
The Potential of Visible Spectroscopy as a Tool for the In-Line Monitoring of Lignin Methylolation

 ,
,  ,
,  ,
,  and
and 
Abstract
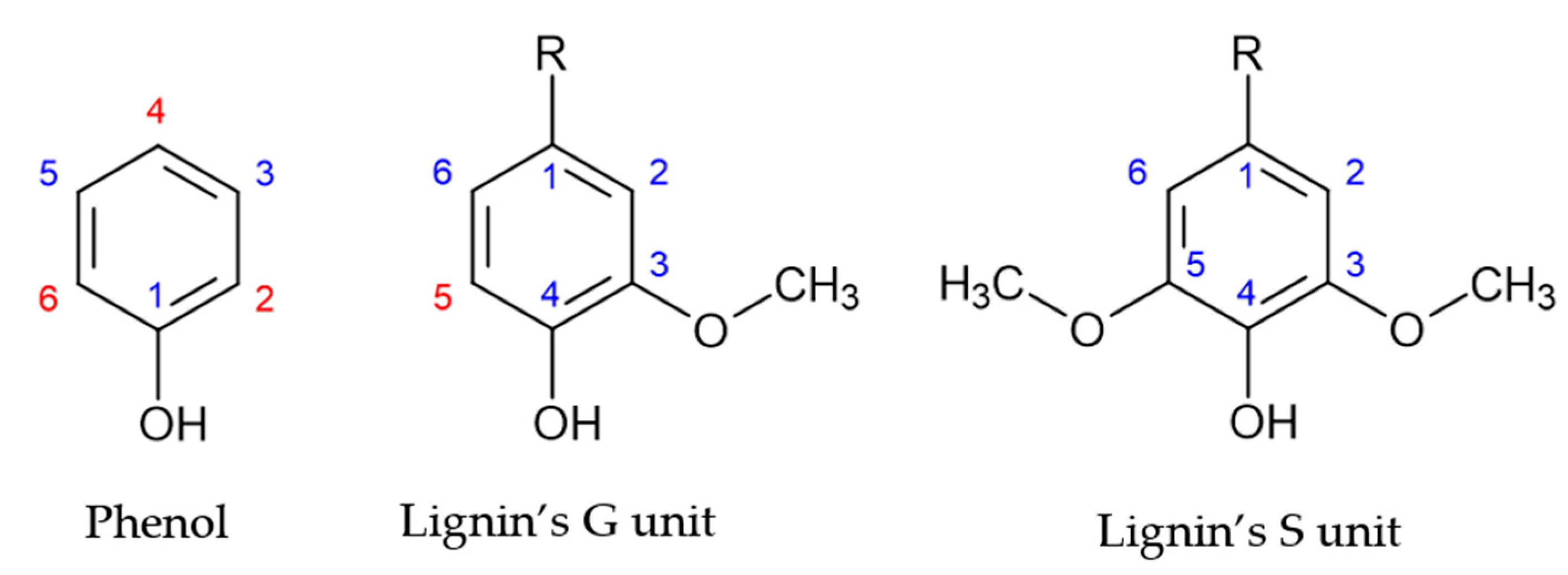
1. Introduction
2. Materials and Methods
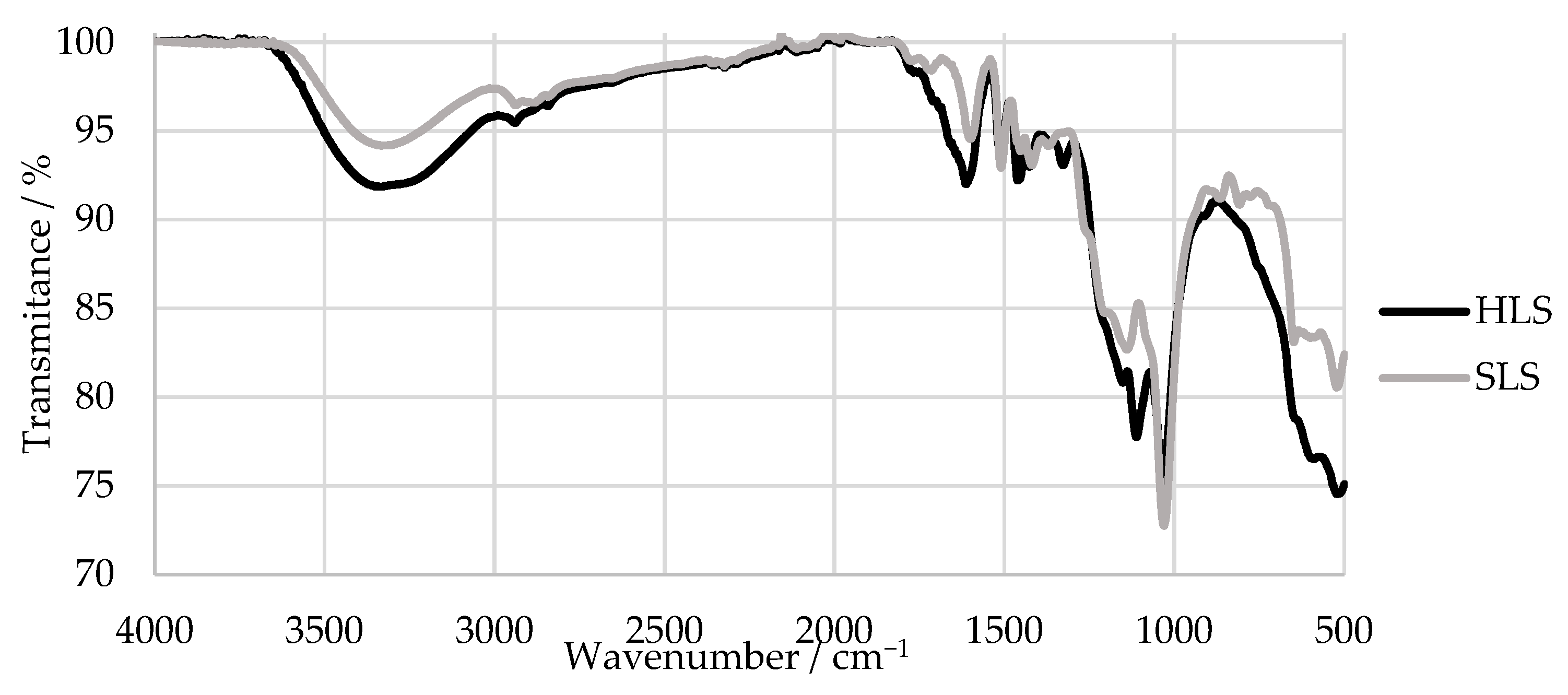
2.1. Physico-Chemical Characterization of the LS Samples
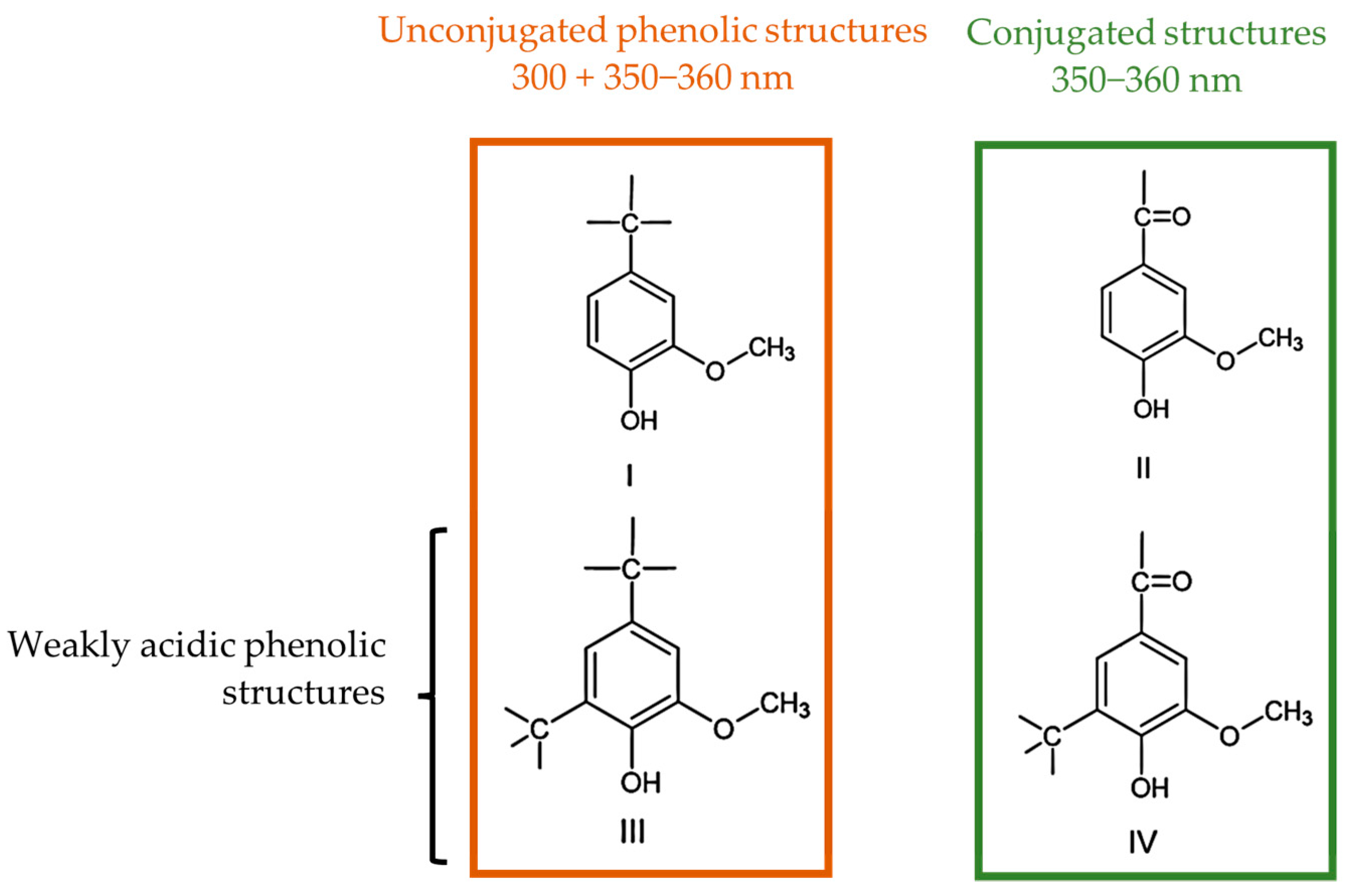
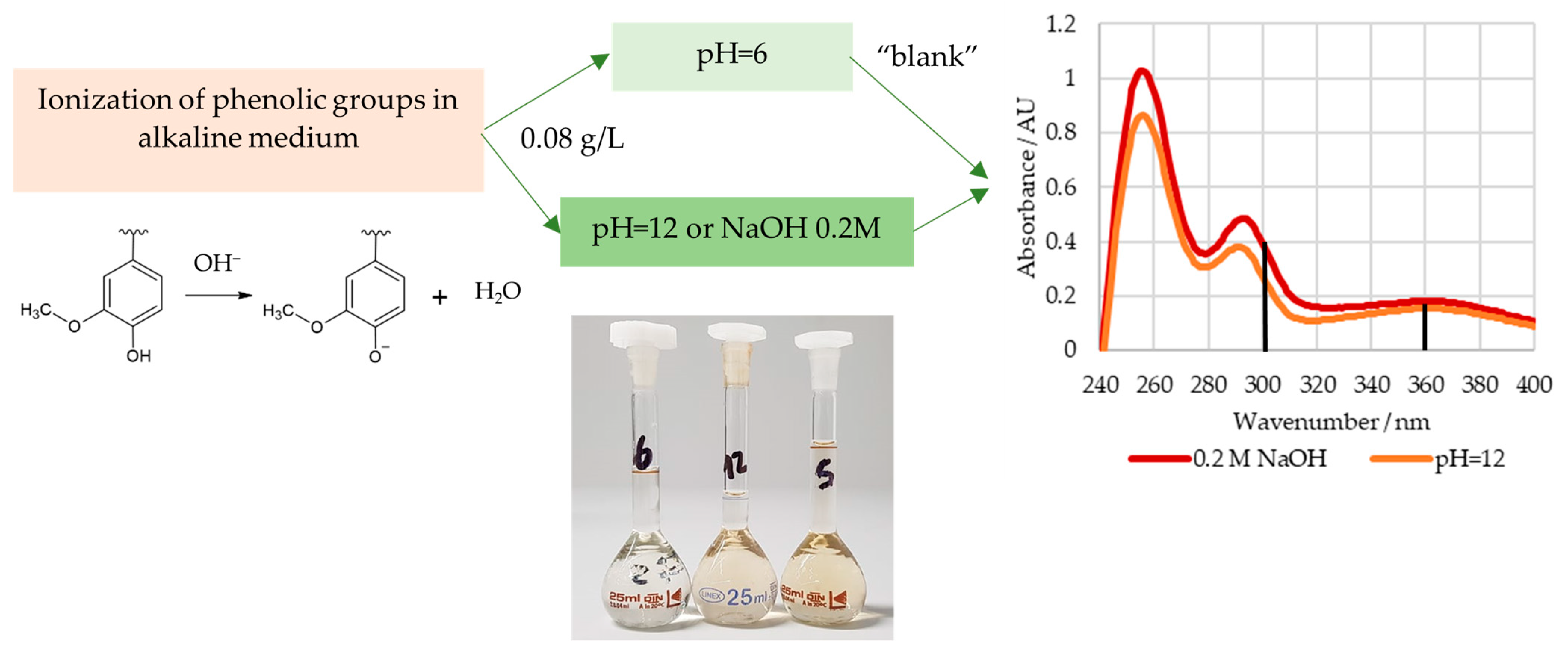
2.1.1. Content of Phenolic Hydroxyl Groups
2.1.2. Fourier Transform Infrared Spectroscopy (FTIR)
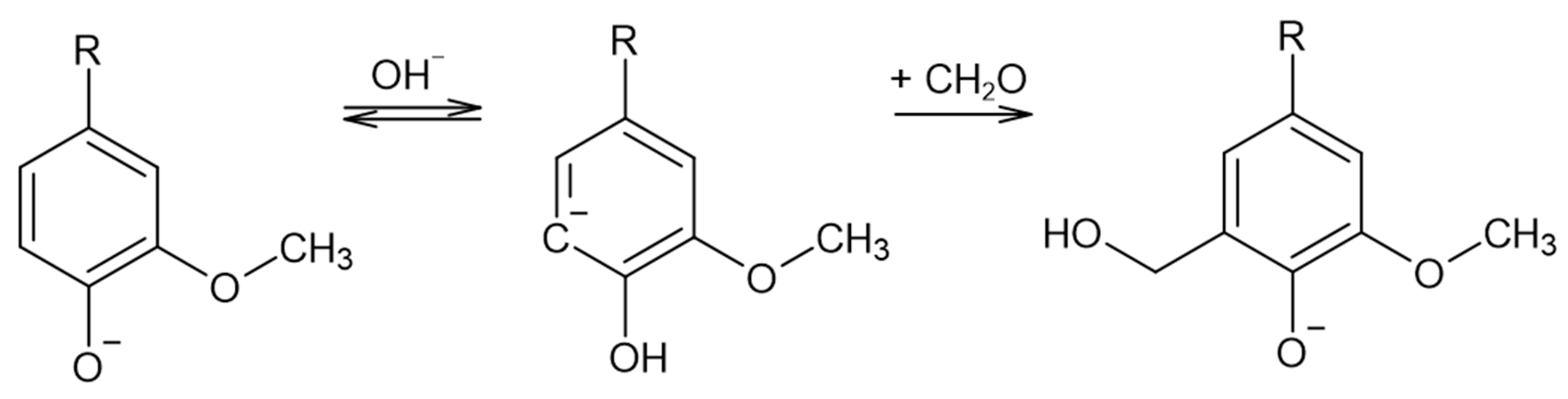
2.1.3. Methylolation of LS
2.2. Monitoring of the Methylolation Reaction
2.2.1. Phenolic Hydroxyl Groups Type I and II
2.2.2. Free Formaldehyde
2.2.3. Determination of the Kinetic Parameters
2.2.4. Spectra Acquisition
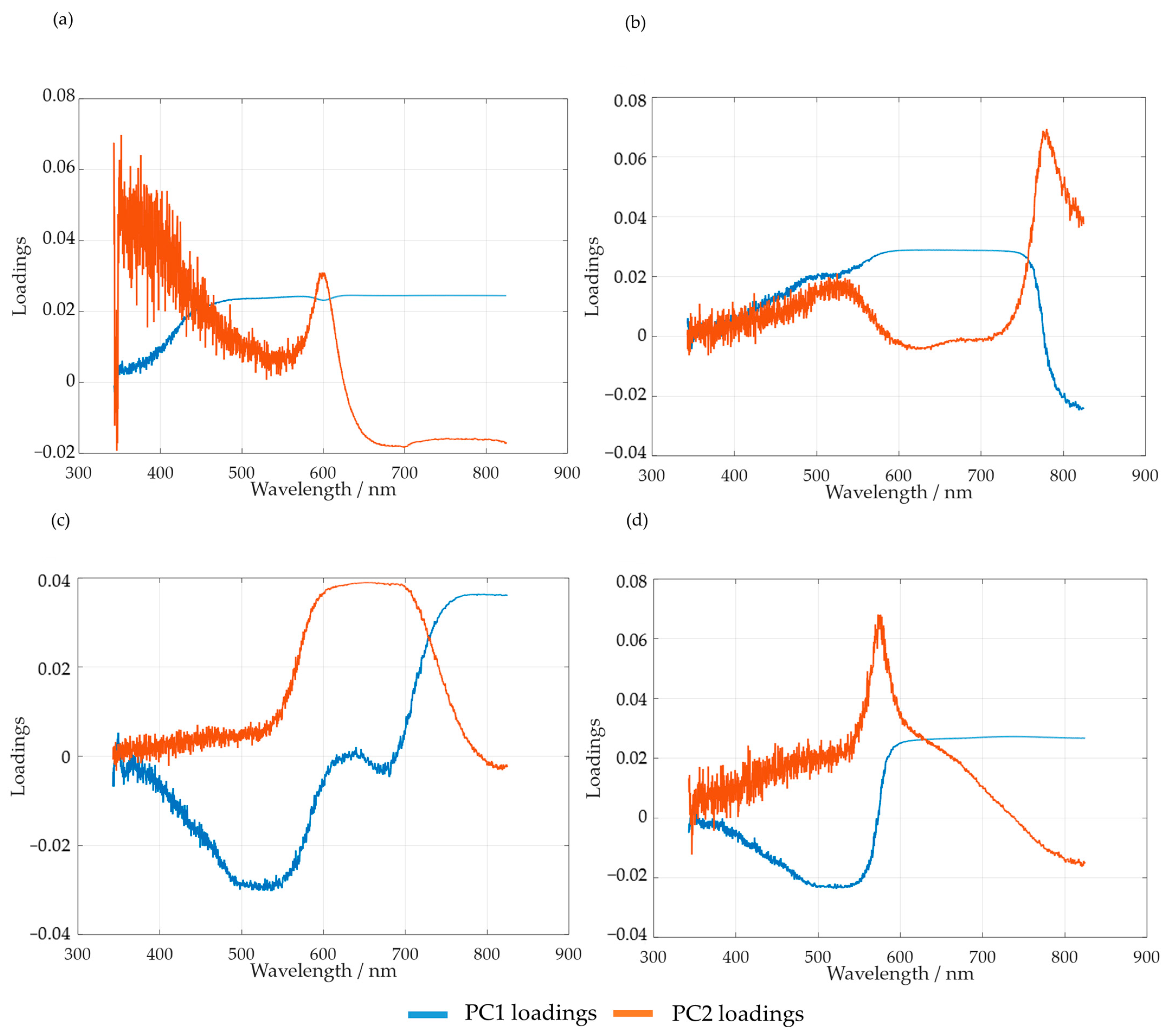
2.2.5. Principal Component Analysis
3. Results and Discussion
3.1. Characterization of the LS Samples
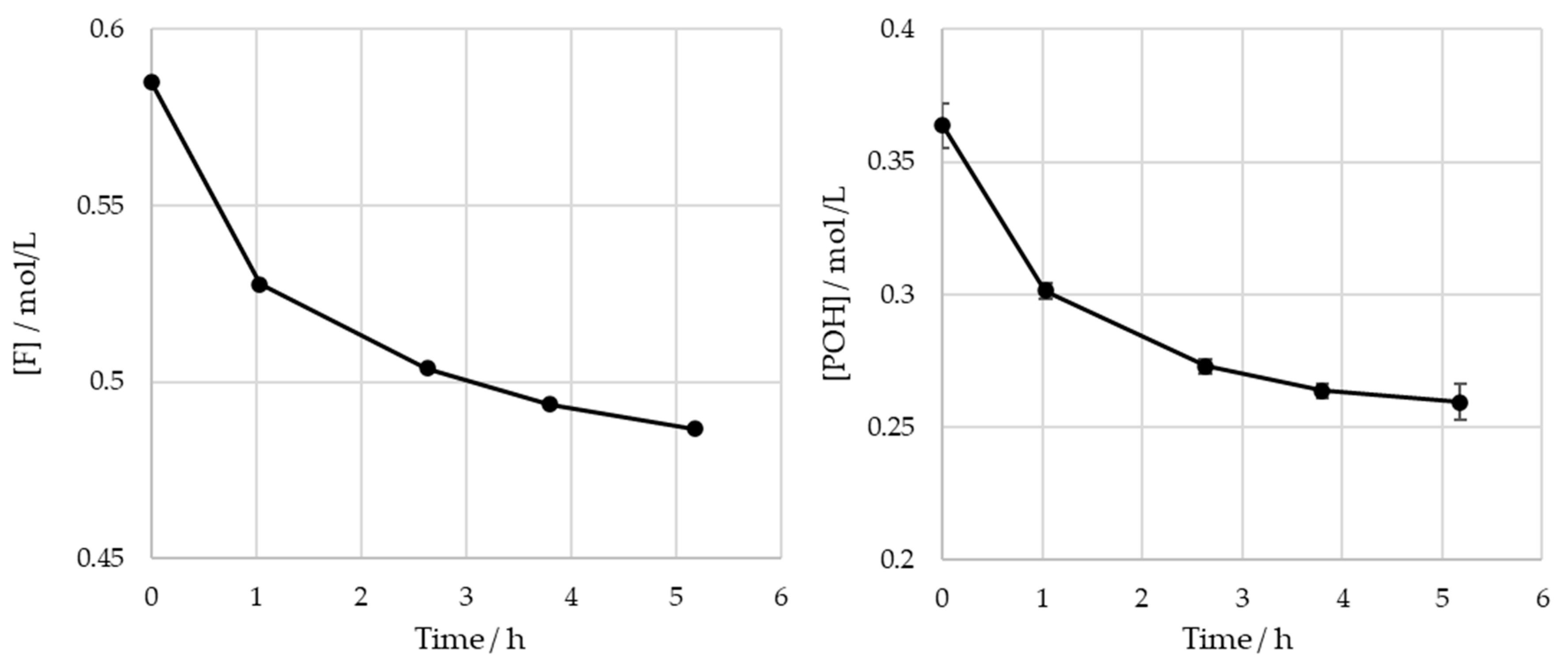
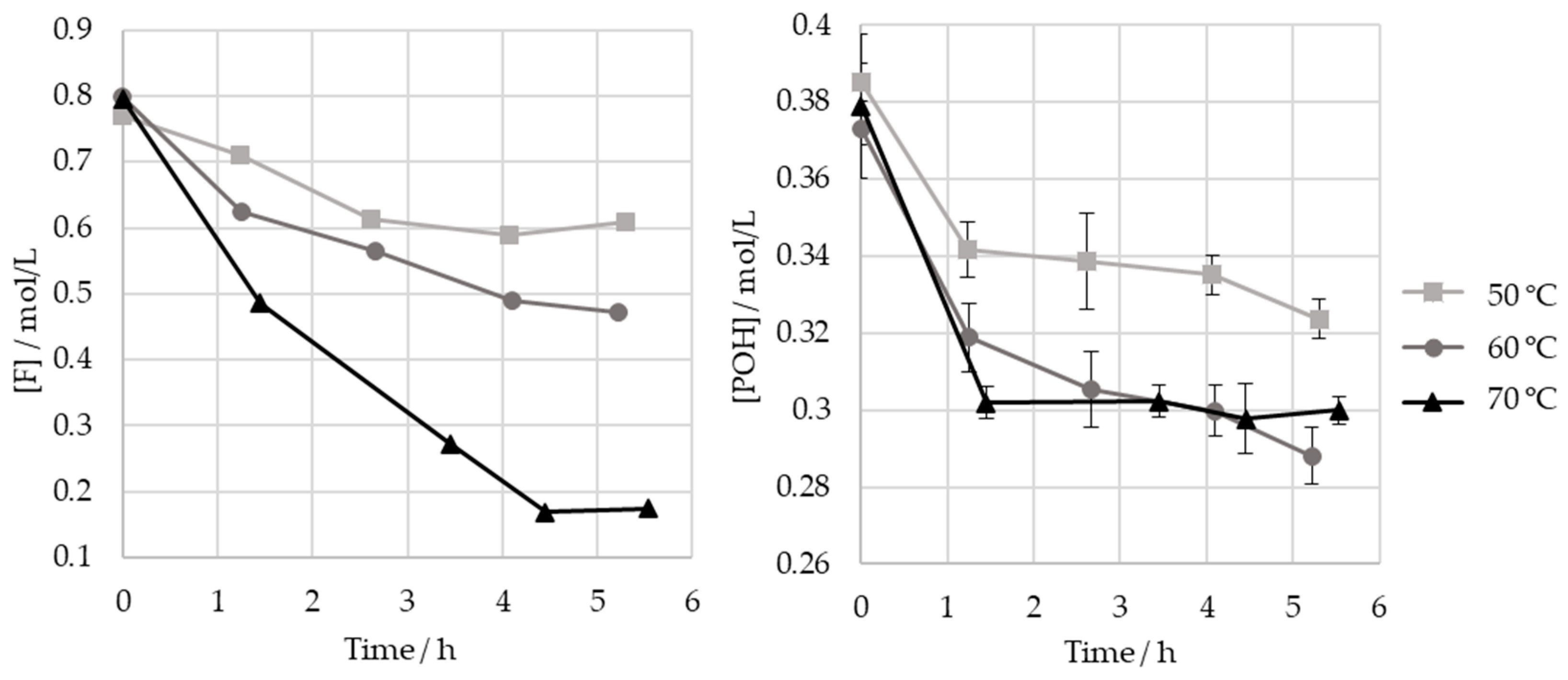
3.2. Monitoring of the Methylolation Reaction
3.2.1. Off-Line Methods
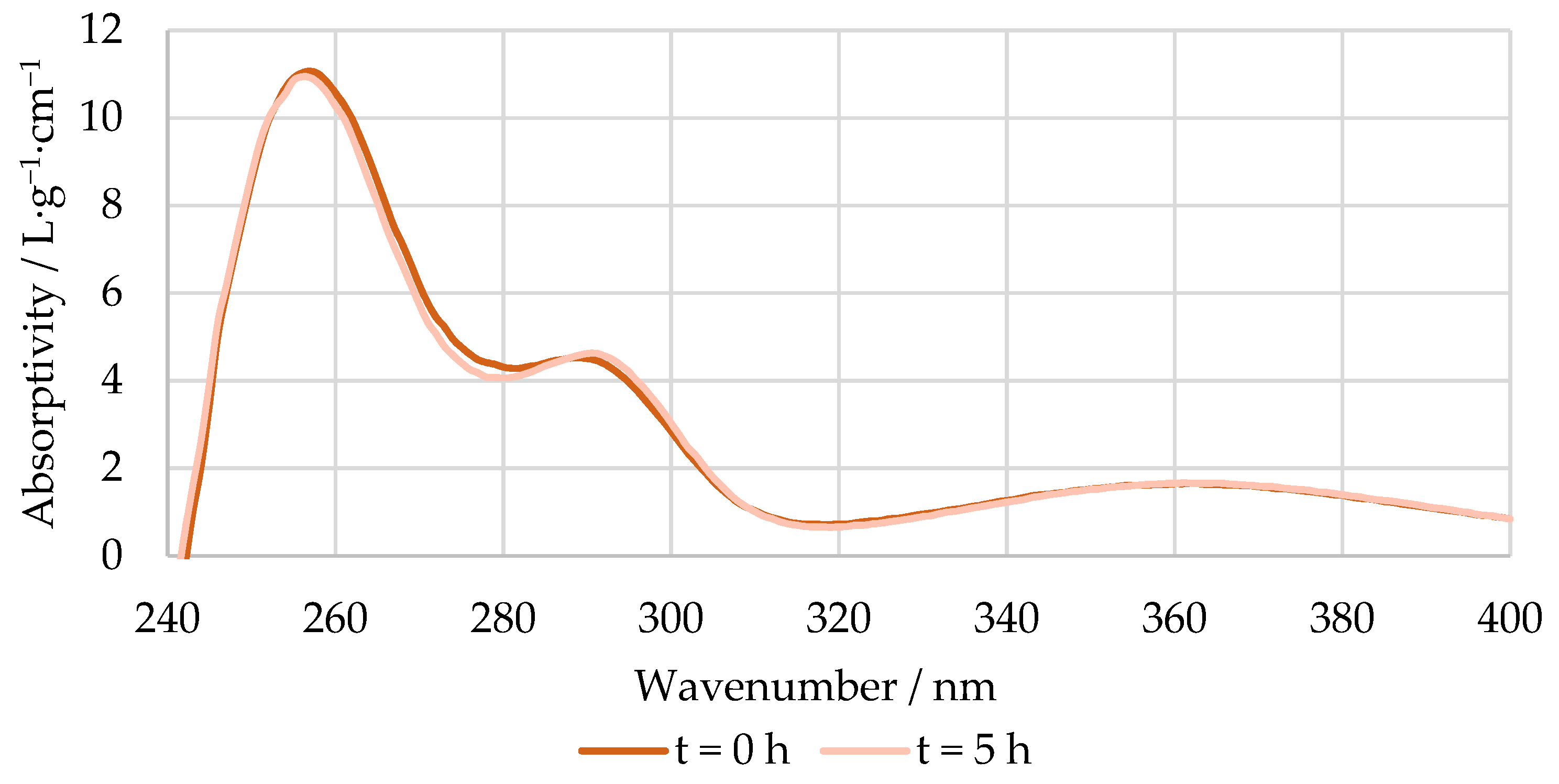
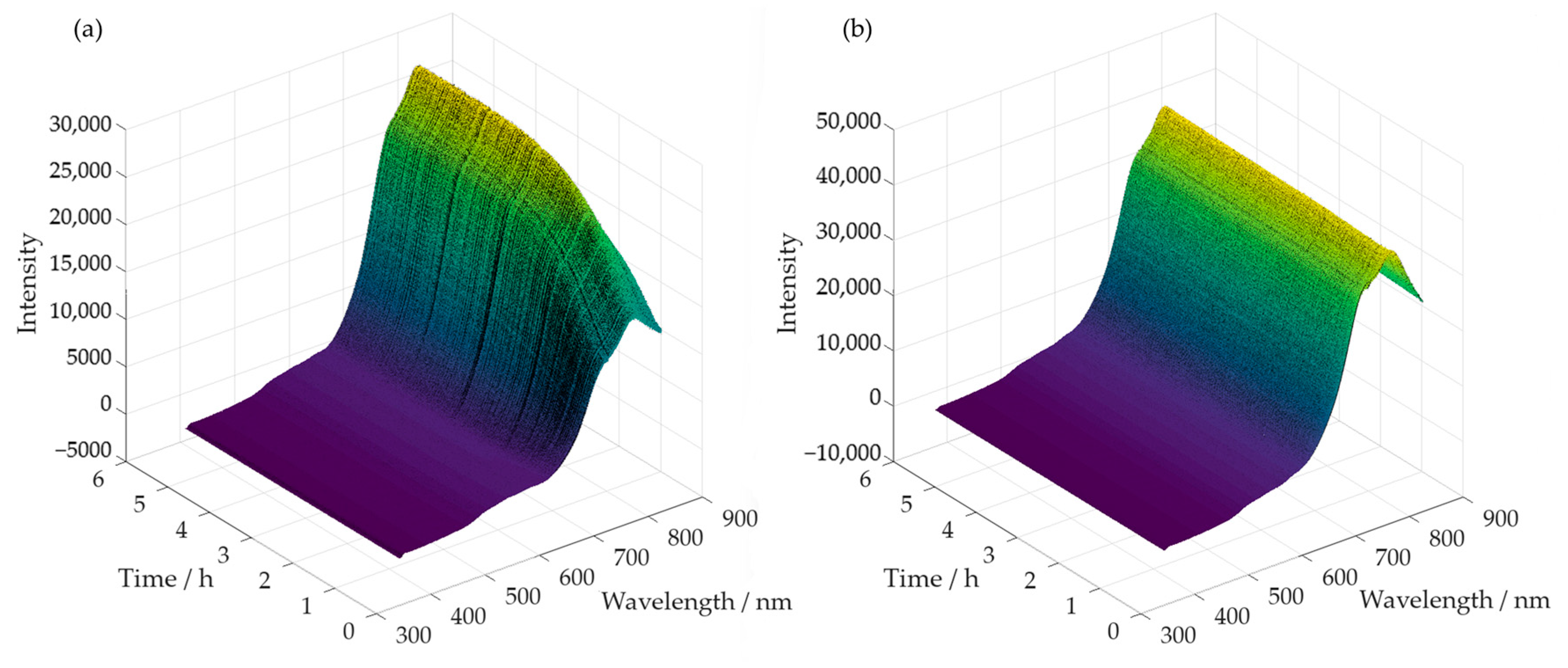
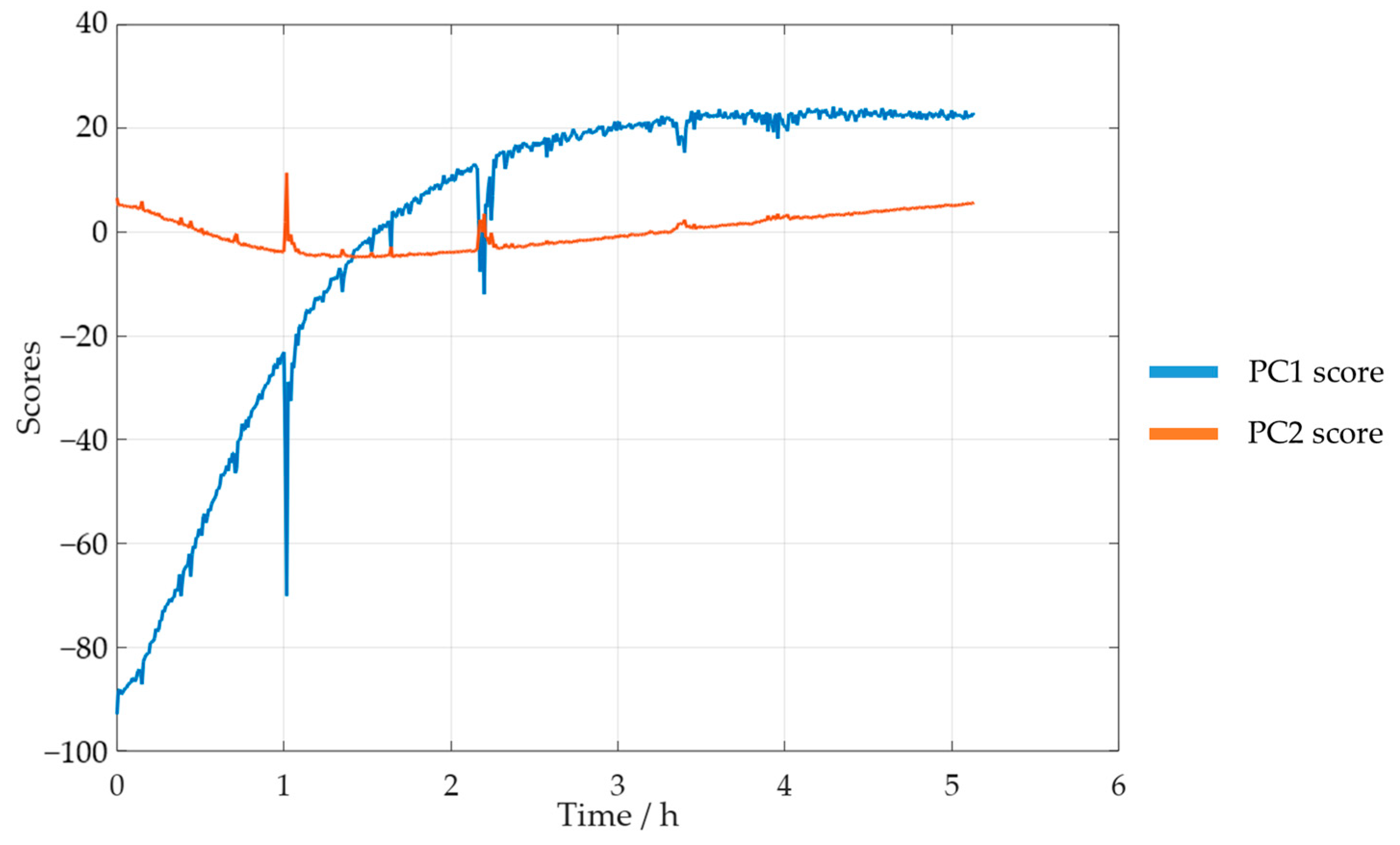
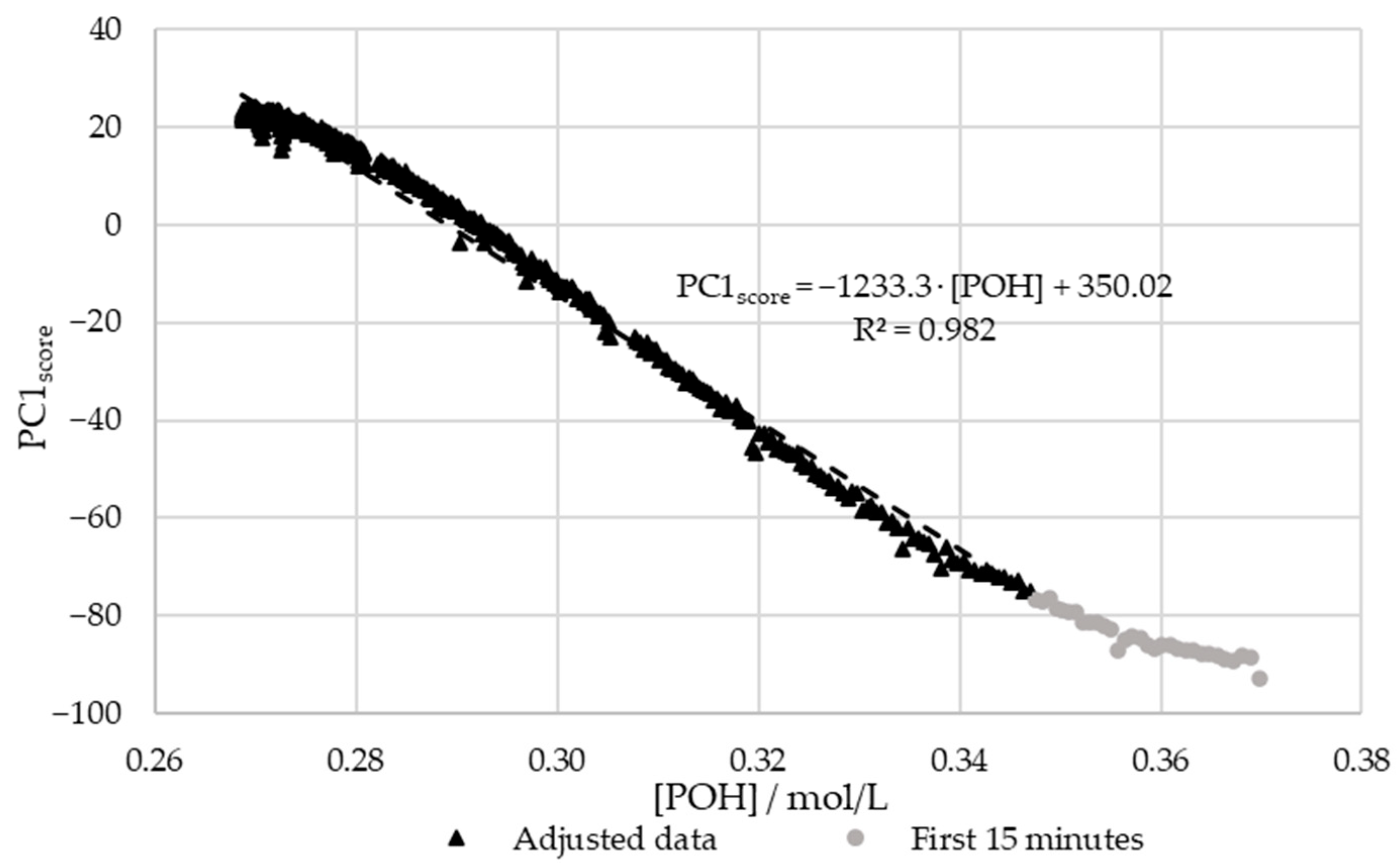
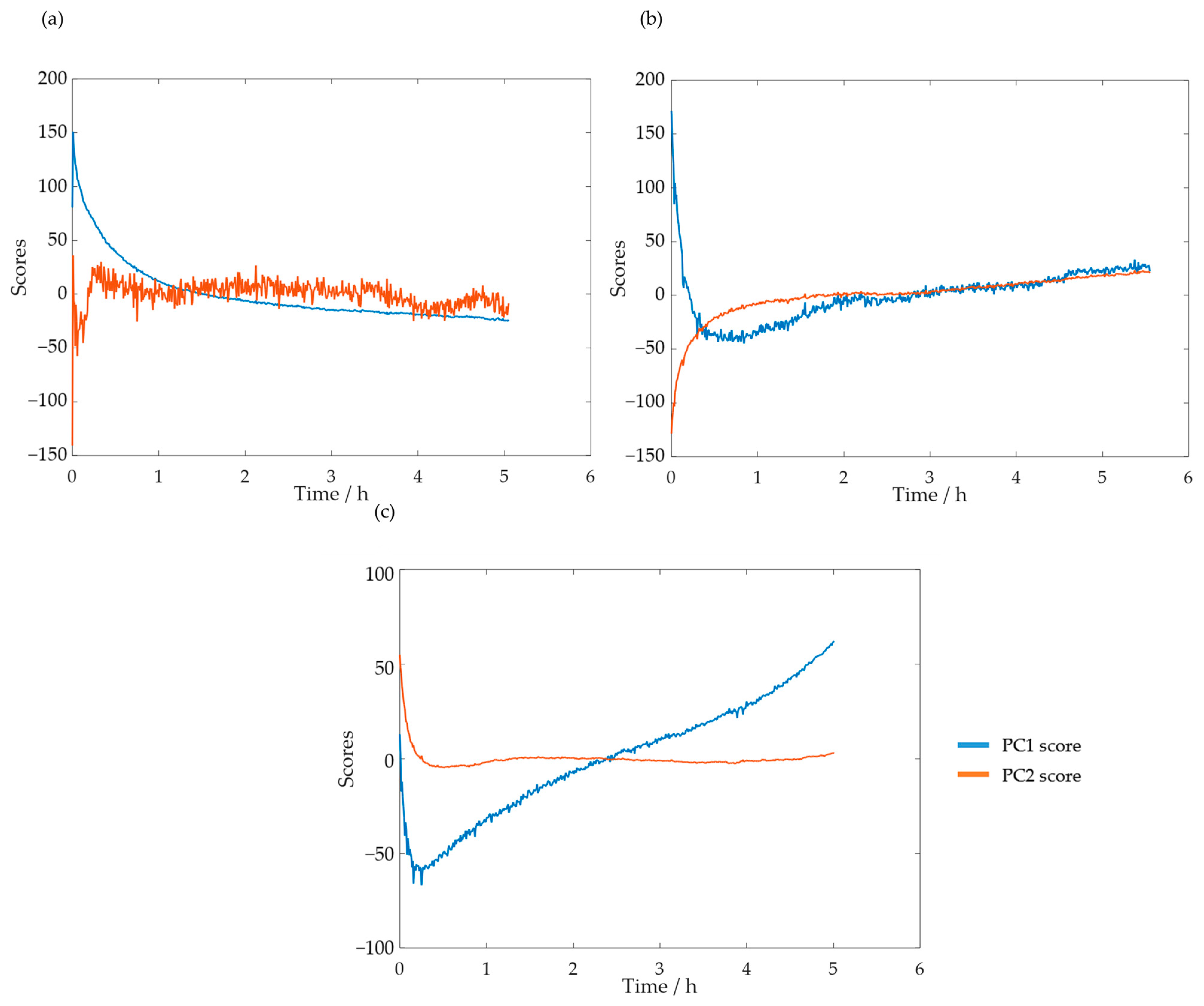
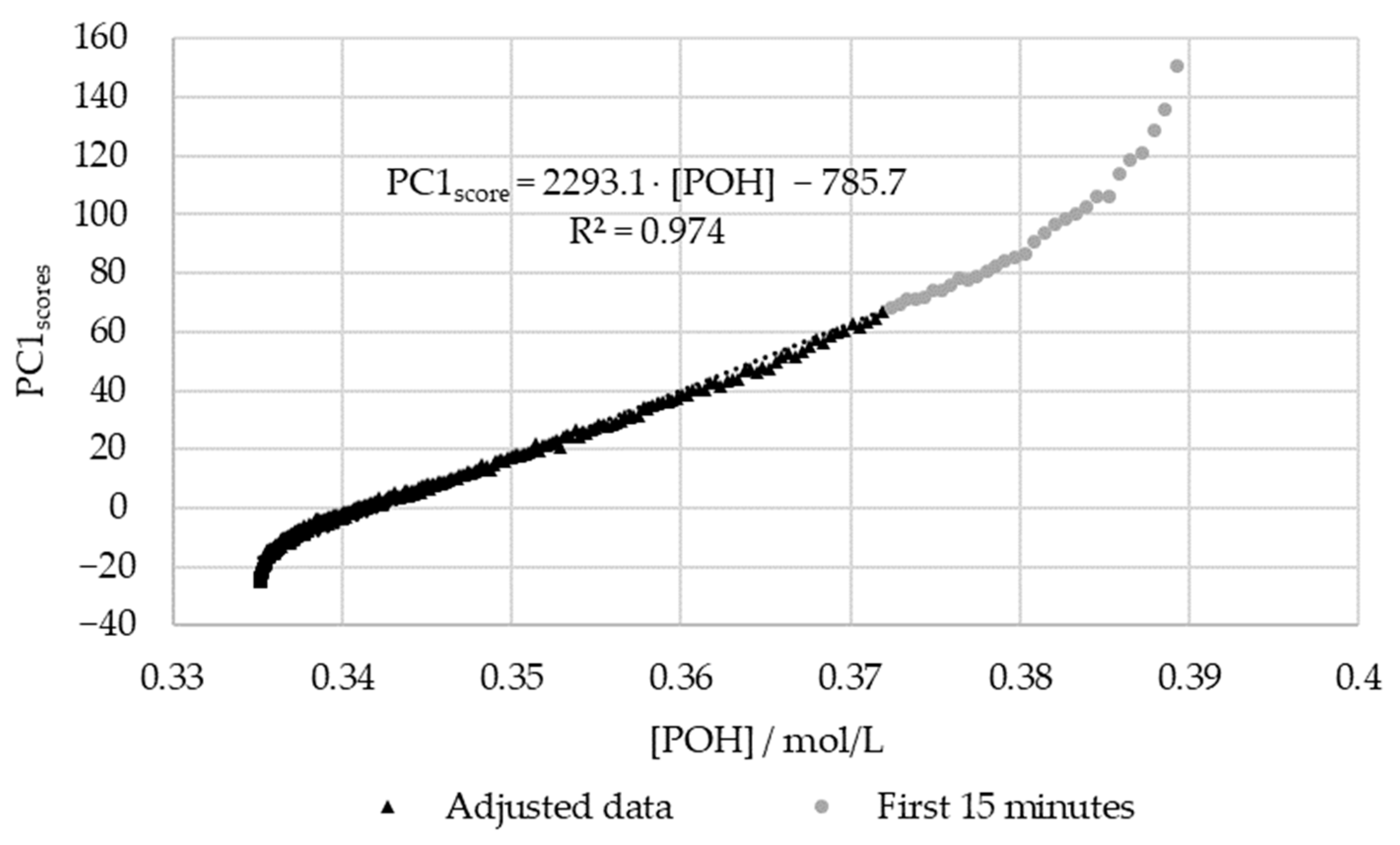
