Natural Fibrous Materials Based on Fungal Mycelium Hyphae as Porous Supports for Shape-Stable Phase-Change Composites

 ,
,  , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Preparation of Microfibrillar cellulose
2.2.2. Strain and Culture Conditions
Solid-State Fermentation of Composite Mycelium/Cellulose Fibers
Liquid-State Surface Fermentation of Pure Mycelium Fibers
2.2.3. Preparation of Phase-Change Composites
2.2.4. Characterization
3. Results and Discussions
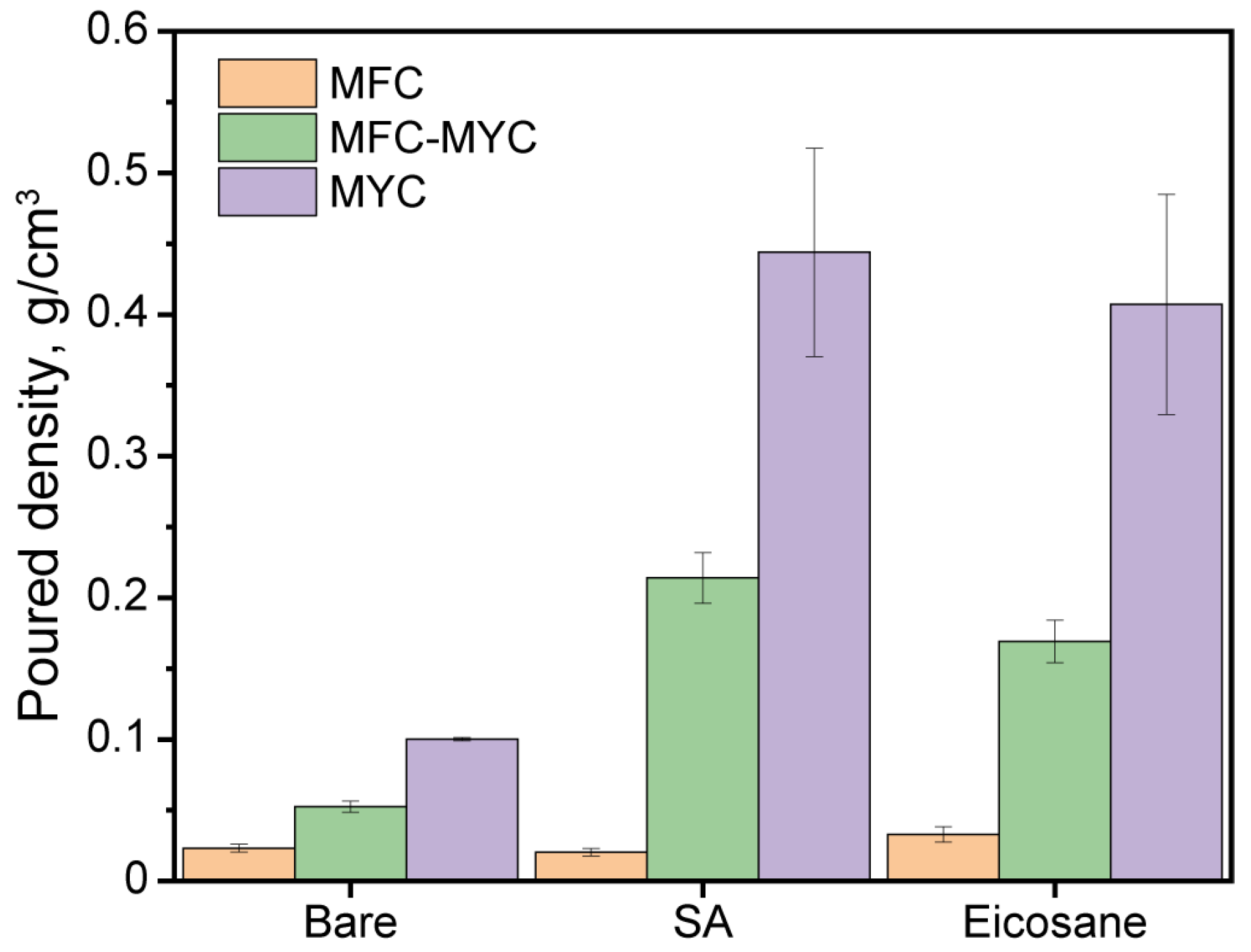
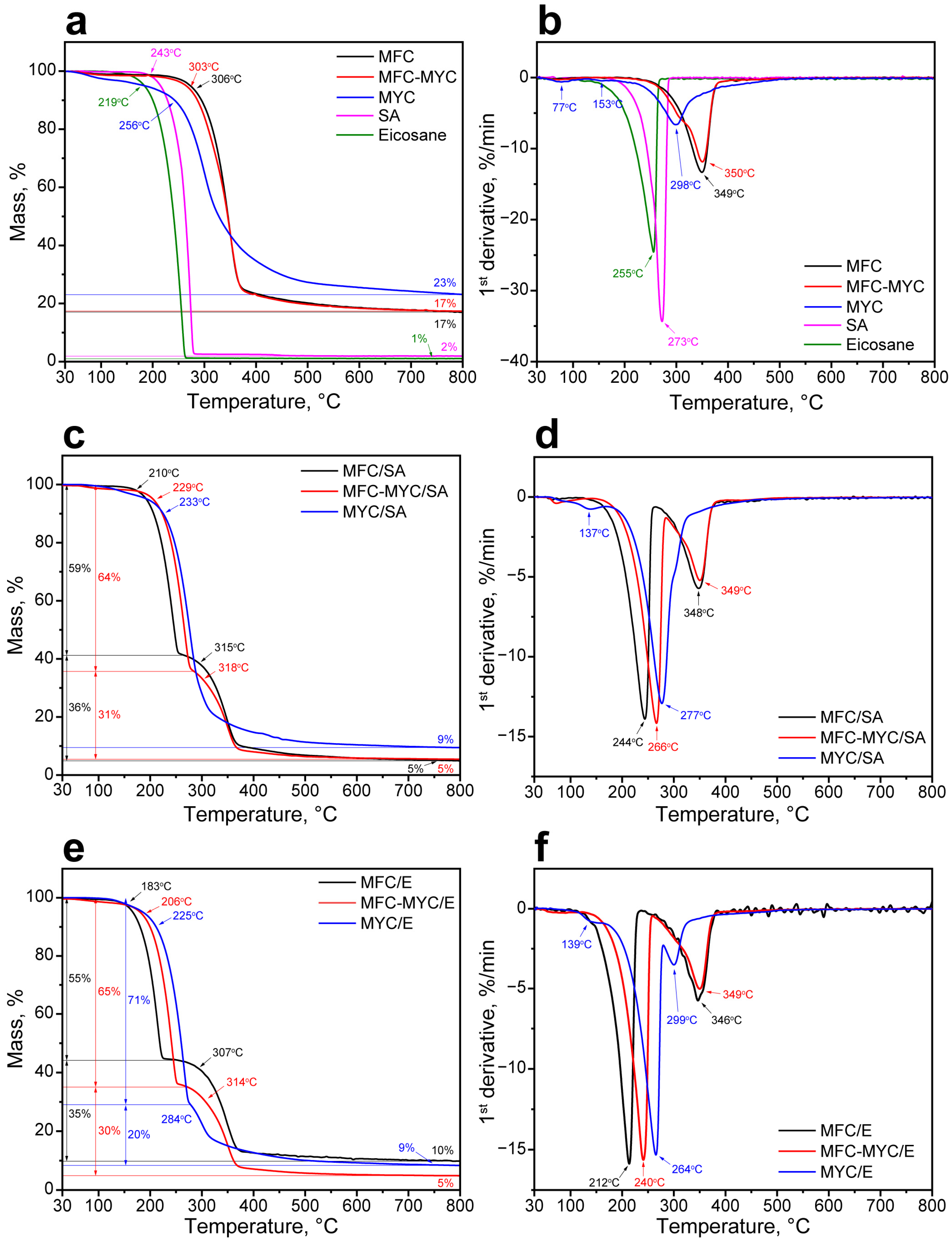
3.1. Preparation and Characterization of the Mycelium-Based Fibers
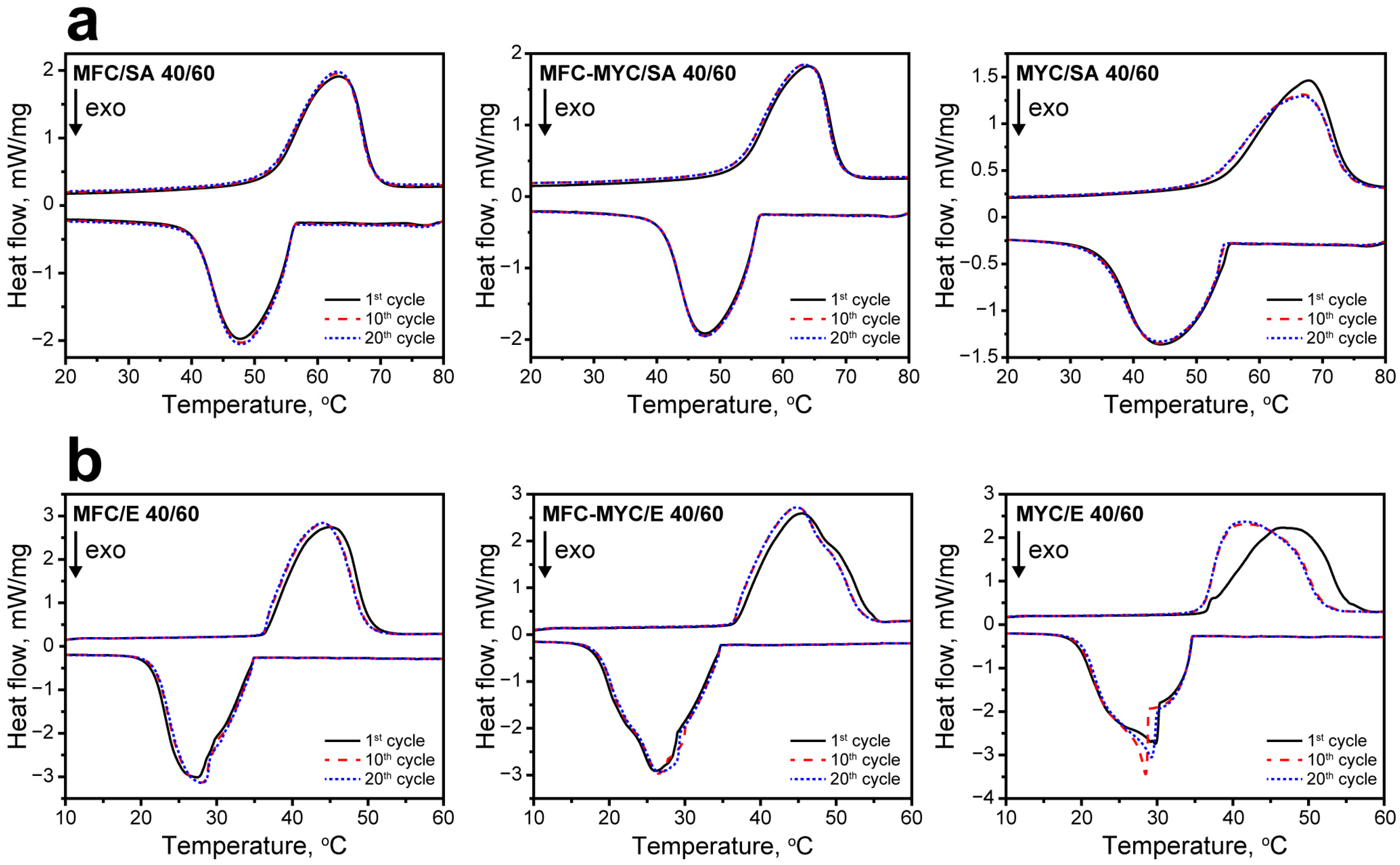
3.2. Preparation and Study of the Phase-Change Composites
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Voronin, D.V.; Mendgaziev, R.I.; Rubtsova, M.I.; Cherednichenko, K.A.; Demina, P.A.; Abramova, A.M.; Shchukin, D.G.; Vinokurov, V. Facile Synthesis of Shape-Stable Phase-Change Composites via the Adsorption of Stearic Acid onto Cellulose Microfibers. Mater. Chem. Front. 2022, 6, 1033–1045. [Google Scholar] [CrossRef]
- Bacakova, L.; Pajorova, J.; Bacakova, M.; Skogberg, A.; Kallio, P.; Kolarova, K.; Svorcik, V. Versatile Application of Nanocellulose: From Industry to Skin Tissue Engineering and Wound Healing. Nanomaterials 2019, 9, 164. [Google Scholar] [CrossRef] [PubMed]
- Salimi, S.; Sotudeh-Gharebagh, R.; Zarghami, R.; Chan, S.Y.; Yuen, K.H. Production of Nanocellulose and Its Applications in Drug Delivery: A Critical Review. ACS Sustain. Chem. Eng. 2019, 7, 15800–15827. [Google Scholar] [CrossRef]
- Bialek, A.; Bialek, M.; Jelinska, M.; Tokarz, A. Fatty Acid Profile of New Promising Unconventional Plant Oils for Cosmetic Use. Int. J. Cosmet. Sci. 2016, 38, 382–388. [Google Scholar] [CrossRef]
