
Preparation of Self-Assembled Nanoparticle–Polymer Hybrids from Modified Silica Nanoparticles and Polystyrene-Block-Polyacrylic Acid Vesicles via the Co-Precipitation Method


Abstract
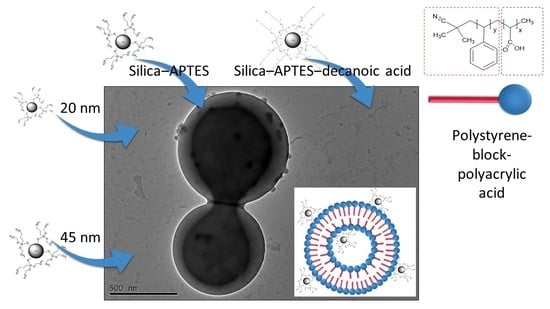
{kind=link}
{kind=link}
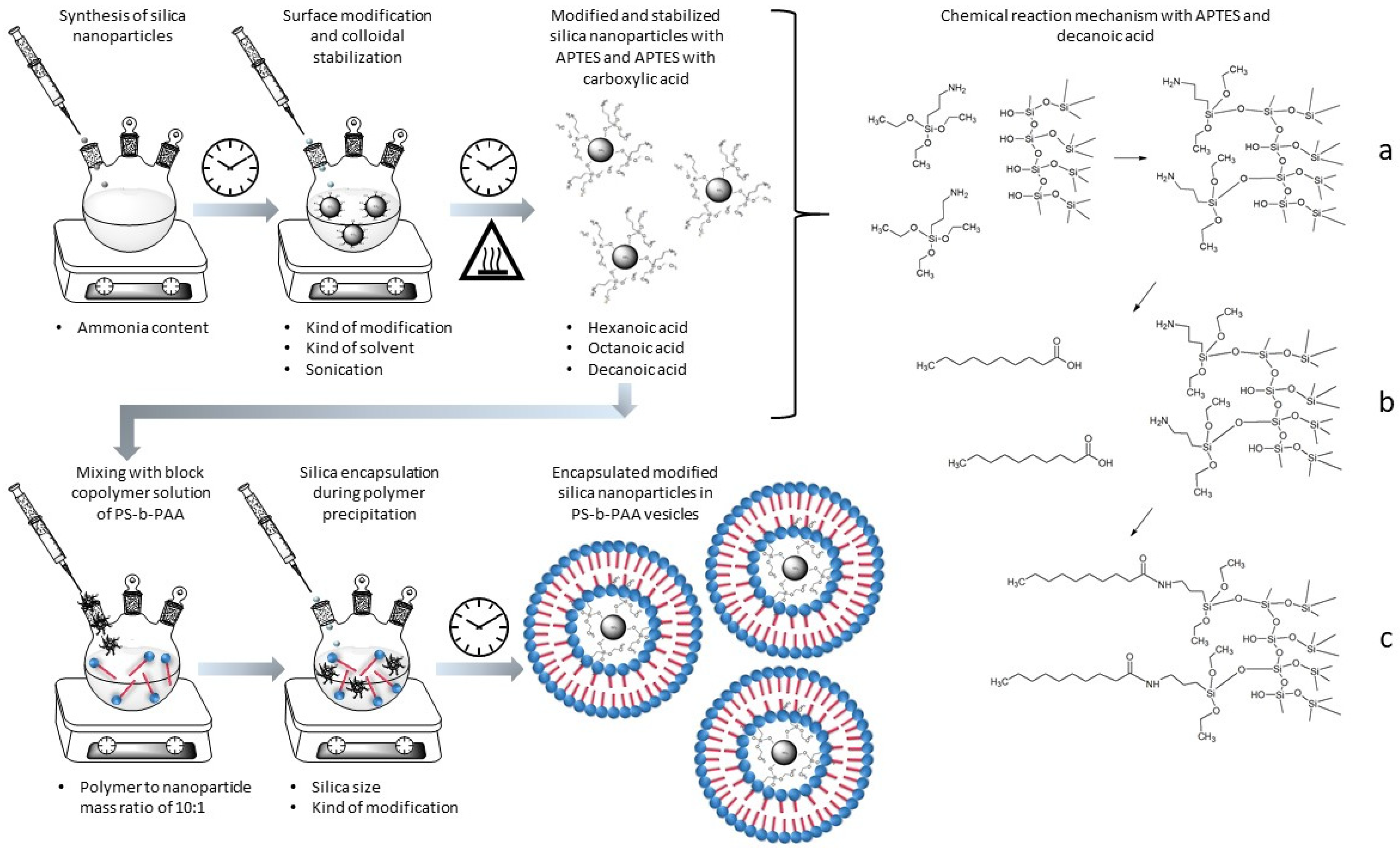{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
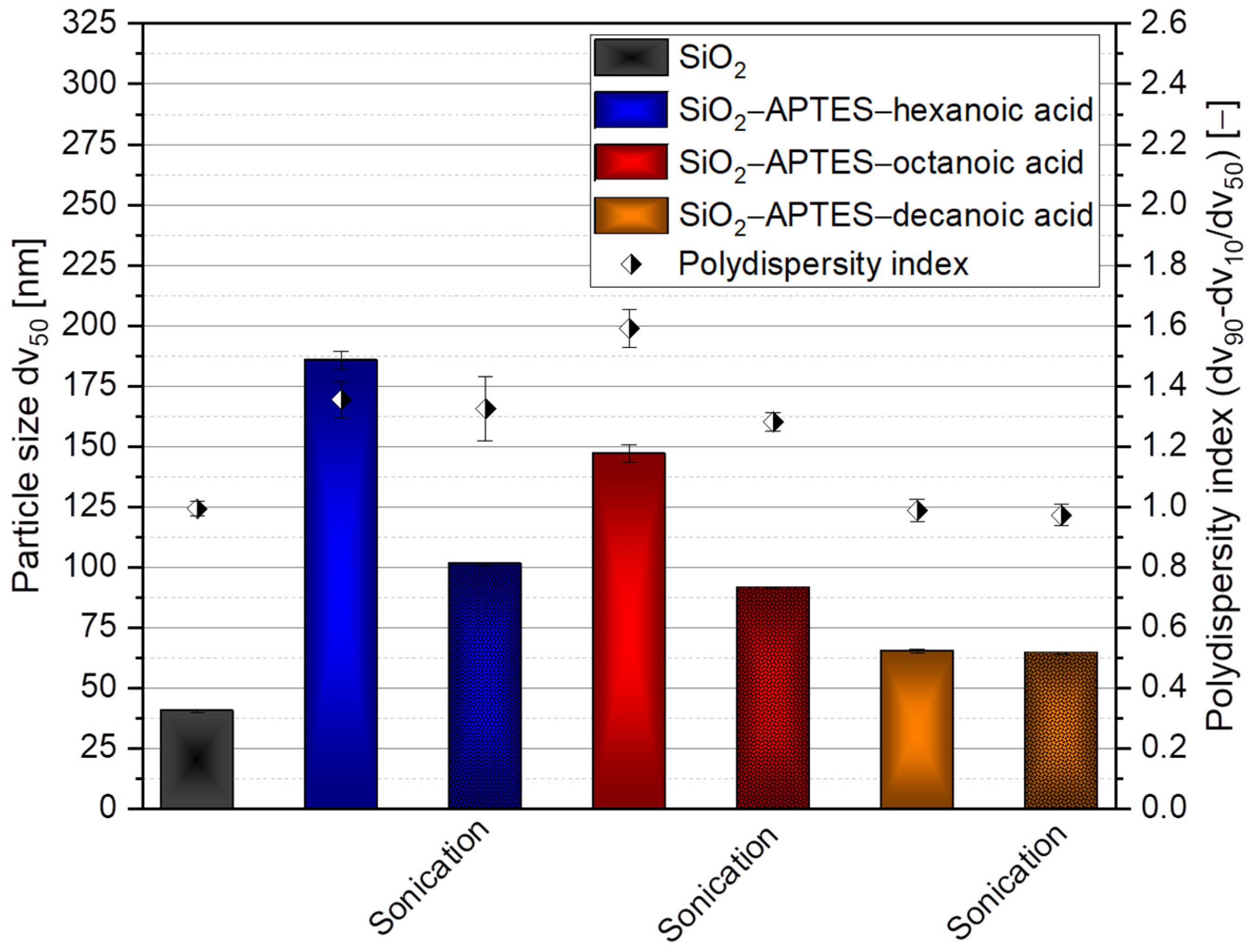{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Silica Synthesis, Modification, and Colloidal Stabilization
2.3. Encapsulation of Modified Silica in PS-b-PAA Vesicles
2.4. Characterization of Particle Size and Surface Modification
2.5. Encapsulation Imaging
3. Results and Discussion
3.1. Silica Synthesis, Modification, and Colloidal Stabilization
3.2. Encapsulation of Modified Silica in PS-b-PAA Vesicles
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mai, Y.; Eisenberg, A. Self-assembly of block copolymers. Chem. Soc. Rev. 2012, 41, 5969–5985. [Google Scholar] [CrossRef]
- Kickelbick, G. Nanokomposite: Anorganisch + organisch: Homogen und doch so heterogen. Chemie Unserer Zeit 2005, 39, 46–53. [Google Scholar] [CrossRef]
- Wang, H.; Chen, L.; Shen, X.; Zhu, L.; He, J.; Chen, H. Unconventional chain-growth mode in the assembly of colloidal gold nanoparticles. Angew. Chem. Int. Ed Engl. 2012, 51, 8021–8025. [Google Scholar] [CrossRef]
- Chen, H.Y.; Abraham, S.; Mendenhall, J.; Delamarre, S.C.; Smith, K.; Kim, I.; Batt, C.A. Encapsulation of single small gold nanoparticles by diblock copolymers. Chemphyschem 2008, 9, 388–392. [Google Scholar] [CrossRef]
- Luchini, A.; Geho, D.H.; Bishop, B.; Tran, D.; Xia, C.; Dufour, R.L.; Jones, C.D.; Espina, V.; Patanarut, A.; Zhou, W.; et al. Smart hydrogel particles: Biomarker harvesting: One-step affinity purification, size exclusion, and protection against degradation. Nano Lett. 2008, 8, 350–361. [Google Scholar] [CrossRef]
- Pyun, J. Selbstorganisation und kolloidale Polymerisation von Polymer-Nanopartikel-Hybriden zu mesoskopischen Ketten. Angew. Chem. 2012, 124, 12576–12578. [Google Scholar] [CrossRef]
- Gaucher, G.; Dufresne, M.-H.; Sant, V.P.; Kang, N.; Maysinger, D.; Leroux, J.-C. Block copolymer micelles: Preparation, characterization and application in drug delivery. J. Control. Release 2005, 109, 169–188. [Google Scholar] [CrossRef]
- Gopinath, S.; Adarsh, N.N.; Nair, P.R.; Mathew, S. Shape-Memory Polymer Nanocomposites of Poly(ε-caprolactone) with the Polystyrene-block-polybutadiene-block-polystyrene-tri-block Copolymer Encapsulated with Metal Oxides. ACS Omega 2021, 6, 6261–6273. [Google Scholar] [CrossRef]
- Singh, T.; Gangil, B.; Ranakoti, L.; Joshi, A. Effect of silica nanoparticles on physical, mechanical, and wear properties of natural fiber reinforced polymer composites. Polym. Compos. 2021, 42, 2396–2407. [Google Scholar] [CrossRef]