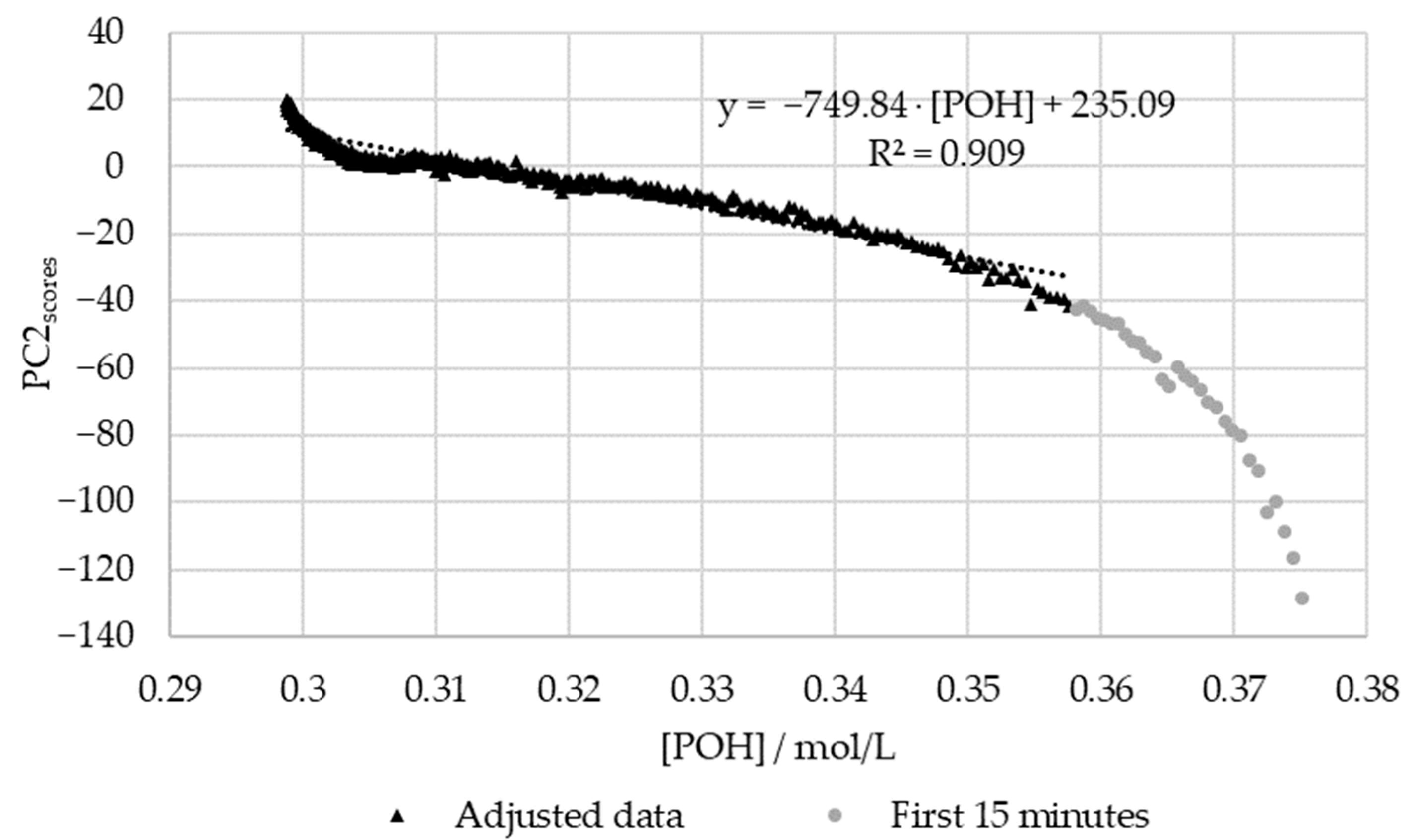
3.2.2. In-Line Method–Visible Spectroscopy
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Li, C.; Zhao, X.; Wang, A.; Huber, G.W.; Zhang, T. Catalytic Transformation of Lignin for the Production of Chemicals and Fuels. Chem. Rev. 2015, 115, 11559–11624. [Google Scholar] [CrossRef] [PubMed]
- Hemmilä, V.; Adamopoulos, S.; Karlsson, O.; Kumar, A. Development of sustainable bio-adhesives for engineered wood panels—A Review. RSC Adv. 2017, 7, 38604–38630. [Google Scholar] [CrossRef]
- El Mansouri, N.-E.; Pizzi, A.; Salvadó, J. Lignin-based wood panel adhesives without formaldehyde. Holz Als Roh Und Werkst. 2007, 65, 65–70. [Google Scholar] [CrossRef]
- Gonçalves, S.; Ferra, J.; Paiva, N.; Martins, J.; Carvalho, L.H.; Magalhães, F.D. Lignosulphonates as an Alternative to Non-Renewable Binders in Wood-Based Materials. Polymers 2021, 13, 4196. [Google Scholar] [CrossRef] [PubMed]
- Lubis, M.A.R.; Labib, A.; Sudarmanto; Akbar, F.; Nuryawan, A.; Antov, P.; Kristak, L.; Papadopoulos, A.N.; Pizzi, A. Influence of Lignin Content and Pressing Time on Plywood Properties Bonded with Cold-Setting Adhesive Based on Poly (Vinyl Alcohol), Lignin, and Hexamine. Polymers 2022, 14, 2111. [Google Scholar] [CrossRef] [PubMed]
