
Recent Advances in Chemically Modified Cellulose and Its Derivatives for Food Packaging Applications: A Review



Abstract
:1. Introduction
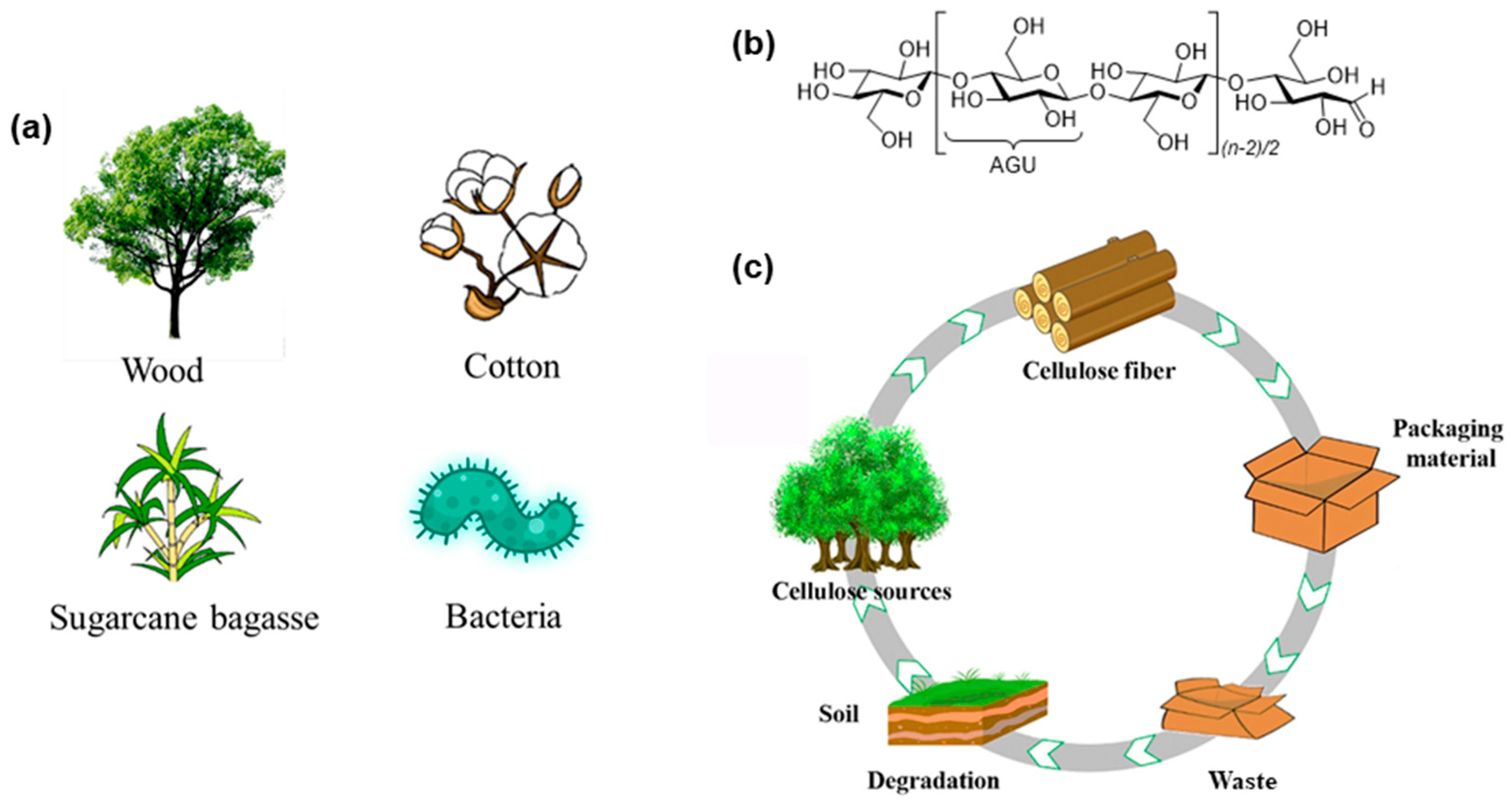
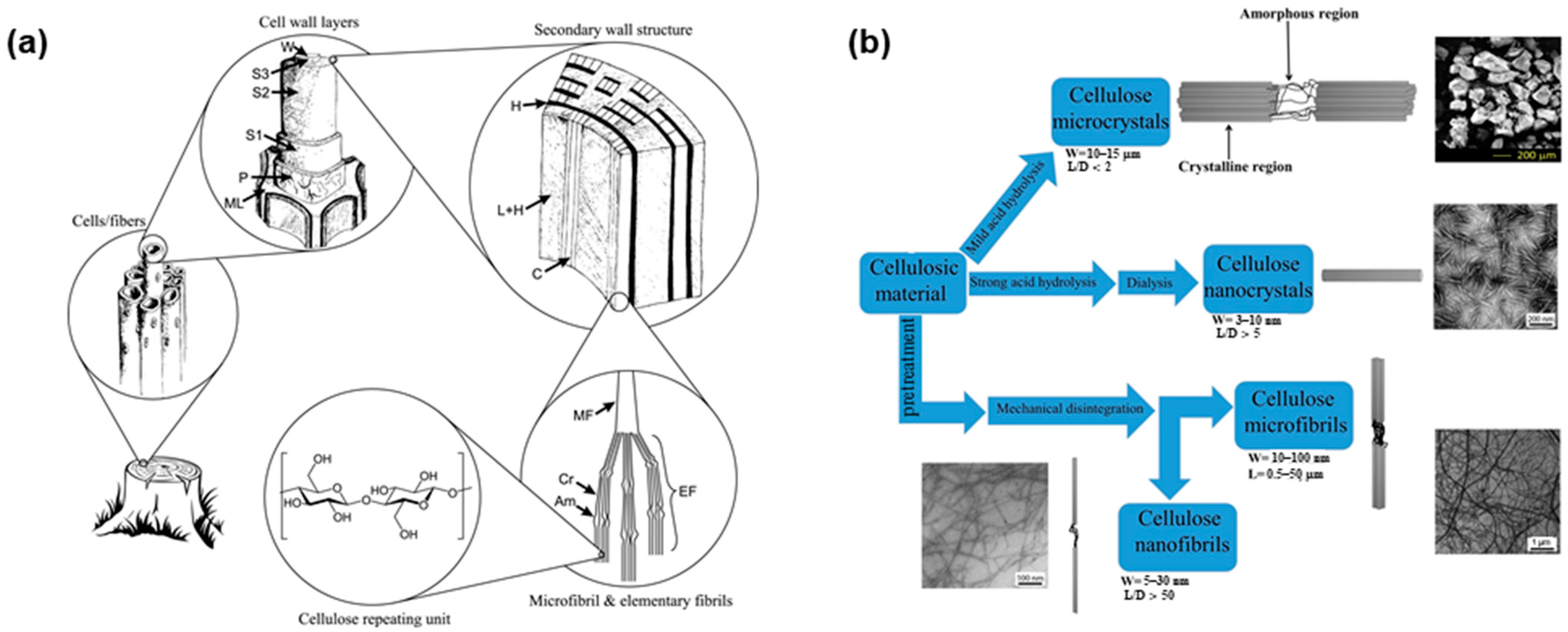
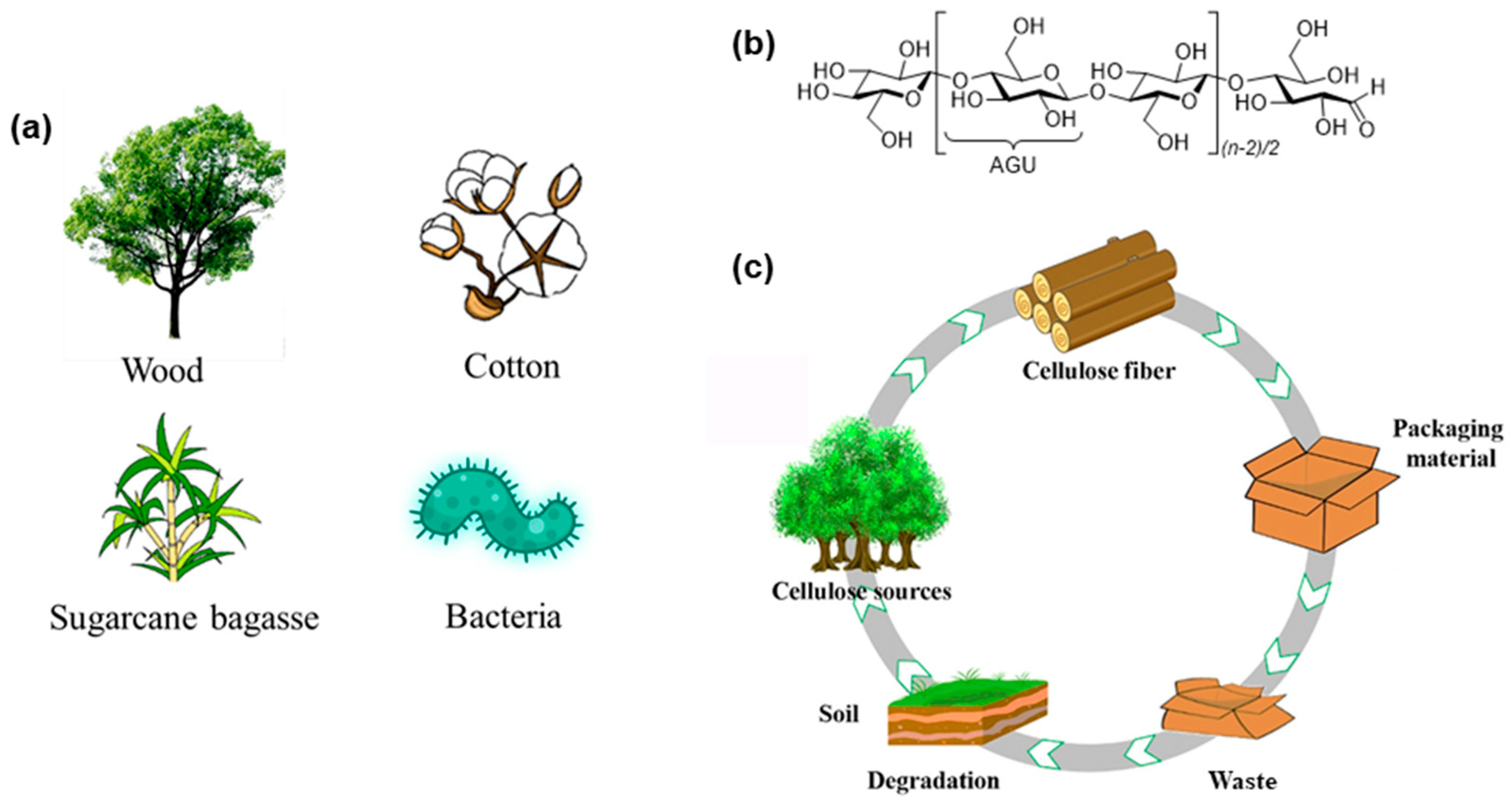
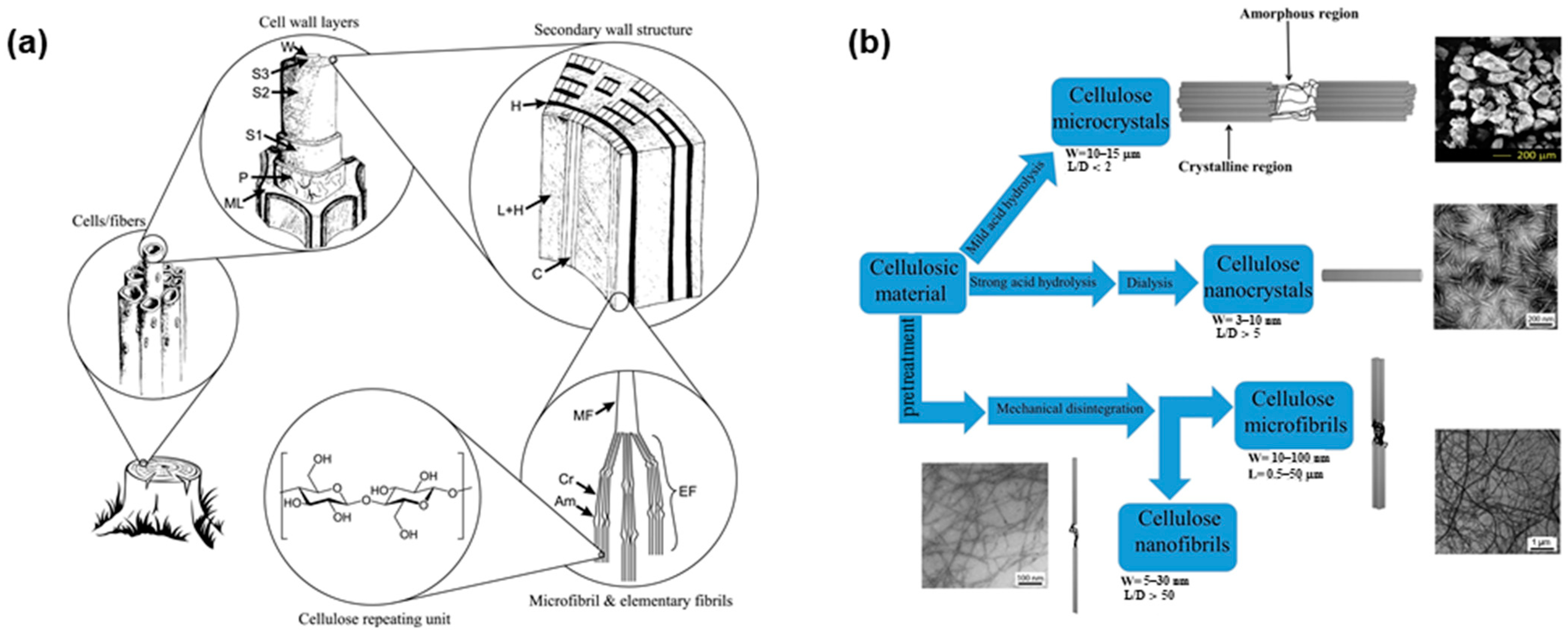