- Roos, C.; Schmidt, M.; Ebenhoch, J.; Baumann, F.; Deubzer, B.; Weis, J. Design and Synthesis of Molecular Reactors for the Preparation of Topologically Trapped Gold Cluster. Adv. Mater. 1999, 11, 761–766. [Google Scholar] [CrossRef]
- Onaca, O.; Enea, R.; Hughes, D.W.; Meier, W. Stimuli-Responsive Polymersomes as Nanocarriers for Drug and Gene Delivery. Macromol. Biosci. 2009, 9, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Yu, Y.; Maysinger, D.; Eisenberg, A. Polycaprolactone-b-poly(ethylene oxide) block copolymer micelles as a novel drug delivery vehicle for neurotrophic agents FK506 and L-685,818. Bioconjug. Chem. 1998, 9, 564–572. [Google Scholar] [CrossRef]
- Meng, F.; Zhong, Z.; Feijen, J. Stimuli-responsive polymersomes for programmed drug delivery. Biomacromolecules 2009, 10, 197–209. [Google Scholar] [CrossRef]
- Litvinchuk, S.; Lu, Z.; Rigler, P.; Hirt, T.D.; Meier, W. Calcein release from polymeric vesicles in blood plasma and PVA hydrogel. Pharm. Res. 2009, 26, 1711–1717. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Xu, J.-J.; Shi, C.-G.; Chen, H.-Y. Multilayer membranes via layer-by-layer deposition of organic polymer protected Prussian blue nanoparticles and glucose oxidase for glucose biosensing. Langmuir 2005, 21, 9630–9634. [Google Scholar] [CrossRef] [PubMed]
- Yaghoubi, Z.; Basiri-Parsa, J. Modification of ultrafiltration membrane by thermo-responsive Bentonite-poly(N-isopropylacrylamide) nanocomposite to improve its antifouling properties. J. Water Process Eng. 2020, 34, 101067. [Google Scholar] [CrossRef]
- Bixner, O.; Kurzhals, S.; Virk, M.; Reimhult, E. Triggered Release from Thermoresponsive Polymersomes with Superparamagnetic Membranes. Materials 2016, 9, 29. [Google Scholar] [CrossRef]
- Li, H.; Yao, Y.; Shi, H.; Lei, Y.; Huang, Y.; Wang, K.; He, X.; Liu, J. A near-infrared light-responsive nanocomposite for photothermal release of H2S and suppression of cell viability. J. Mater. Chem. B 2019, 7, 5992–5997. [Google Scholar] [CrossRef]
- Soto, G.D.; Meiorin, C.; Actis, D.G.; Mendoza Zélis, P.; Moscoso Londoño, O.; Muraca, D.; Mosiewicki, M.A.; Marcovich, N.E. Magnetic nanocomposites based on shape memory polyurethanes. Eur. Polym. J. 2018, 109, 8–15. [Google Scholar] [CrossRef]
- Nitin, N.; LaConte, L.E.W.; Zurkiya, O.; Hu, X.; Bao, G. Functionalization and peptide-based delivery of magnetic nanoparticles as an intracellular MRI contrast agent. J. Biol. Inorg. Chem. 2004, 9, 706–712. [Google Scholar] [CrossRef]
