova, E.A.; Korotaev, M.S. Synthesis of amphiphilic (meth)acrylates with oligo(ethylene glycol) and (or) oligo(propylene glycol) blocks by the esterification of (meth)acrylic acid. J. Appl. Polym. Sci. 2021, 138, 50982. [Google Scholar] [CrossRef]
- Moad, G.; Chong, Y.K.; Postma, A.; Rizzardo, E.; Thang, S.H. Advances in RAFT polymerization: The synthesis of polymers with defined end-groups. Polymer 2005, 46, 8458–8468. [Google Scholar] [CrossRef]
- Sivokhin, A.Р.; Kazantsev, O.A. Temperature Dependence of the Rayleigh Ratio for Toluene: Thermoresponsive Polymers Characterization. ChemistrySelect 2021, 6, 9499–9502. [Google Scholar] [CrossRef]
- Lutz, J.-F. Thermo-Switchable Materials Prepared Using the OEGMA-Platform. Adv. Mater. 2011, 23, 2237–2243. [Google Scholar] [CrossRef]
- Hartlieb, M. Photo-Iniferter RAFT Polymerization. Macromol. Rapid Commun. 2022, 43, 2100514. [Google Scholar] [CrossRef]
- Matsumoto, M.; Takenaka, M.; Sawamoto, M.; Terashima, T. Self-assembly of amphiphilic block pendant polymers as microphase separation materials and folded flower micelles. Polym. Chem. 2019, 10, 4954–4961. [Google Scholar] [CrossRef]