2. Cellulose Sources, Extraction Methods, and Size, Crystallinity and Functionality of Fibers
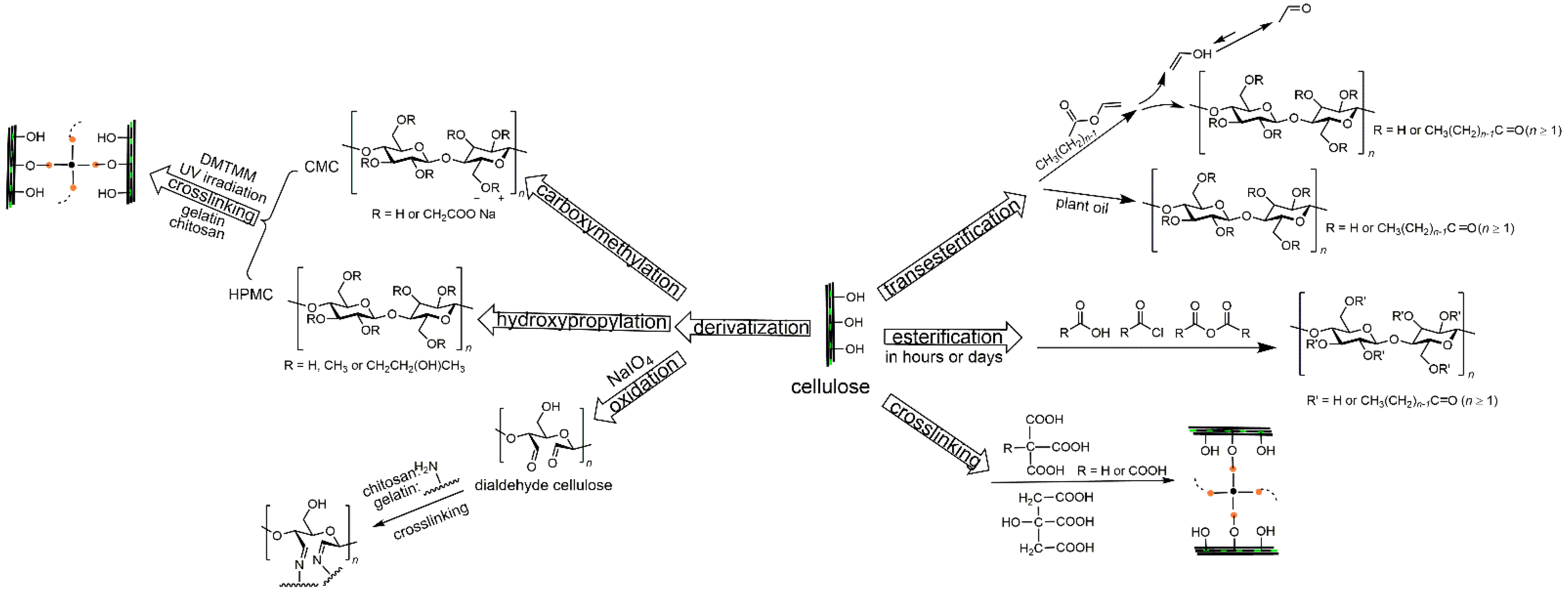