- Meng, L.; Watson, B.W.; Qin, Y. Hybrid conjugated polymer/magnetic nanoparticle composite nanofibers through cooperative non-covalent interactions. Nanoscale Adv. 2020, 2, 2462–2470. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Yuan, J.; Ma, X.; Wang, S.; Liu, Y. Polymeric nanocomposite membranes for water treatment: A review. Environ. Chem. Lett. 2019, 17, 1539–1551. [Google Scholar] [CrossRef]
- Ren, S.; Chang, L.-Y.; Lim, S.-K.; Zhao, J.; Smith, M.; Zhao, N.; Bulović, V.; Bawendi, M.; Gradecak, S. Inorganic-organic hybrid solar cell: Bridging quantum dots to conjugated polymer nanowires. Nano Lett. 2011, 11, 3998–4002. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, S.; Kickelbick, G. Double Reversible Networks: Improvement of Self-Healing in Hybrid Materials via Combination of Diels–Alder Cross-Linking and Hydrogen Bonds. Macromolecules 2018, 51, 6099–6110. [Google Scholar] [CrossRef]
- Luo, L.; Eisenberg, A. Thermodynamic Size Control of Block Copolymer Vesicles in Solution. Langmuir 2001, 17, 6804–6811. [Google Scholar] [CrossRef]
- Maskos, M.; Harris, J.R. Double-Shell Vesicles, Strings of Vesicles and Filaments Found in Crosslinked Micellar Solutions of Poly(1,2-butadiene)-block-poly(ethylene oxide) Diblock Copolymers. Macromol. Rapid Commun. 2001, 22, 271–273. [Google Scholar] [CrossRef]
- Choucair, A.A.; Kycia, A.H.; Eisenberg, A. Kinetics of Fusion of Polystyrene- b -poly(acrylic acid) Vesicles in Solution. Langmuir 2003, 19, 1001–1008. [Google Scholar] [CrossRef]
- Qu, F.; Liu, N.; Bu, W. Vesicle fusion intermediates obtained from the self-assembly of a cationic platinum(ii) complex with sulfonate terminated polystyrenes. RSC Adv. 2014, 4, 9750. [Google Scholar] [CrossRef]
- Burke, S.E.; Eisenberg, A. Kinetics and Mechanisms of the Sphere-to-Rod and Rod-to-Sphere Transitions in the Ternary System PS310-b-PAA52/Dioxane/Water. Langmuir 2001, 17, 6705–6714. [Google Scholar] [CrossRef]
- Israelachvili, J.N.; Mitchell, D.J.; Ninham, B.W. Theory of self-assembly of hydrocarbon amphiphiles into micelles and bilayers. J. Chem. Soc. Faraday Trans. 2 1976, 72, 1525. [Google Scholar] [CrossRef]
- Leibler, L. Theory of Microphase Separation in Block Copolymers. Macromolecules 1980, 13, 1602–1617. [Google Scholar] [CrossRef]
- Blanazs, A.; Armes, S.P.; Ryan, A.J. Self-Assembled Block Copolymer Aggregates: From Micelles to Vesicles and their Biological Applications. Macromol. Rapid Commun. 2009, 30, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Peinemann, K.-V.; Abetz, V.; Simon, P.F.W. Asymmetric superstructure formed in a block copolymer via phase separation. Nat. Mater. 2007, 6, 992–996. [Google Scholar] [CrossRef] [PubMed]
- Guida, V. Thermodynamics and kinetics of vesicles formation processes. Adv. Colloid Interface Sci. 2010, 161, 77–88. [Google Scholar] [CrossRef]