- Hemmilä, V.; Adamopoulos, S.; Hosseinpourpia, R.; Ahmed, S.A. Ammonium Lignosulfonate Adhesives for Particleboards with pMDI and Furfuryl Alcohol as Crosslinkers. Polymers 2019, 11, 1633. [Google Scholar] [CrossRef]
- Ferreira, A.M.; Pereira, J.; Almeida, M.; Ferra, J.; Paiva, N.; Martins, J.; Magalhães, F.D.; Carvalho, L.H. Biosourced Binder for Wood Particleboards Based on Spent Sulfite Liquor and Wheat Flour. Polymers 2018, 10, 1070. [Google Scholar] [CrossRef]
- Ferreira, A.; Pereira, J.; Almeida, M.; Ferra, J.; Paiva, N.; Martins, J.; Magalhães, F.; Carvalho, L. Low-cost natural binder for particleboards production: Study of manufacture conditions and stability. Int. J. Adhes. Adhes. 2019, 93, 102325. [Google Scholar] [CrossRef]
- Yuan, Y.; Guo, M.H.; Liu, F.Y. Preparation and Evaluation of Green Composites Using Modified Ammonium Lignosulfonate and Polyethylenimine as a Binder. BioResources 2013, 9, 836–848. [Google Scholar] [CrossRef]
- Ji, X.; Guo, M. Preparation and properties of a chitosan-lignin wood adhesive. Int. J. Adhes. Adhes. 2018, 82, 8–13. [Google Scholar] [CrossRef]
- Rhazi, N.; Oumam, M.; Sesbou, A.; Hannache, H.; Charrier-El Bouhtoury, F. Physico-mechanical properties of plywood bonded with ecological adhesives from Acacia mollissima tannins and lignosulfonates. Eur. Phys. J. Appl. Phys. 2017, 78, 34813. [Google Scholar] [CrossRef]