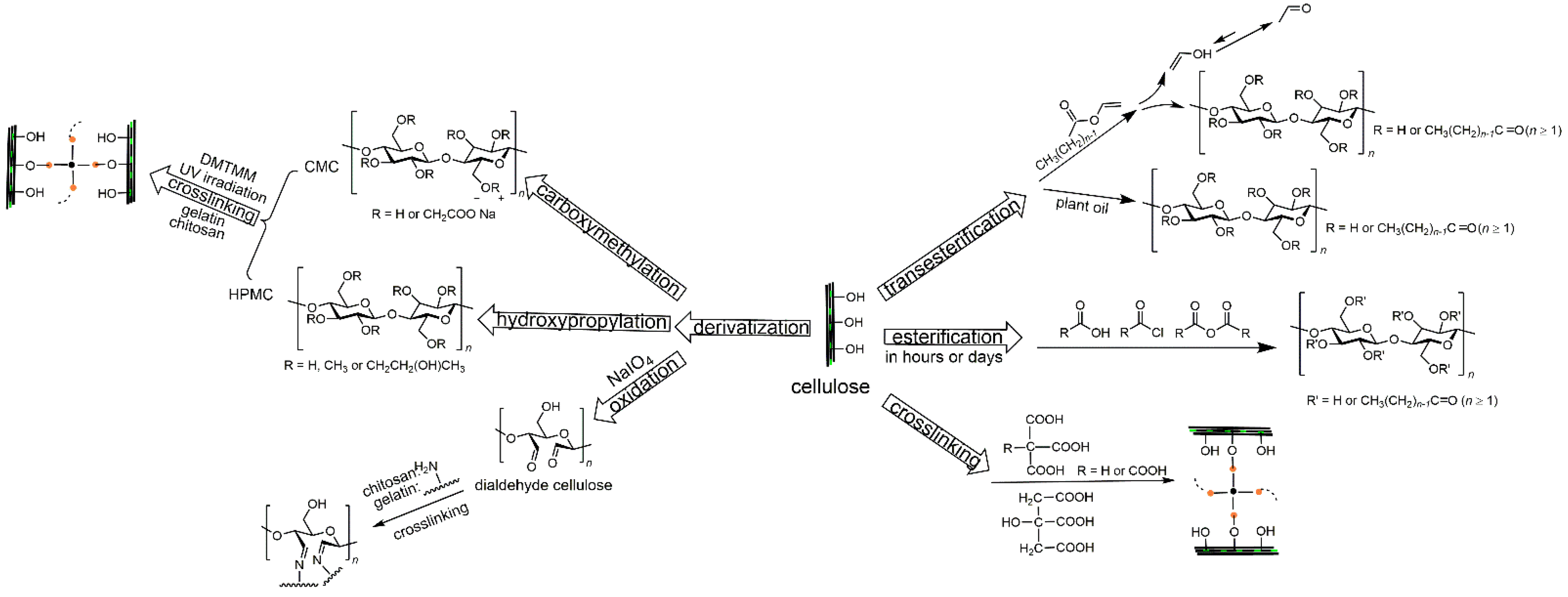
3. Esterification
3.1. Esterification with Acylants Bearing a Short Substituent Chain (C2–C6)
3.1.1. Synthesis of CESs