- Leng, J.; Egelhaaf, S.U.; Cates, M.E. Kinetics of the Micelle-to-Vesicle Transition: Aqueous Lecithin-Bile Salt Mixtures. Biophys. J. 2003, 85, 1624–1646. [Google Scholar] [CrossRef] [PubMed]
- Pochan, D.J.; Chen, Z.; Cui, H.; Hales, K.; Qi, K.; Wooley, K.L. Toroidal triblock copolymer assemblies. Science 2004, 306, 94–97. [Google Scholar] [CrossRef]
- Yu, H.; Qiu, X.; Nunes, S.P.; Peinemann, K.-V. Biomimetic block copolymer particles with gated nanopores and ultrahigh protein sorption capacity. Nat. Commun. 2014, 5, 4110. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, Y.; Du, J. Evolution of diverse higher-order membrane structures of block copolymer vesicles. Polym. Chem. 2019, 10, 3020–3029. [Google Scholar] [CrossRef]
- Geng, Z.; Xiong, B.; Wang, L.; Wang, K.; Ren, M.; Zhang, L.; Zhu, J.; Yang, Z. Moebius strips of chiral block copolymers. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Zhang, L.; Eisenberg, A. Thermodynamic vs Kinetic Aspects in the Formation and Morphological Transitions of Crew-Cut Aggregates Produced by Self-Assembly of Polystyrene- b -poly(acrylic acid) Block Copolymers in Dilute Solution. Macromolecules 1999, 32, 2239–2249. [Google Scholar] [CrossRef]
- Hashimoto, T.; Mitsumura, N.; Yamaguchi, D.; Takenaka, M.; Morita, H.; Kawakatsu, T.; Doi, M. Nonequilibrium helical-domain morphology in diblock copolymer systems. Polymer 2001, 42, 8477–8481. [Google Scholar] [CrossRef]
- Du, J.; O’Reilly, R.K. Advances and challenges in smart and functional polymer vesicles. Soft Matter 2009, 5, 3544. [Google Scholar] [CrossRef]
- Smart, T.; Lomas, H.; Massignani, M.; Flores-Merino, M.; Perez, L.; Battaglia, G. Block Copolymer Nanostructures. Nano Today [Online]. 38–46. Available online: https://www.observatorioplastico.com/ficheros/noticias/30133944nano1.pdf (accessed on 27 June 2022).
- Yu, Y.; Eisenberg, A. Control of Morphology through Polymer−Solvent Interactions in Crew-Cut Aggregates of Amphiphilic Block Copolymers. J. Am. Chem. Soc. 1997, 119, 8383–8384. [Google Scholar] [CrossRef]
- Zhang, L.; Eisenberg, A. Multiple Morphologies of “Crew-Cut” Aggregates of Polystyrene-b-poly(acrylic acid) Block Copolymers. Science 1995, 268, 1728–1731. [Google Scholar] [CrossRef]
- Zhang, L.; Eisenberg, A. Structures of “crew-cut” aggregates of polystyrene- b -poly(acrylic acid) diblock copolymers. Macromol. Symp. 1997, 113, 221–232. [Google Scholar] [CrossRef]
- Chen, Q.; Schönherr, H.; Vancso, G.J. Encapsulation and release of molecular cargos via temperature-induced vesicle-to-micelle transitions. Small 2010, 6, 2762–2768. [Google Scholar] [CrossRef] [PubMed]
- Shioi, A.; Hatton, T.A. Model for Formation and Growth of Vesicles in Mixed Anionic/Cationic (SOS/CTAB) Surfactant Systems. Langmuir 2002, 18, 7341–7348. [Google Scholar] [CrossRef]