- Tribot, A.; Amer, G.; Alio, M.A.; de Baynast, H.; Delattre, C.; Pons, A.; Mathias, J.-D.; Callois, J.-M.; Vial, C.; Michaud, P.; et al. Wood-lignin: Supply, extraction processes and use as bio-based material. Eur. Polym. J. 2019, 112, 228–240. [Google Scholar] [CrossRef]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The Catalytic Valorization of Lignin for the Production of Renewable Chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Gellerstedt, G.; Henriksson, G. Lignins: Major Sources, Structure and Properties. In Monomers, Polymers and Composites from Renewable Resources; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Younesi-Kordkheili, H.; Pizzi, A.; Honarbakhsh-Raouf, A.; Nemati, F. The effect of soda bagasse lignin modified by ionic liquids on properties of the urea–formaldehyde resin as a wood adhesive. J. Adhes. 2017, 93, 914–925. [Google Scholar] [CrossRef]
- Lourençon, T.V.; Alakurtti, S.; Virtanen, T.; Jääskeläinen, A.-S.; Liitiä, T.; Hughes, M.; Magalhaes, W.; Muniz, G.I.; Tamminen, T. Phenol-formaldehyde resins with suitable bonding strength synthesized from “less-reactive” hardwood lignin fractions. Holzforschung 2020, 74, 175–183. [Google Scholar] [CrossRef]