with and without Solvents
3.1.2. CESs in Food Packaging Applications
3.1.3. CES-Based Food Packaging Containing Various Additives
3.2. Esterification with Acylants Bearing Medium (C8–C12) or Long Substituent Chains (>C12)
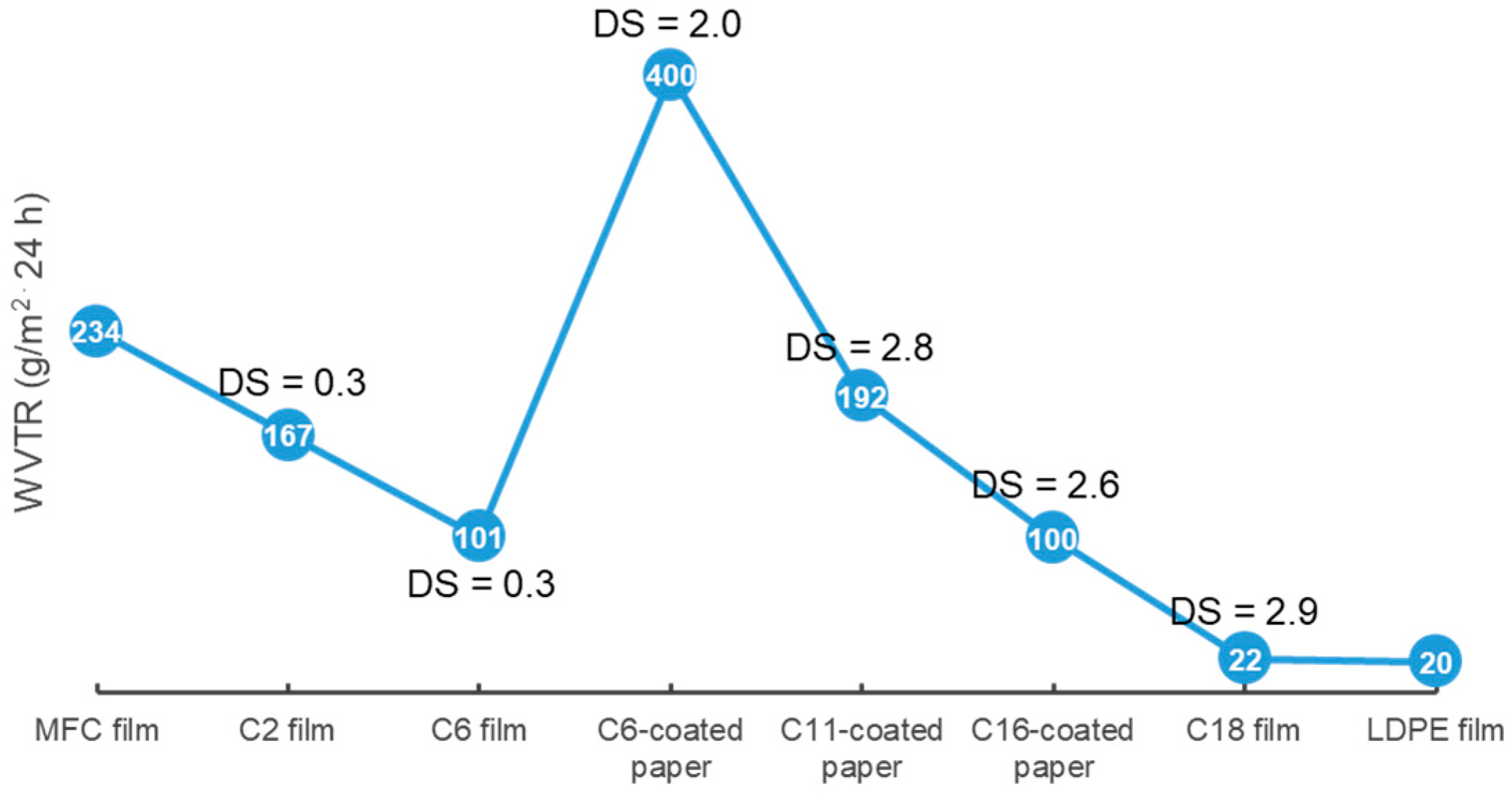
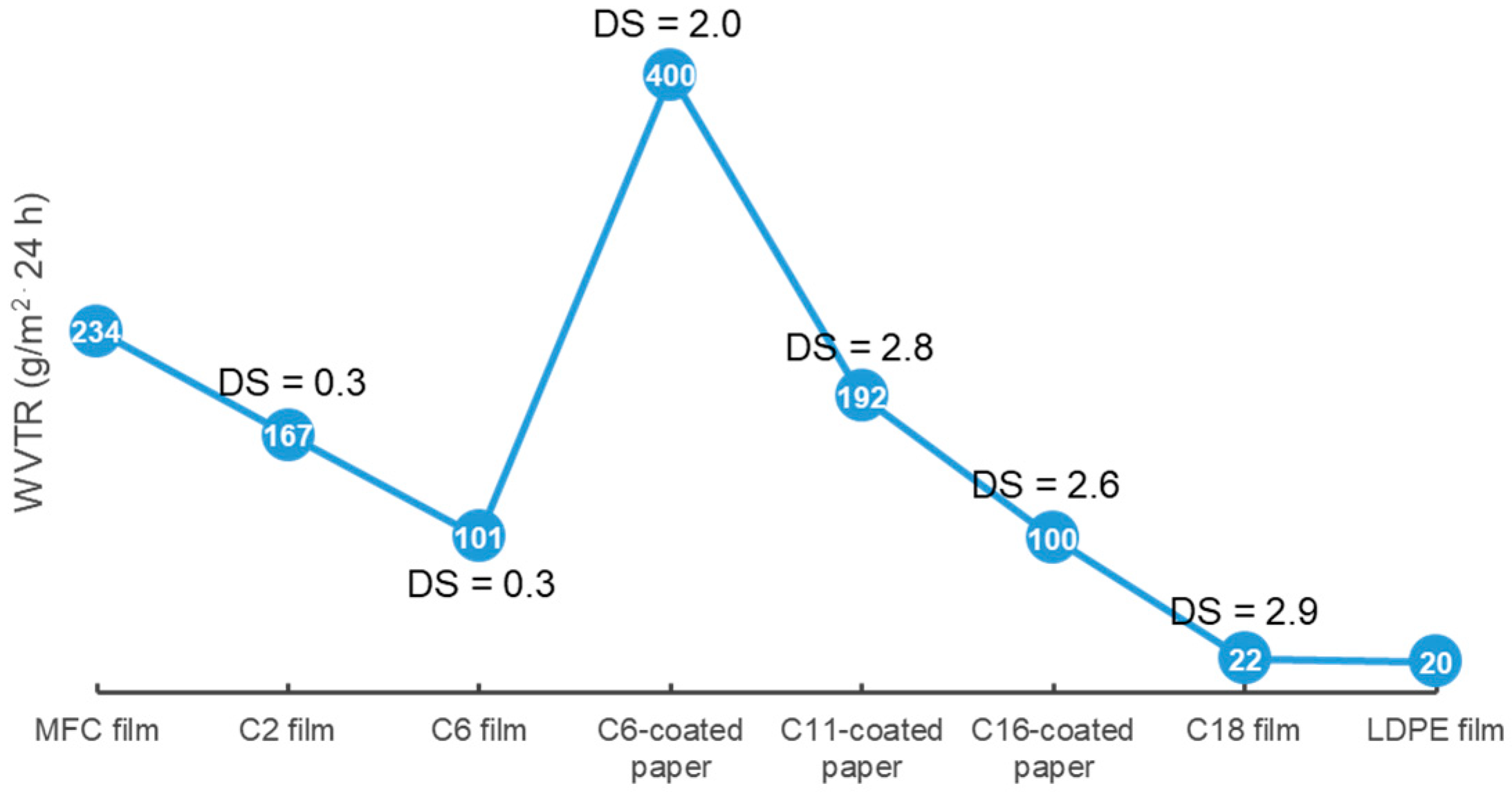