- Cui, H.; Chen, Z.; Zhong, S.; Wooley, K.L.; Pochan, D.J. Block copolymer assembly via kinetic control. Science 2007, 317, 647–650. [Google Scholar] [CrossRef]
- Matthews, G. Synaptic Vesicle Exocytosis. Neuron 2002, 35, 1013–1014. [Google Scholar] [CrossRef]
- Mondal Roy, S.; Sarkar, M. Membrane fusion induced by small molecules and ions. J. Lipids 2011, 2011, 528784. [Google Scholar] [CrossRef]
- Chernomordik, L.V.; Melikyan, G.B.; Chizmadzhev, Y.A. Biomembrane fusion: A new concept derived from model studies using two interacting planar lipid bilayers. Biochim. Biophys. Acta (BBA) Rev. Biomembr. 1987, 906, 309–352. [Google Scholar] [CrossRef]
- Wang, J.; Li, W.; Zhu, J. Encapsulation of inorganic nanoparticles into block copolymer micellar aggregates: Strategies and precise localization of nanoparticles. Polymer 2014, 55, 1079–1096. [Google Scholar] [CrossRef]
- Priestley, R.; Prud’homme, R.K. (Eds.) Polymer Colloids: Formation, Characterization and Applications; Royal Society of Chemistry: Cambridge, UK, 2020; ISBN 1788014170. [Google Scholar]
- Kang, Y.; Taton, T.A. Controlling Shell Thickness in Core−Shell Gold Nanoparticles via Surface-Templated Adsorption of Block Copolymer Surfactants. Macromolecules 2005, 38, 6115–6121. [Google Scholar] [CrossRef]
- Kim, B.-S.; Qiu, J.-M.; Wang, J.-P.; Taton, T.A. Magnetomicelles: Composite nanostructures from magnetic nanoparticles and cross-linked amphiphilic block copolymers. Nano Lett. 2005, 5, 1987–1991. [Google Scholar] [CrossRef] [PubMed]
- Darwish, M.S.A.; Mostafa, M.H.; Al-Harbi, L.M. Polymeric Nanocomposites for Environmental and Industrial Applications. Int. J. Mol. Sci. 2022, 23, 1023. [Google Scholar] [CrossRef]
- Dantas de Oliveira, A.; Augusto Gonçalves Beatrice, C. Polymer Nanocomposites with Different Types of Nanofiller. In Nanocomposites—Recent Evolutions; Sivasankaran, S., Ed.; IntechOpen: London, UK, 2019; ISBN 978-1-78985-011-6. [Google Scholar]
- Zhu, J.; Hayward, R.C. Spontaneous generation of amphiphilic block copolymer micelles with multiple morphologies through interfacial Instabilities. J. Am. Chem. Soc. 2008, 130, 7496–7502. [Google Scholar] [CrossRef]
- Feng, Y.; Yu, J.; Sun, D.; Ren, W.; Shao, C.; Sun, R. Solvent-induced in-situ self-assembly lignin nanoparticles to reinforce conductive nanocomposite organogels as anti-freezing and anti-dehydration flexible strain sensors. Chem. Eng. J. 2022, 433, 133202. [Google Scholar] [CrossRef]
- Hansen, C.R.; Westerlund, F.; Moth-Poulsen, K.; Ravindranath, R.; Valiyaveettil, S.; Bjørnholm, T. Polymer-templated self-assembly of a 2-dimensional gold nanoparticle network. Langmuir 2008, 24, 3905–3910. [Google Scholar] [CrossRef]
- Cheng, J.Y.; Ross, C.A.; Smith, H.I.; Thomas, E.L. Templated Self-Assembly of Block Copolymers: Top-Down Helps Bottom-Up. Adv. Mater. 2006, 18, 2505–2521. [Google Scholar] [CrossRef]