- Pirtsul, A.E.; Mendgaziev, R.I.; Komlev, A.S.; Cherednichenko, K.A.; Vinokurov, V.A.; Voronin, D.V. Composite Fibers with Phase-Change Properties as Thermoregulating Additives to Dry Mortar Mixes with the Possibility of Bimodal Heating. Chem. Technol. Fuels Oils 2023, 59, 449–458. [Google Scholar] [CrossRef]
- Kedzior, S.A.; Gabriel, V.A.; Dubé, M.A.; Cranston, E.D. Nanocellulose in Emulsions and Heterogeneous Water-Based Polymer Systems: A Review. Adv. Mater. 2021, 33, e2002404. [Google Scholar] [CrossRef]
- Islam, M.R.; Tudryn, G.; Bucinell, R.; Schadler, L.; Picu, R.C. Morphology and Mechanics of Fungal Mycelium. Sci. Rep. 2017, 7, 13070. [Google Scholar] [CrossRef] [PubMed]
- Filipova, I.; Irbe, I.; Spade, M.; Skute, M.; Dāboliņa, I.; Baltiņa, I.; Vecbiskena, L. Mechanical and Air Permeability Performance of Novel Biobased Materials from Fungal Hyphae and Cellulose Fibers. Materials 2021, 14, 136. [Google Scholar] [CrossRef]
- Elsacker, E. Mycelium Matters: An Interdisciplinary Exploration of the Fabrication and Properties of Mycelium-Based Materials. Ph.D. Thesis, Vrije Universiteit Brussel, Ixelles, Belgium, 2021. [Google Scholar]
- Holt, G.A.; McIntyre, G.; Flagg, D.; Bayer, E.; Wanjura, J.D.; Pelletier, M.G. Fungal Mycelium and Cotton Plant Materials in the Manufacture of Biodegradable Molded Packaging Material: Evaluation Study of Select Blends of Cotton Byproducts. J. Biobased Mater. Bioenergy 2012, 6, 431–439. [Google Scholar] [CrossRef]
- Bruscato, C.; Malvessi, E.; Brandalise, R.N.; Camassola, M. High Performance of Macrofungi in the Production of Mycelium-Based Biofoams Using Sawdust—Sustainable Technology for Waste Reduction. J. Clean. Prod. 2019, 234, 225–232. [Google Scholar] [CrossRef]
- Karana, E.; Blauwhoff, D.; Hultink, E.J.; Camere, S. When the Material Grows: A Case Study on Designing (with) Mycelium-Based Materials. Int. J. Des. 2018, 12, 119–136. [Google Scholar]