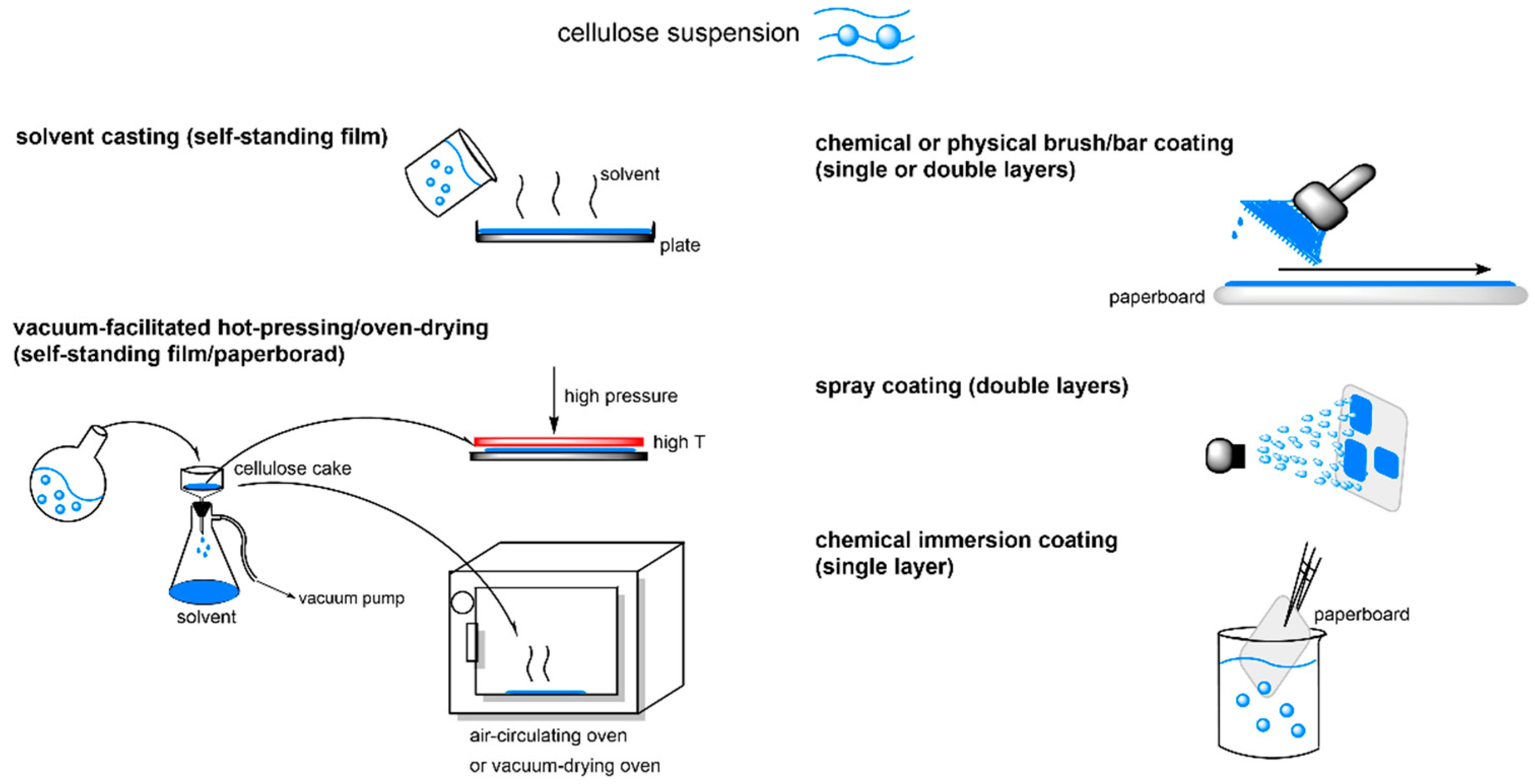
3.2.1. Influence of Substituent Length and DS on Surface Hydrophobicity and WVTR of Cellulose Esters with