- Chen, Y.; Cho, J.; Young, A.; Taton, T.A. Enhanced stability and bioconjugation of photo-cross-linked polystyrene-shell, Au-core nanoparticles. Langmuir 2007, 23, 7491–7497. [Google Scholar] [CrossRef]
- Kang, Y.; Erickson, K.J.; Taton, T.A. Plasmonic nanoparticle chains via a morphological, sphere-to-string transition. J. Am. Chem. Soc. 2005, 127, 13800–13801. [Google Scholar] [CrossRef]
- Kockmann, A.; Hesselbach, J.; Schilde, C.; Kwade, A.; Garnweitner, G. Verbesserung von Kunstharzbeschichtungen durch Nanopartikel mit maßgeschneiderter Oberflächenmodifizierung. Chemie Ingenieur Technik 2016, 88, 958–966. [Google Scholar] [CrossRef]
- Parashar, P. Electrical behaviour of discontinuous silver films deposited on compatible Polystyrene/Poly (2-vinylpyridine) composite. J. Mater. Sci. Mater. Electron. 2012, 23, 468–473. [Google Scholar] [CrossRef]
- Bonacchi, S.; Genovese, D.; Juris, R.; Montalti, M.; Prodi, L.; Rampazzo, E.; Zaccheroni, N. Glänzende Aussichten für lumineszierende Siliciumdioxidnanopartikel. Angew. Chem. 2011, 123, 4142–4152. [Google Scholar] [CrossRef]
- Bogush, G.H.; Tracy, M.A.; Zukoski, C.F. Preparation of monodisperse silica particles: Control of size and mass fraction. J. Non-Cryst. Solids 1988, 104, 95–106. [Google Scholar] [CrossRef]
- Zhu, S.-G.; Xiang, J.-J.; Li, X.-L.; Shen, S.-R.; Lu, H.-B.; Zhou, J.; Xiong, W.; Zhang, B.-C.; Nie, X.-M.; Zhou, M.; et al. Poly(l-lysine)-modified silica nanoparticles for the delivery of antisense oligonucleotides. Biotechnol. Appl. Biochem. 2004, 39, 179–187. [Google Scholar] [CrossRef]
- Kneuer, C.; Sameti, M.; Bakowsky, U.; Schiestel, T.; Schirra, H.; Schmidt, H.; Lehr, C.M. A nonviral DNA delivery system based on surface modified silica-nanoparticles can efficiently transfect cells in vitro. Bioconjug. Chem. 2000, 11, 926–932. [Google Scholar] [CrossRef]
- Said, A.M.; Zeidan, M.S.; Bassuoni, M.T.; Tian, Y. Properties of concrete incorporating nano-silica. Constr. Build. Mater. 2012, 36, 838–844. [Google Scholar] [CrossRef]
- Kockmann, A.; Hesselbach, J.; Zellmer, S.; Kwade, A.; Garnweitner, G. Facile surface tailoring of metal oxide nanoparticles via a two-step modification approach. RSC Adv. 2015, 5, 60993–60999. [Google Scholar] [CrossRef]
- Byun, H.; Hu, J.; Pakawanit, P.; Srisombat, L.; Kim, J.-H. Polymer particles filled with multiple colloidal silica via in situ sol-gel process and their thermal property. Nanotechnology 2017, 28, 25601. [Google Scholar] [CrossRef]
- Ghamarpoor, R.; Jamshidi, M. Synthesis of vinyl-based silica nanoparticles by sol-gel method and their influences on network microstructure and dynamic mechanical properties of nitrile rubber nanocomposites. Sci. Rep. 2022, 12, 15286. [Google Scholar] [CrossRef]