- Hattori, G.; Hirai, Y.; Sawamoto, M.; Terashima, T. Self-assembly of PEG/dodecyl-graft amphiphilic copolymers in water: Consequences of the monomer sequence and chain flexibility on uniform micelles. Polym. Chem. 2017, 8, 7248–7259. [Google Scholar] [CrossRef]
- Zehm, D.; Laschewsky, A.; Heunemann, P.; Gradzielski, M.; Prévost, S.; Liang, H.; Rabe, J.P.; Lutz, J.-F. Synthesis and self-assembly of amphiphilic semi-brush and dual brush block copolymers in solution and on surfaces. Polym. Chem. 2011, 2, 137–147. [Google Scholar] [CrossRef]
- Sivokhin, A.; Orekhov, D.; Kazantsev, O.; Otopkova, K.; Sivokhina, O.; Chesnokov, Y.; Smirnov, M.; Ovchinnikov, A.; Makhov, I. High-molecular weight bottlebrushes via continuous flow photoiniferter polymerization of macromonomers. Polym. Chem. 2023, 14, 3186–3195. [Google Scholar] [CrossRef]
- Rasines Mazo, A.; Tran, T.N.; Zhang, W.; Meng, Y.; Reyhani, A.; Pascual, S.; Fontaine, L.; Qiao, G.G.; Piogé, S. Blue LED light-activated RAFT polymerization of PEG acrylate with high chain-end fidelity for efficient PEGylation. Polym. Chem. 2020, 11, 5238–5248. [Google Scholar] [CrossRef]
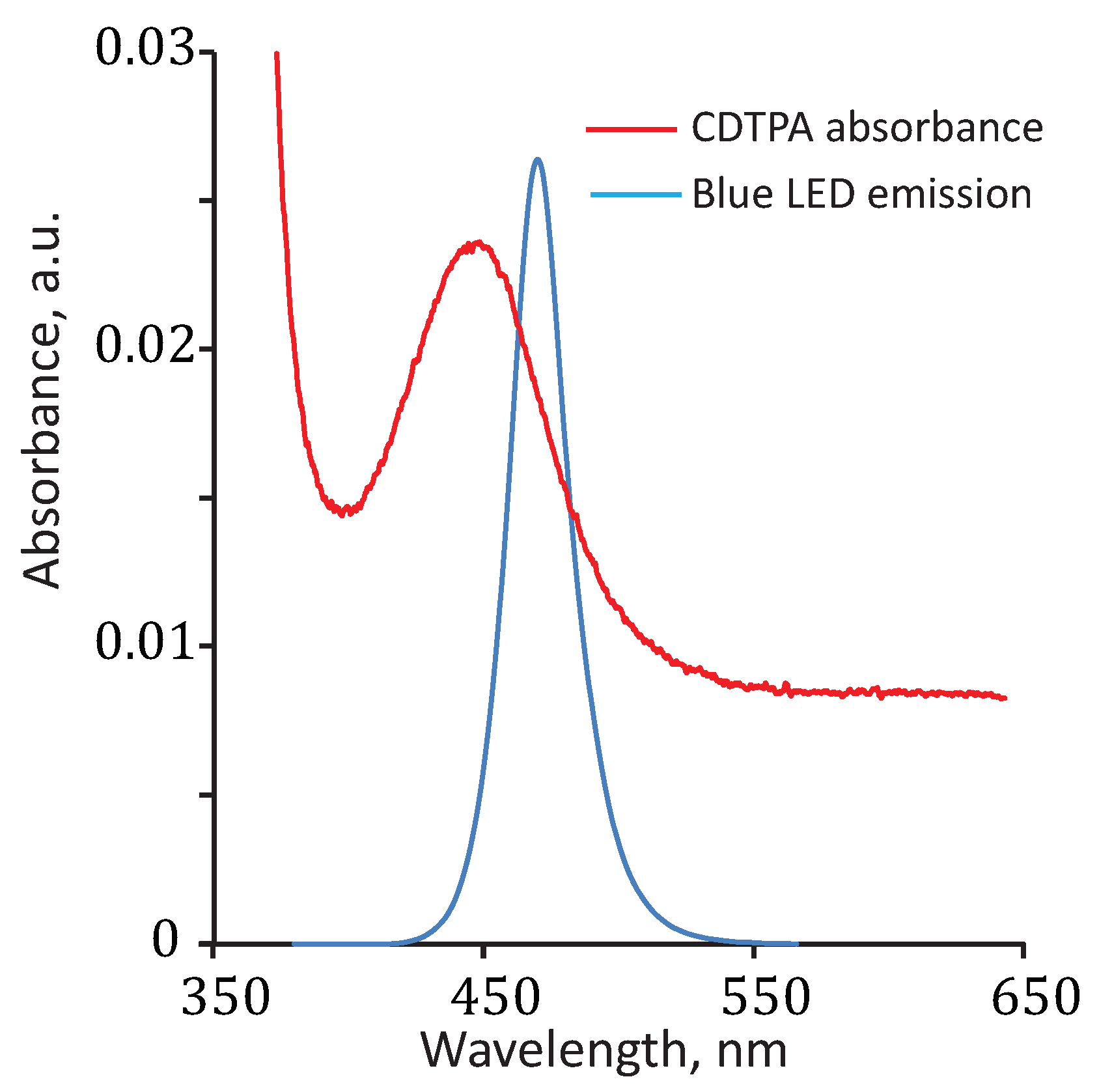
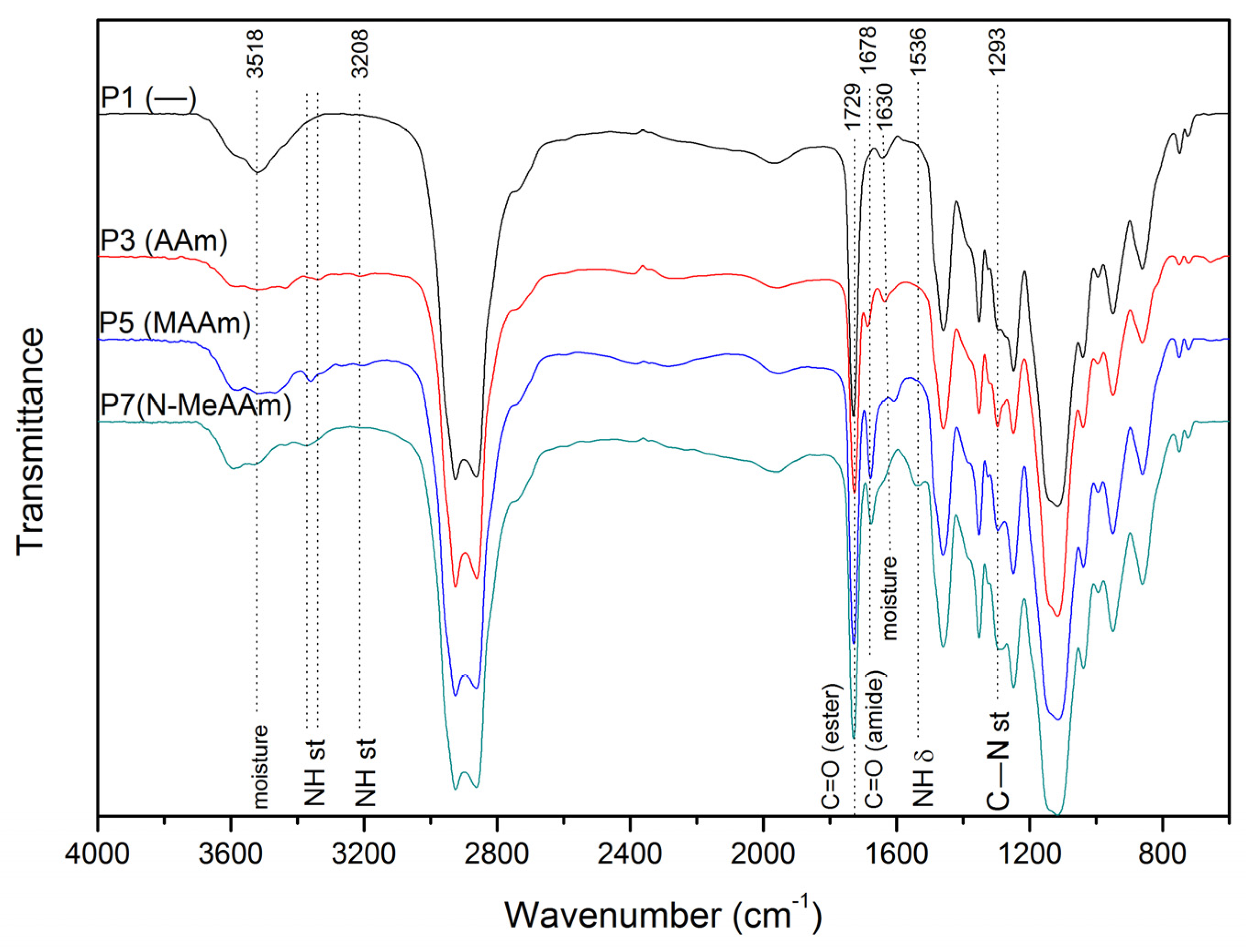
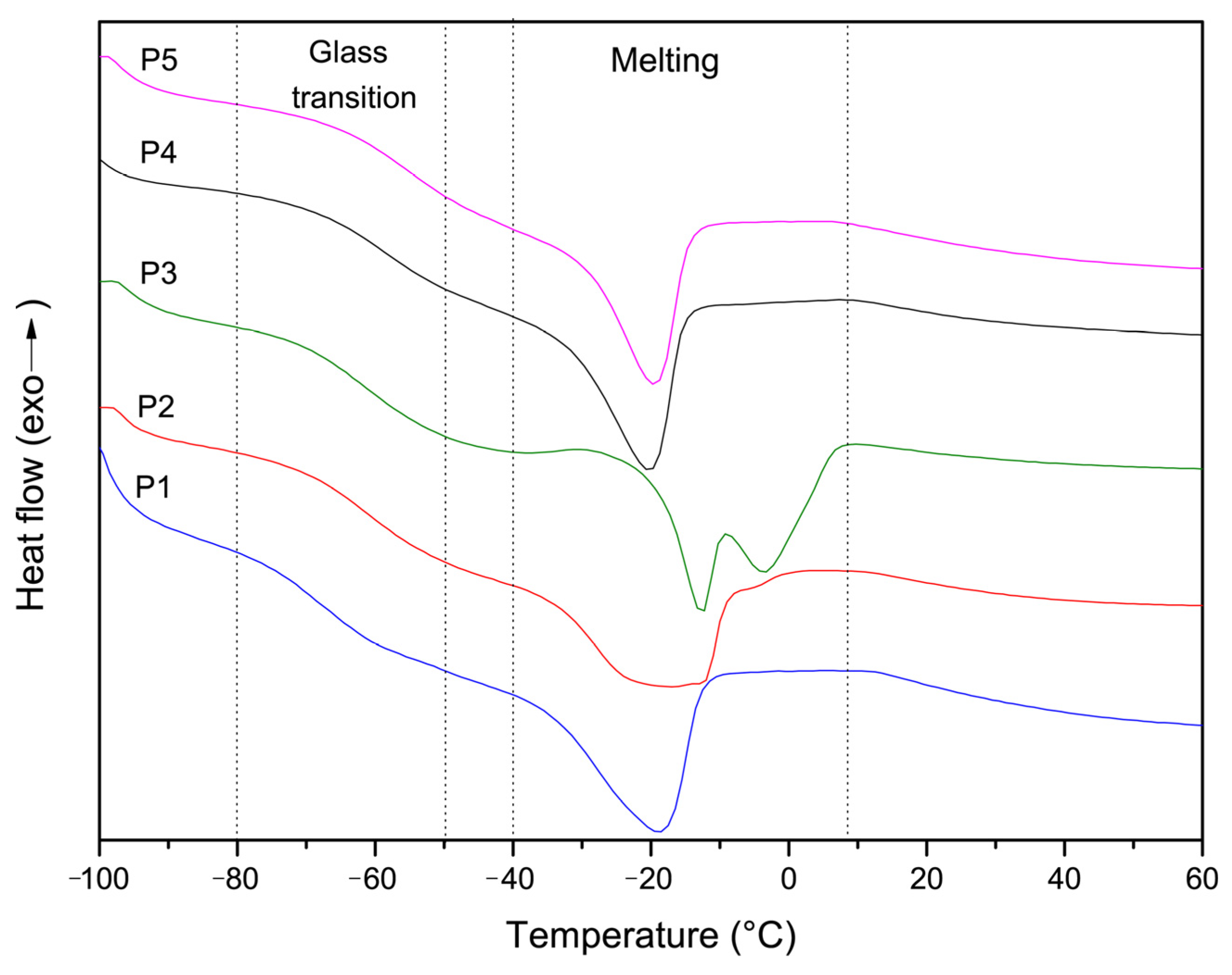