- Jones, M.; Mautner, A.; Luenco, S.; Bismarck, A.; John, S. Engineered Mycelium Composite Construction Materials from Fungal Biorefineries: A Critical Review. Mater. Des. 2020, 187, 108397. [Google Scholar] [CrossRef]
- Attias, N.; Danai, O.; Abitbol, T.; Tarazi, E.; Ezov, N.; Pereman, I.; Grobman, Y.J. Mycelium Bio-Composites in Industrial Design and Architecture: Comparative Review and Experimental Analysis. J. Clean. Prod. 2020, 246, 119037. [Google Scholar] [CrossRef]
- Elsacker, E.; Vandelook, S.; Van Wylick, A.; Ruytinx, J.; De Laet, L.; Peeters, E. A Comprehensive Framework for the Production of Mycelium-Based Lignocellulosic Composites. Sci. Total Environ. 2020, 725, 138431. [Google Scholar] [CrossRef]
- Yang, Z.J.; Zhang, F.; Still, B.; White, M.; Amstislavski, P. Physical and Mechanical Properties of Fungal Mycelium-Based Biofoam. J. Mater. Civ. Eng. 2017, 29, 04017030. [Google Scholar] [CrossRef]
- Zimele, Z.; Irbe, I.; Grinins, J.; Bikovens, O.; Verovkins, A.; Bajare, D. Novel Mycelium-Based Biocomposites (MBB) as Building Materials. J. Renew. Mater. 2020, 8, 1067–1076. [Google Scholar] [CrossRef]
- Antinori, M.E.; Contardi, M.; Suarato, G.; Armirotti, A.; Bertorelli, R.; Mancini, G.; Debellis, D.; Athanassiou, A. Advanced Mycelium Materials as Potential Self-Growing Biomedical Scaffolds. Sci. Rep. 2021, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ruggeri, M.; Miele, D.; Contardi, M.; Vigani, B.; Boselli, C.; Icaro Cornaglia, A.; Rossi, S.; Suarato, G.; Athanassiou, A.; Sandri, G. Mycelium-Based Biomaterials as Smart Devices for Skin Wound Healing. Front. Bioeng. Biotechnol. 2023, 11, 1225722. [Google Scholar] [CrossRef] [PubMed]
- Rakitina, M.A.; Sayfutdinova, A.R.; Kozhevnikova, E.Y.; Voronin, D.V.; Vinokurov, V.A. Evaluation of Optimal Fungi Strains for Development of Mycelium-Based Biopolymeric Matrices. Chem. Technol. Fuels Oils 2023, 58, 1005–1010. [Google Scholar] [CrossRef]