- Alonso, M.; Oliet, M.; Rodríguez, F.; García, J.; Gilarranz, M.; Rodríguez, J. Modification of ammonium lignosulfonate by phenolation for use in phenolic resins. Bioresour. Technol. 2005, 96, 1013–1018. [Google Scholar] [CrossRef]
- Hu, L.; Pan, H.; Zhou, Y.; Zhang, M. Methods to improve lignin’s reactivity as a phenol substitute and as replacement for other phenolic compounds: A brief review. Bioresources 2011, 6, 3515–3525. [Google Scholar] [CrossRef]
- Zuluaga, C.L.; Du, J.; Chang, H.-M.; Jameel, H.; Gonzalez, R.W. Lignin Modifications and Perspectives towards Applications of Phenolic Foams: A Review. BioResources 2018, 13, 9158–9179. [Google Scholar] [CrossRef]
- El Mansouri, N.-E.; Farriol, X.; Salvadó, J. Structural modification and characterization of lignosulfonate by a reaction in an alkaline medium for its incorporation into phenolic resins. J. Appl. Polym. Sci. 2006, 102, 3286–3292. [Google Scholar] [CrossRef]
- Younesi-Kordkheili, H.; Pizzi, A. A comparison between lignin modified by ionic liquids and glyoxalated lignin as modifiers of urea-formaldehyde resin. J. Adhes. 2017, 93, 1120–1130. [Google Scholar] [CrossRef]
- Gilca, I.A.; Ghitescu, R.E.; Puitel, A.C.; Popa, V.I. Preparation of lignin nanoparticles by chemical modification. Iran. Polym. J. 2014, 23, 355–363. [Google Scholar] [CrossRef]