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | [MOEGM]0 /[AOEGM]0 /[M]0 | Comonomer (M) | Solvent | Flow Rate, mL/min | Time, min | Conversion, % | Composition a, m1:m2:m3 (mol) | Mn, th b | Mn c | Mw c | Đ c (Mw/Mn) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | 50:50:0 | - | DMSO | 9 | 137 | 91 | 49:51:0 | 95,400 | 8900 | 11,400 | 1.28 |
| P2 | 45:45:10 | AAm | THF | 9 | 115 | 54 | 47.5:46.3:6.2 | 53,800 | 8500 | 10,500 | 1.23 |
| P3 | 40:40:20 | AAm | THF | 9 | 111 | 42 | 43.2:44.3:12.4 | 39,800 | 8200 | 9900 | 1.21 |
| P4 | 45:45:10 | MAAm | THF | 9 | 102 | 62 | 47.6:45.2:7.2 | 61,200 | 8100 | 9900 | 1.22 |
| P5 | 40:40:20 | MAAm | THF | 9 | 111 | 50 | 44.5:41.6:13.8 | 46,600 | 6600 | 7800 | 1.18 |
| P6 | 45:45:10 | N-MeAAm | THF | 8 | 124 | 55 | 48.3:46.4:5.3 | 54,500 | 9500 | 11,700 | 1.23 |
| P7 | 40:40:20 | N-MeAAm | THF | 7 | 145 | 41 | 45.3:44.0:10.7 | 40,150 | 9600 | 11,700 | 1.22 |
| Copolymer ID | Tg, °C | Tm, °C |
|---|---|---|
| P1 (MOEGM-AOEGM) | −68.8 | −18.8 |
| P2 (MOEGM-AOEGM-AAm) | −61.1 | −16.8 |
| P3 (MOEGM-AOEGM-AAm) | −62.2 | −12.6/−3.8 |
| P4 (MOEGM-AOEGM-MAAm) | −63.7 | −20.2 |
| P5 (MOEGM-AOEGM-MAAm) | −60.1 | −19.4 |
| Copolymer ID | Cp, °C | CMC (mg/mL) | Loading Capacity (mg Pyrene/g Polymer) |
|---|---|---|---|