- Cherednichenko, K.; Sayfutdinova, A.; Rimashevskiy, D.; Malik, B.; Panchenko, A.; Kopitsyna, M.; Ragnaev, S.; Vinokurov, V.; Voronin, D.; Kopitsyn, D. Composite Bone Cements with Enhanced Drug Elution. Polymers 2023, 15, 3757. [Google Scholar] [CrossRef]
- Cherednichenko, K.A.; Sayfutdinova, A.R.; Kraynov, A.; Anikushin, B.; Ignatiev, V.; Rubtsova, M.I.; Konstantinova, S.A.; Shchukin, D.G.; Vinokurov, V.A. A Rapid Synthesis of Nanofibrillar Cellulose/Polystyrene Composite via Ultrasonic Treatment. Ultrason. Sonochem. 2022, 90, 106180. [Google Scholar] [CrossRef]
- Sayfutdinova, A.; Samofalova, I.; Barkov, A.; Cherednichenko, K.; Rimashevskiy, D.; Vinokurov, V. Structure and Properties of Cellulose/Mycelium Biocomposites. Polymers 2022, 14, 1519. [Google Scholar] [CrossRef] [PubMed]
- Livne, A.; Mijowska, S.C.; Polishchuk, I.; Mashikoane, W.; Katsman, A.; Pokroy, B. A Fungal Mycelium Templates the Growth of Aragonite Needles. J. Mater. Chem. B 2019, 7, 5725–5731. [Google Scholar] [CrossRef] [PubMed]
- Kuribayashi, T.; Lankinen, P.; Hietala, S.; Mikkonen, K.S. Dense and Continuous Networks of Aerial Hyphae Improve Flexibility and Shape Retention of Mycelium Composite in the Wet State. Compos. Part A Appl. Sci. Manuf. 2022, 152, 106688. [Google Scholar] [CrossRef]
- Appels, F.V.W.; Camere, S.; Montalti, M.; Karana, E.; Jansen, K.M.B.; Dijksterhuis, J.; Krijgsheld, P.; Wösten, H.A.B. Fabrication Factors Influencing Mechanical, Moisture- and Water-Related Properties of Mycelium-Based Composites. Mater. Des. 2019, 161, 64–71. [Google Scholar] [CrossRef]
- Santos, I.S.; Nascimento, B.L.; Marino, R.H.; Sussuchi, E.M.; Matos, M.P.; Griza, S. Influence of Drying Heat Treatments on the Mechanical Behavior and Physico-Chemical Properties of Mycelial Biocomposite. Compos. Part B Eng. 2021, 217, 108870. [Google Scholar] [CrossRef]