- Alonso, M.V.; Rodríguez, J.J.; Oliet, M.; Rodríguez, F.; García, J.; Gilarranz, M.A. Characterization and structural modification of ammonic lignosulfonate by methylolation. J. Appl. Polym. Sci. 2001, 82, 2661–2668. [Google Scholar] [CrossRef]
- Peng, W.; Riedl, B.; Barry, A.O. Study on the kinetics of lignin methylolation. J. Appl. Polym. Sci. 1993, 48, 1757–1763. [Google Scholar] [CrossRef]
- Bernar, P.; Gonçalves, A.; Mandelli, D.; Ferreira, M.; Schuchardt, U. Principal Component Analysis of the Hydroxymethylation of Sugarcane Lignin: A Time-Depending Study by FTlR. J. Wood Chem. Technol. 1999, 19, 151–165. [Google Scholar] [CrossRef]
- Taverna, M.E.; Felissia, F.; Area, M.C.; Estenoz, D.A.; Nicolau, V.V. Hydroxymethylation of technical lignins from South American sources with potential use in phenolic resins. J. Appl. Polym. Sci. 2019, 136, 47712. [Google Scholar] [CrossRef]
- Van Der Klashorst, G.H. The Modification of Lignin at Positions 2 and 6 of the Phenylpropanoid Nucleii—Part II1: Hydroxymethylation of Lignin Model Compounds. J. Wood Chem. Technol. 1988, 8, 209–220. [Google Scholar] [CrossRef]
- Malutan, T.; Nicu, R.; Popa, V.I. Contribution to the Study of Hydroxymetylation Reaction of Alkali Lignin. Bioresources 2008, 3, 13–20. [Google Scholar]