- Shchipunov, Y.A.; Karpenko, T.y.Y. Hybrid polysaccharide-silica nanocomposites prepared by the sol-gel technique. Langmuir 2004, 20, 3882–3887. [Google Scholar] [CrossRef]
- Chujo, Y.; Matsuki, H.; Kure, S.; Saegusa, T.; Yazawa, T. Control of pore size of porous silica by means of pyrolysis of an organic–inorganic polymer hybrid. J. Chem. Soc. Chem. Commun. 1994, A27, 635–636. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, W.; Wang, S. Synthesis of Well-Defined Silica and Pd/Silica Nanotubes through a Surface Sol−Gel Process on a Self-Assembled Chelate Block Copolymer. J. Phys. Chem. C 2010, 114, 15640–15644. [Google Scholar] [CrossRef]
- Kickelbick, G. Hybrid Materials—Past, Present and Future. Hybrid Mater. 2014, 1, 39–51. [Google Scholar] [CrossRef]
- Wang, H.; Patil, A.J.; Liu, K.; Petrov, S.; Mann, S.; Winnik, M.A.; Manners, I. Fabrication of Continuous and Segmented Polymer/Metal Oxide Nanowires Using Cylindrical Micelles and Block Comicelles as Templates. Adv. Mater. 2009, 21, 1805–1808. [Google Scholar] [CrossRef]
- Corma, A. From Microporous to Mesoporous Molecular Sieve Materials and Their Use in Catalysis. Chem. Rev. 1997, 97, 2373–2420. [Google Scholar] [CrossRef] [PubMed]
- Luque, R.; Mariana Balu, A.; Manuel Campelo, J.; Dolores Gracia, M.; Losada, E.; Pineda, A.; Angel Romero, A.; Carlos Serrano-Ruiz, J. Catalytic applications of mesoporous silica-based materials. In Catalysis; Spivey, J.J., Gupta, M., Eds.; Royal Society of Chemistry: Cambridge, UK, 2012; pp. 253–280. ISBN 978-1-84973-375-5. [Google Scholar]
- Guliants, V.V.; Carreon, M.A.; Lin, Y.S. Ordered mesoporous and macroporous inorganic films and membranes. J. Membr. Sci. 2004, 235, 53–72. [Google Scholar] [CrossRef]
- Vallet-Regí, M.; Balas, F.; Arcos, D. Mesoporous materials for drug delivery. Angew. Chem. Int. Ed Engl. 2007, 46, 7548–7558. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Fryxell, G.E.; Wang, L.-Q.; Kim, A.Y.; Liu, J.; Kemner, K.M. Functionalized Monolayers on Ordered Mesoporous Supports. Science 1997, 276, 923–926. [Google Scholar] [CrossRef]
- Kockmann, A.; Porsiel, J.C.; Saadat, R.; Garnweitner, G. Impact of nanoparticle surface modification on the mechanical properties of polystyrene-based nanocomposites. RSC Adv. 2018, 8, 11109–11118. [Google Scholar] [CrossRef] [PubMed]
- Stöber, W.; Fink, A.; Bohn, E. Controlled growth of monodisperse silica spheres in the micron size range. J. Colloid Interface Sci. 1968, 26, 62–69. [Google Scholar] [CrossRef]