- Tudryn, G.J.; Smith, L.C.; Freitag, J.; Bucinell, R.; Schadler, L.S. Processing and Morphology Impacts on Mechanical Properties of Fungal Based Biopolymer Composites. J. Polym. Environ. 2018, 26, 1473–1483. [Google Scholar] [CrossRef]
- Attias, N.; Danai, O.; Tarazi, E.; Pereman, I.; Grobman, Y.J. Implementing Bio-Design Tools to Develop Mycelium-Based Products. Des. J. 2019, 22, 1647–1657. [Google Scholar] [CrossRef]
- Antinori, M.E.; Ceseracciu, L.; Mancini, G.; Heredia-Guerrero, J.A.; Athanassiou, A. Fine-Tuning of Physicochemical Properties and Growth Dynamics of Mycelium-Based Materials. ACS Appl. Bio Mater. 2020, 3, 1044–1051. [Google Scholar] [CrossRef]
- Cartabia, M.; Girometta, C.E.; Milanese, C.; Baiguera, R.M.; Buratti, S.; Branciforti, D.S.; Vadivel, D.; Girella, A.; Babbini, S.; Savino, E.; et al. Collection and Characterization of Wood Decay Fungal Strains for Developing Pure Mycelium Mats. J. Fungi 2021, 7, 1008. [Google Scholar] [CrossRef]
- Elsacker, E.; Zhang, M.; Dade-Robertson, M. Fungal Engineered Living Materials: The Viability of Pure Mycelium Materials with Self-Healing Functionalities. Adv. Funct. Mater. 2023, 33, 2301875. [Google Scholar] [CrossRef]
- Oostra, J.; Le Comte, E.P.; Van Den Heuvel, J.C.; Tramper, J.; Rinzema, A. Intra-Particle Oxygen Diffusion Limitation in Solid-State Fermentation. Biotechnol. Bioeng. 2001, 75, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Powrie, W.D.; Chiu Wu, H.; Molund, V.P. Browning Reaction Systems as Sources of Mutagens and Antimutagens. Environ. Health Perspect. 1986, 67, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Martins, S.; Jongen, W.; Boekel, V.; Martinus, A. A Review of Maillard Reaction in Food and Implications to Kinetic Modelling. Trends Food Sci. Technol. 2001, 11, 364–373. [Google Scholar] [CrossRef]
- Attias, N.; Reid, M.; Mijowska, S.C.; Dobryden, I.; Isaksson, M.; Pokroy, B.; Grobman, Y.J.; Abitbol, T. Biofabrication of Nanocellulose–Mycelium Hybrid Materials. Adv. Sustain. Syst. 2021, 5, 2000196. [Google Scholar] [CrossRef]