- Pizzi, A. Lignin-Based Wood Adhesives. In Advanced Wood Adhesives Technology; Marcel Dekker: New York, NY, USA, 1994; pp. 219–242. [Google Scholar]
- Ogata, Y.; Kawasaki, A. The Kinetics of the Reaction of Formaldehyde with Ammonia. Bull. Chem. Soc. Jpn. 1964, 37, 514–519. [Google Scholar] [CrossRef]
- Khalili, K.N.M.; Peinder, P.; Donkers, J.; Gosselink, R.J.A.; Bruijnincx, P.C.A.; Weckhuysen, B.M. Monitoring Molecular Weight Changes during Technical Lignin Depolymerization by Operando Attenuated Total Reflectance Infrared Spectroscopy and Chemometrics. ChemSusChem 2021, 14, 5517–5524. [Google Scholar] [CrossRef]
- Cateto, C.; Barreiro, M.; Rodrigues, A. Monitoring of lignin-based polyurethane synthesis by FTIR-ATR. Ind. Crop. Prod. 2008, 27, 168–174. [Google Scholar] [CrossRef]
- Liu, L.-Y.; Bessler, K.; Chen, S.; Cho, M.; Hua, Q.; Renneckar, S. In-Situ Real-Time Monitoring of Hydroxyethyl Modification in Obtaining Uniform Lignin Derivatives. Eur. Polym. J. 2021, 142, 110082. [Google Scholar] [CrossRef]
- Marques, A.P. Caracterização e Transformação de Lenhosulfonatos de Eucalyptus Globulus. Doctoral Thesis, Universidade de Aveiro, Aveiro, Portugal, 2011. [Google Scholar]
- ISO 1762:2019; Paper, Board, Pulps and Cellulose Nanomaterials—Determination of Residue (Ash Content) on Ignition at 525 °C. ISO: Geneva, Switzerland, 2019.