| P1 | 56.1 | 1.35 × 10−3 | 22.1 |
| P2 | 55.7 | 1.52 × 10−3 | 23.3 |
| P3 | 39.7 | 1.55 × 10−3 | 23.4 |
| P4 | 55.8 | 1.13 × 10−3 | 21.7 |
| P5 | 59.0 | 1.26 × 10−3 | 21.0 |
| P6 | 55.9 | 1.43 × 10−3 | 21.7 |
| P7 | 53.7 | 1.26 × 10−3 | 20.3 |
| ID | Hydrodynamic Radius, Rh, nm a | b | mole·cm3·g−2 b | b | mole·cm3·g−2 b | Nagg | |
|---|---|---|---|---|---|---|---|
| Acetonitrile | Water | ||||||
| P1 | 8.4 | 5.8 | 224,500 | 0.42 | 223,300 | 0.31 | 1.0 |
| P3 | 6.3; 21.0 | 6.7; 25 | 168,600 | 0.78 | 406,400 | 0.19 | 2.4 |
| P2 | 5.4; 19.6 | 6.8; 26 | 150,500 | 0.05 | 413,300 | −0.13 | 2.7 |
| P4 | 6.3 | 5.0 | 95,600 | 1.45 | 166,200 | −0.49 | 1.7 |
| P5 | 6.2 | 3.7 | 80,000 | 1.61 | 144,400 | −0.38 | 1.8 |
| P6 | 6.2 | 5.0 | 104,700 | 1.07 | 180,200 | −0.03 | 1.7 |
| P7 | 6.8 | 5.8 | 106,500 | −0.50 | 172,600 | −0.31 | 1.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sivokhin, A.; Orekhov, D.; Kazantsev, O.; Otopkova, K.; Sivokhina, O.; Chuzhaykin, I.; Ovchinnikov, A.; Zamyshlyayeva, O.; Pavlova, I.; Ozhogina, O.; et al. Amide-Containing Bottlebrushes via Continuous-Flow Photoiniferter Reversible Addition–Fragmentation Chain Transfer Polymerization: Micellization Behavior. Polymers 2024, 16, 134. https://doi.org/10.3390/polym16010134