- Tarigond, H.; Reddy, R.M.; Maheswari, C.U.; Reddy, E.S. Effect of Iron Scrap Additives in Stearic Acid as PCM for Thermal Energy Storage System. J. Therm. Anal. Calorim. 2020, 141, 2497–2510. [Google Scholar] [CrossRef]
- Li, Z.; Yuan, J. Phase Change Microcapsules with High Encapsulation Efficiency Using Janus Silica Particles as Stabilizers and Their Application in Cement. Constr. Build. Mater. 2021, 307, 124971. [Google Scholar] [CrossRef]
- Vennapusa, J.R.; Konala, A.; Dixit, P.; Chattopadhyay, S. Caprylic Acid Based PCM Composite with Potential for Thermal Buffering and Packaging Applications. Mater. Chem. Phys. 2020, 253, 123453. [Google Scholar] [CrossRef]
- Siddhan, P.; Jassal, M.; Agrawal, A.K. Core Content and Stability Ofn-Octadecane-Containing Polyurea Microencapsules Produced by Interfacial Polymerization. J. Appl. Polym. Sci. 2007, 106, 786–792. [Google Scholar] [CrossRef]
- Maithya, O.M.; Li, X.; Feng, X.; Sui, X.; Wang, B. Microencapsulated Phase Change Material via Pickering Emulsion Stabilized by Graphene Oxide for Photothermal Conversion. J. Mater. Sci. 2020, 55, 7731–7742. [Google Scholar] [CrossRef]
- Xie, B.; Liu, G.; Jiang, S.; Zhao, Y.; Wang, D. Crystallization Behaviors of n -Octadecane in Confined Space: Crossover of Rotator Phase from Transient to Metastable Induced by Surface Freezing. J. Phys. Chem. B 2008, 112, 13310–13315. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Wang, X.; Wu, D. Microencapsulation of N-Octadecane Phase Change Material with Calcium Carbonate Shell for Enhancement of Thermal Conductivity and Serving Durability: Synthesis, Microstructure, and Performance Evaluation. Appl. Energy 2014, 114, 632–643. [Google Scholar] [CrossRef]