- Lin, S.Y. Ultraviolet Spectrophotometry. In Methods in Lignin Chemistry; Lin, S.Y., Dence, C.W., Eds.; Springer: Berlin, Germany, 1992. [Google Scholar] [CrossRef]
- Marques, A.P.; Evtuguin, D.V.; Magina, S.; Amado, F.M.L.; Prates, A. Chemical Composition of Spent Liquors from Acidic Magnesium–Based Sulphite Pulping of Eucalyptus globulus. J. Wood Chem. Technol. 2009, 29, 322–336. [Google Scholar] [CrossRef]
- Gärtner, A.; Gellerstedt, G.; Tamminen, T. Determination of phenolic hydroxyl groups in residual lignin using a modified UV-method. Nord. Pulp Pap. Res. J. 1999, 14, 163–170. [Google Scholar] [CrossRef]
- Zakis, G.F. Functional Analysis of Lignins and Their Derivatives; TAPPI PRESS: Atlanta, GA, USA, 1994. [Google Scholar]
- Alonso Rubio, M. Formulación y Curado de Resinas Fenol-Formaldehído Tipo “Resol” Con Sustitución Parcial Del Fenol Por Lignosulfonatos Modificados. Ph.D. Thesis, Universidad Complutense de Madrid, Madrid, Spain, 2002. [Google Scholar]
- Ferreira, A.M.; Pereira, J.; Almeida, M.; Ferra, J.; Paiva, N.; Martins, J.; Carvalho, L.H.; Magalhães, F.D. Effect of spent sulfite liquor on urea-formaldehyde resin performance. J. Appl. Polym. Sci. 2019, 136, 47389. [Google Scholar] [CrossRef]
- Yan, M.; Yang, D.; Deng, Y.; Chen, P.; Zhou, H.; Qiu, X. Influence of pH on the behavior of lignosulfonate macromolecules in aqueous solution. Colloids Surf. A Physicochem. Eng. Asp. 2010, 371, 50–58. [Google Scholar] [CrossRef]
- ISO 11402:2004; Phenolic, Amino and Condensation Resins—Determination of Free-Formaldehyde Content. ISO: Geneva, Switzerland, 2004.
- Fogler, H.S. Elements of Chemical Reaction Engineering, 4th ed.; Prentice Hall: Hoboken, NJ, USA, 2006. [Google Scholar]
- Mansouri, M.; Harkat, M.-F.; Nounou, H.N.; Nounou, M.N. PCA and PLS-Based Generalized Likelihood Ratio for Fault Detection. In Data-Driven and Model-Based Methods for Fault Detection and Diagnosis; Elsevier: Amsterdam, The Netherlands, 2020; pp. 11–48. [Google Scholar]
- Carlson, R.; Carlson, J. Principal Properties. In Design and Optimization in Organic Synthesis; Rutan, S., Walczak, B., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; Volume 24, pp. 351–401. [Google Scholar]
- Kherif, F.; Latypova, A. Principal Component Analysis. In Machine Learning; Elsevier: Amsterdam, The Netherlands, 2020; pp. 209–225. [Google Scholar]
- Yuan, Q.; Lennox, B. Improved model predictive control using PCA. IFAC Proc. Vol. 2006, 39, 103–108. [Google Scholar] [CrossRef]
- Magina, S.P. New Polymeric Formulations Based on Lignosulphonates. Ph.D. Thesis, University of Aveiro, Aveiro, Portugal, 2021. [Google Scholar]
- Resende, M. Desenvolvimento de Uma Resina Fenol-Formaldeído Para Impregnação de Papel Kraft Para Produção de Laminados de Alta Pressão. Master’s Thesis, Faculty of Engineering of the University of Porto, Porto, Portugal, 2018. [Google Scholar]
- Marques, A.P.; Evtuguin, D.V.; Magina, S.; Amado, F.M.L.; Prates, A. Structure of Lignosulphonates from Acidic Magnesium-Based Sulphite Pulping of Eucalyptus globulus. J. Wood Chem. Technol. 2009, 29, 337–357. [Google Scholar] [CrossRef]
- Faix, O. Fourier Transform Infrared Spectroscopy. In Methods in Lignin Chemistry; Lin, S.Y., Dence, C.W., Eds.; Springer: Berlin, Germany, 1992. [Google Scholar] [CrossRef]
- Hemmilä, V.; Hosseinpourpia, R.; Adamopoulos, S.; Eceiza, A. Characterization of Wood-based Industrial Biorefinery Lignosulfonates and Supercritical Water Hydrolysis Lignin. Waste Biomass. Valorization 2020, 11, 5835–5845. [Google Scholar] [CrossRef]
- Bellamy, L.J. The Infrared Spectra of Complex Molecules—Advances in Infrared Group Frequencies, 2nd ed.; Chapman & Hall: London, UK, 1980; Volume 2. [Google Scholar]
- Higuchi, M.; Nohno, S.; Morita, M.; Tohmura, S.-I. Kinetics of the hydroxymethylation of phenol II: Values of rate parameters and results of simulation experiments. J. Wood Sci. 1999, 45, 306–312. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | HLS | SLS |
|---|---|---|
| Density (g/cm3) | 1.308 | - |
| Viscosity (cP) | 32 | - |
| pH | 2.48 | - |
| Dry matter (%) | 54.7 | 95.8 |
| LS content (%) | 33.5 | 72 |
| Carbohydrate content (%) | 12.6 b | 8.8 c |
| LS content (%) a | 61.2 | 75.1 |
| Ash content (%) a | 12.2 | 15.7 |
| Phenolic Hydroxyl Groups (%) on a Dry LS Basis | HLS | SLS |
|---|---|---|
| [OH]I+II+III+IV | 2.69 | 2.23 |
| [OH]I+II | 1.76 | 1.37 |
| [OH]III+IV | 0.93 | 0.86 |
| Wavenumber (cm−1) | Band Origin and Comments | HLS | SLS |
|---|---|---|---|
| 3420–3250 | O-H stretch (phenolic and aliphatic OH) | 3332 | 3332 |
| 3000–2842 | C-H stretch in methyl and methylene groups | 2940 | 2938 |
| 2850–2840 | C-H stretching (OCH3) | 2844 | 2843 |
| 2000–1650 | Several bands from overtones and combinations (substituted benzene rings) | 1766 | 1769 |
| 1738–1709 | C=O stretch in unconjugated ketones, carbonyls and in ester groups (frequently of carbohydrate origin) | 1704 | 1714 |
| 1605–1593 | Aromatic skeletal vibrations; C=O stretch | 1613 | 1601 |
| 1515–1505 | Aromatic skeletal vibrations | 1515 | 1510 |
| 1470–1460 | C-H deformations (asymmetric in CH3 and CH2) | 1461 | 1452 |
| 1430–1422 | Aromatic skeletal vibrations and C-H in-plane deformation | 1426 | 1418 |
| 1370–1365 | Aliphatic C-H stretch in CH3, not in OCH3; phenolic OH | 1367 | 1370 |
| 1330–1325 | S ring and G ring substituted in C5 | 1328 | - |
| 1270–1266 | G ring; C=O stretch | - | 1261 |
| 1260–1150 | Sulphonic acids | 1209 | 1205 |
| 1166 | C=O in conjugated ester groups | 1151 | - |
| 1140 | Aromatic C-H in-plane deformation; typical for G units | - | 1140 |
| 1128–1125 | Aromatic C-H in-plane deformation (S units); secondary alcohols; C=O stretch | 1111 | - |
| 1080–1010 | Characteristic LS peak at 1037 cm−1; sulfonic acids; deformation of aromatic C-H and C-O in primary alcohols; C=O stretch unconjugated | 1034 | 1030 |
| 858–853 | C-H out-of-plane in positions 2, 5, and 6 of G units | - | 865 |
| 832–817 | C-H out-of-plane in positions 2, 5, and 6 of G units | - | 809 |
| 700–600 | Sulphonic acids | 644 | 648 |
| Sample | Lignin | Formaldehyde | ||||
|---|---|---|---|---|---|---|
| Dry Mass/g | Concentration of Pure Lignin/g/L | [POH]/mol/L | Mass/g | Mass Ratio F:LS (Impure) | [F]/mol/L | |
| SLS | 95.8 | 280 | 0.36 | 16.3 | 0.17 | 0.59 |
| HLS | 115 | 280 | 0.38 | 19.5 | 0.17 | 0.79 |
| Temperature/°C | 50 | 60 | 70 |
|---|---|---|---|
| F consumed/mol/L | 0.16 | 0.33 | 0.62 |
| [POH] consumed/mol/L | 0.062 | 0.085 | 0.080 |
| Sample | SLS | HLS | HLS |
|---|---|---|---|
| Temperature/°C | 60 | 60 | 50 |
| /L mol−1 h−1 | 1.53 | 1.32 | 2.02 |
| /L mol−1 h−1 | - | 0.14 | 0.06 |
| 71 | 79 | 86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonçalves, S.; Martins, J.; Paiva, N.T.; Paiva, D.; Carvalho, L.H.; Magalhães, F.D. The Potential of Visible Spectroscopy as a Tool for the In-Line Monitoring of Lignin Methylolation. Polymers 2023, 15, 178. https://doi.org/10.3390/polym15010178
Gonçalves S, Martins J, Paiva NT, Paiva D, Carvalho LH, Magalhães FD. The Potential of Visible Spectroscopy as a Tool for the In-Line Monitoring of Lignin Methylolation. Polymers. 2023; 15(1):178. https://doi.org/10.3390/polym15010178
Chicago/Turabian StyleGonçalves, Sofia, Jorge Martins, Nádia T. Paiva, Diana Paiva, Luísa H. Carvalho, and Fernão D. Magalhães. 2023. "The Potential of Visible Spectroscopy as a Tool for the In-Line Monitoring of Lignin Methylolation" Polymers 15, no. 1: 178. https://doi.org/10.3390/polym15010178
APA StyleGonçalves, S., Martins, J., Paiva, N. T., Paiva, D., Carvalho, L. H., & Magalhães, F. D. (2023). The Potential of Visible Spectroscopy as a Tool for the In-Line Monitoring of Lignin Methylolation. Polymers, 15(1), 178. https://doi.org/10.3390/polym15010178