Sivokhin A, Orekhov D, Kazantsev O, Otopkova K, Sivokhina O, Chuzhaykin I, Ovchinnikov A, Zamyshlyayeva O, Pavlova I, Ozhogina O, et al. Amide-Containing Bottlebrushes via Continuous-Flow Photoiniferter Reversible Addition–Fragmentation Chain Transfer Polymerization: Micellization Behavior. Polymers. 2024; 16(1):134. https://doi.org/10.3390/polym16010134
Chicago/Turabian StyleSivokhin, Alexey, Dmitry Orekhov, Oleg Kazantsev, Ksenia Otopkova, Olga Sivokhina, Ilya Chuzhaykin, Alexey Ovchinnikov, Olga Zamyshlyayeva, Irina Pavlova, Olga Ozhogina, and et al. 2024. "Amide-Containing Bottlebrushes via Continuous-Flow Photoiniferter Reversible Addition–Fragmentation Chain Transfer Polymerization: Micellization Behavior" Polymers 16, no. 1: 134. https://doi.org/10.3390/polym16010134
APA StyleSivokhin, A., Orekhov, D., Kazantsev, O., Otopkova, K., Sivokhina, O., Chuzhaykin, I., Ovchinnikov, A., Zamyshlyayeva, O., Pavlova, I., Ozhogina, O., & Chubenko, M. (2024). Amide-Containing Bottlebrushes via Continuous-Flow Photoiniferter Reversible Addition–Fragmentation Chain Transfer Polymerization: Micellization Behavior. Polymers, 16(1), 134. https://doi.org/10.3390/polym16010134