- Zhu, Y.; Liang, S.; Chen, K.; Gao, X.; Chang, P.; Tian, C.; Wang, J.; Huang, Y. Preparation and Properties of Nanoencapsulated N-Octadecane Phase Change Material with Organosilica Shell for Thermal Energy Storage. Energy Convers. Manag. 2015, 105, 908–917. [Google Scholar] [CrossRef]
- Frone, A.N.; Berlioz, S.; Chailan, J.-F.; Panaitescu, D.M. Morphology and Thermal Properties of PLA–Cellulose Nanofibers Composites. Carbohydr. Polym. 2013, 91, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Poletto, M.; Ornaghi, H.; Zattera, A. Native Cellulose: Structure, Characterization and Thermal Properties. Materials 2014, 7, 6105–6119. [Google Scholar] [CrossRef]
- Jones, M.; Bhat, T.; Kandare, E.; Thomas, A.; Joseph, P.; Dekiwadia, C.; Yuen, R.; John, S.; Ma, J.; Wang, C.H. Thermal Degradation and Fire Properties of Fungal Mycelium and Mycelium-Biomass Composite Materials. Sci. Rep. 2018, 8, 17583. [Google Scholar] [CrossRef]
- Werner, K.; Pommer, L.; Broström, M. Thermal Decomposition of Hemicelluloses. J. Anal. Appl. Pyrolysis 2014, 110, 130–137. [Google Scholar] [CrossRef]
- Girometta, C.; Dondi, D.; Baiguera, R.M.; Bracco, F.; Branciforti, D.S.; Buratti, S.; Lazzaroni, S.; Savino, E. Characterization of Mycelia from Wood-Decay Species by TGA and IR Spectroscopy. Cellulose 2020, 27, 6133–6148. [Google Scholar] [CrossRef]
- Menczel, J.D. Thermogravimetric Analysis of Fibers. In Thermal Analysis of Textiles and Fibers; Elsevier: Amsterdam, The Netherlands, 2020; pp. 71–79. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Support Type | PCM | |
|---|---|---|
| Stearic Acid | Eicosane | |
| MFC | MFC/SA | MFC/E |
| MFC–MYC | MFC–MYC/SA | MFC–MYC/E |
| MYC | MYC/SA | MYC/E |
| Sample | 1st Cycle | 20th Cycle | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TM, °C | ΔHM, J/g | TC, °C | ΔHC, J/g | Ea, % | TM, °C | ΔHM, J/g | TC, °C | ΔHC, J/g | Ea, % | |
| SA | 61 | 178 | 51 | 182 | - | - | - | - | - | - |