- Lee, K.; Look, J.L.; Harris, M.T.; McCormick, A.V. Assessing Extreme Models of the Stober Synthesis Using Transients under a Range of Initial Composition. J. Colloid Interface Sci. 1997, 194, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Matsoukas, T.; Gulari, E. Dynamics of growth of silica particles from ammonia-catalyzed hydrolysis of tetra-ethyl-orthosilicate. J. Colloid Interface Sci. 1988, 124, 252–261. [Google Scholar] [CrossRef]
- Ibrahim, I.A.; Zikry, A.A.F.; Sharaf, M.A. Preparation of spherical silica nanoparticles: Stober silica. J. Am. Sci. 2010, 6, 985–989. [Google Scholar]
- Cooper, E.A.; Knutson, K. Fourier transform infrared spectroscopy investigations of protein structure. Pharm. Biotechnol. 1995, 7, 101–143. [Google Scholar] [CrossRef]
- Wu, Z.; Xiang, H.; Kim, T.; Chun, M.-S.; Lee, K. Surface properties of submicrometer silica spheres modified with aminopropyltriethoxysilane and phenyltriethoxysilane. J. Colloid Interface Sci. 2006, 304, 119–124. [Google Scholar] [CrossRef]
- Schiavon, M.A.; Redondo, S.U.A.; Pina, S.R.O.; Yoshida, I.V.P. Investigation on kinetics of thermal decomposition in polysiloxane networks used as precursors of silicon oxycarbide glasses. J. Non-Cryst. Solids 2002, 304, 92–100. [Google Scholar] [CrossRef]
- Carl Roth GmbH + Co KG. Sicherheitsdatenblatt: Ethanol > 99.8%, Vergällt; Carl Roth GmbH + Co KG: Karlsruhe, Germany, 2015. [Google Scholar]
- Carl Roth GmbH + Co KG. Sicherheitsdatenblatt: Tetrahydrofuran ROTISOLV HPLC, Nicht Stabilisiert; Carl Roth GmbH + Co KG: Karlsruhe, Germany, 2016. [Google Scholar]
- Herbst, F.; Döhler, D.; Michael, P.; Binder, W.H. Self-healing polymers via supramolecular forces. Macromol. Rapid Commun. 2013, 34, 203–220. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mann, J.; Garnweitner, G.; Schilde, C. Preparation of Self-Assembled Nanoparticle–Polymer Hybrids from Modified Silica Nanoparticles and Polystyrene-Block-Polyacrylic Acid Vesicles via the Co-Precipitation Method. Polymers 2023, 15, 444. https://doi.org/10.3390/polym15020444
Mann J, Garnweitner G, Schilde C. Preparation of Self-Assembled Nanoparticle–Polymer Hybrids from Modified Silica Nanoparticles and Polystyrene-Block-Polyacrylic Acid Vesicles via the Co-Precipitation Method. Polymers. 2023; 15(2):444. https://doi.org/10.3390/polym15020444
Chicago/Turabian StyleMann, Jil, Georg Garnweitner, and Carsten Schilde. 2023. "Preparation of Self-Assembled Nanoparticle–Polymer Hybrids from Modified Silica Nanoparticles and Polystyrene-Block-Polyacrylic Acid Vesicles via the Co-Precipitation Method" Polymers 15, no. 2: 444. https://doi.org/10.3390/polym15020444
APA StyleMann, J., Garnweitner, G., & Schilde, C. (2023). Preparation of Self-Assembled Nanoparticle–Polymer Hybrids from Modified Silica Nanoparticles and Polystyrene-Block-Polyacrylic Acid Vesicles via the Co-Precipitation Method. Polymers, 15(2), 444. https://doi.org/10.3390/polym15020444