| MFC/SA | 63 | 105 | 48 | 106 | 59 | 63 | 105 | 48 | 107 | 59 |
| MFC–MYC/SA | 64 | 106 | 48 | 109 | 59 | 63 | 103 | 48 | 105 | 58 |
| MYC/SA | 68 | 95 | 44 | 97 | 55 | 67 | 91 | 44 | 93 | 53 |
| Eicosane | 41 | 236 | 28 | 236 | - | - | - | - | - | - |
| MFC/E | 45 | 134 | 27 | 137 | 57 | 44 | 136 | 28 | 137 | 58 |
| MFC–MYC/E | 45 | 138 | 26 | 142 | 59 | 45 | 137 | 27 | 140 | 59 |
| MYC/E | 48 | 136 | 30 | 139 | 58 | 41 | 139 | 29 | 141 | 59 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sayfutdinova, A.R.; Cherednichenko, K.A.; Rakitina, M.A.; Dubinich, V.N.; Bardina, K.A.; Rubtsova, M.I.; Petrova, D.A.; Vinokurov, V.A.; Voronin, D.V. Natural Fibrous Materials Based on Fungal Mycelium Hyphae as Porous Supports for Shape-Stable Phase-Change Composites. Polymers 2023, 15, 4504. https://doi.org/10.3390/polym15234504
Sayfutdinova AR, Cherednichenko KA, Rakitina MA, Dubinich VN, Bardina KA, Rubtsova MI, Petrova DA, Vinokurov VA, Voronin DV. Natural Fibrous Materials Based on Fungal Mycelium Hyphae as Porous Supports for Shape-Stable Phase-Change Composites. Polymers. 2023; 15(23):4504. https://doi.org/10.3390/polym15234504
Chicago/Turabian StyleSayfutdinova, Adeliya R., Kirill A. Cherednichenko, Maria A. Rakitina, Valeria N. Dubinich, Kristina A. Bardina, Maria I. Rubtsova, Daria A. Petrova, Vladimir A. Vinokurov, and Denis V. Voronin. 2023. "Natural Fibrous Materials Based on Fungal Mycelium Hyphae as Porous Supports for Shape-Stable Phase-Change Composites" Polymers 15, no. 23: 4504. https://doi.org/10.3390/polym15234504
APA StyleSayfutdinova, A. R., Cherednichenko, K. A., Rakitina, M. A., Dubinich, V. N., Bardina, K. A., Rubtsova, M. I., Petrova, D. A., Vinokurov, V. A., & Voronin, D. V. (2023). Natural Fibrous Materials Based on Fungal Mycelium Hyphae as Porous Supports for Shape-Stable Phase-Change Composites. Polymers, 15(23), 4504. https://doi.org/10.3390/polym15234504