Medium (CEMs) and Long Substitution Chains (CELs)
3.2.2. Influence of Substituent Length on OTR of CEMs and CELs
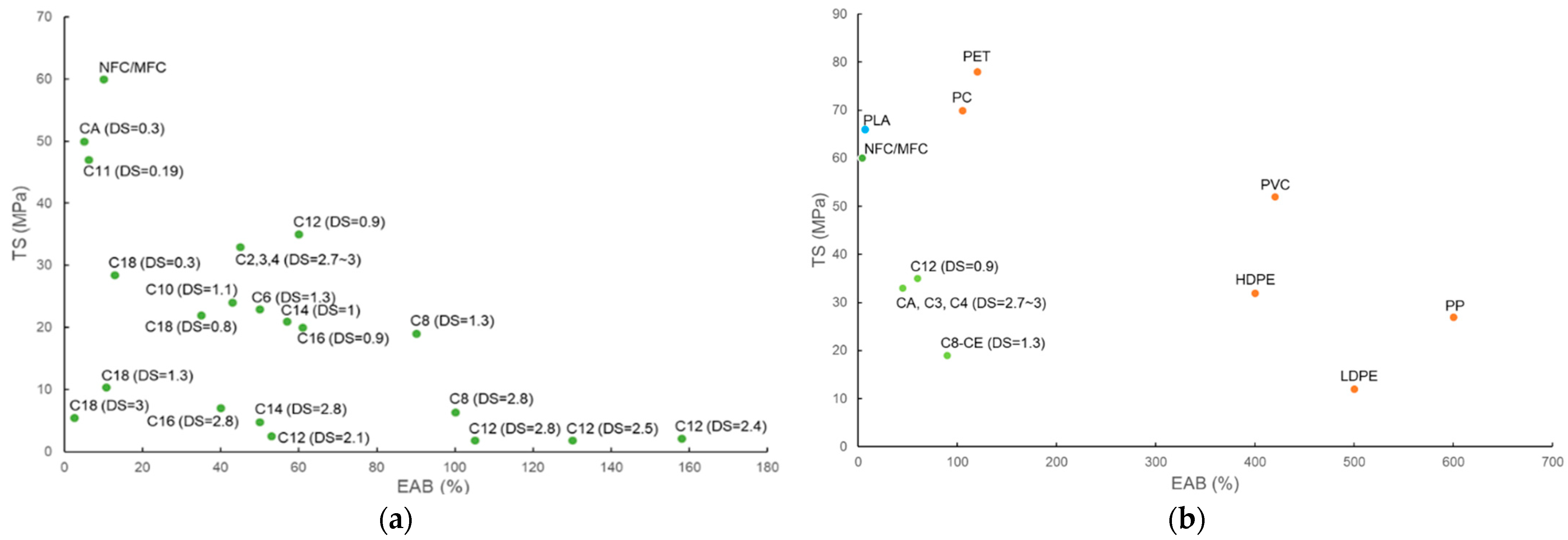
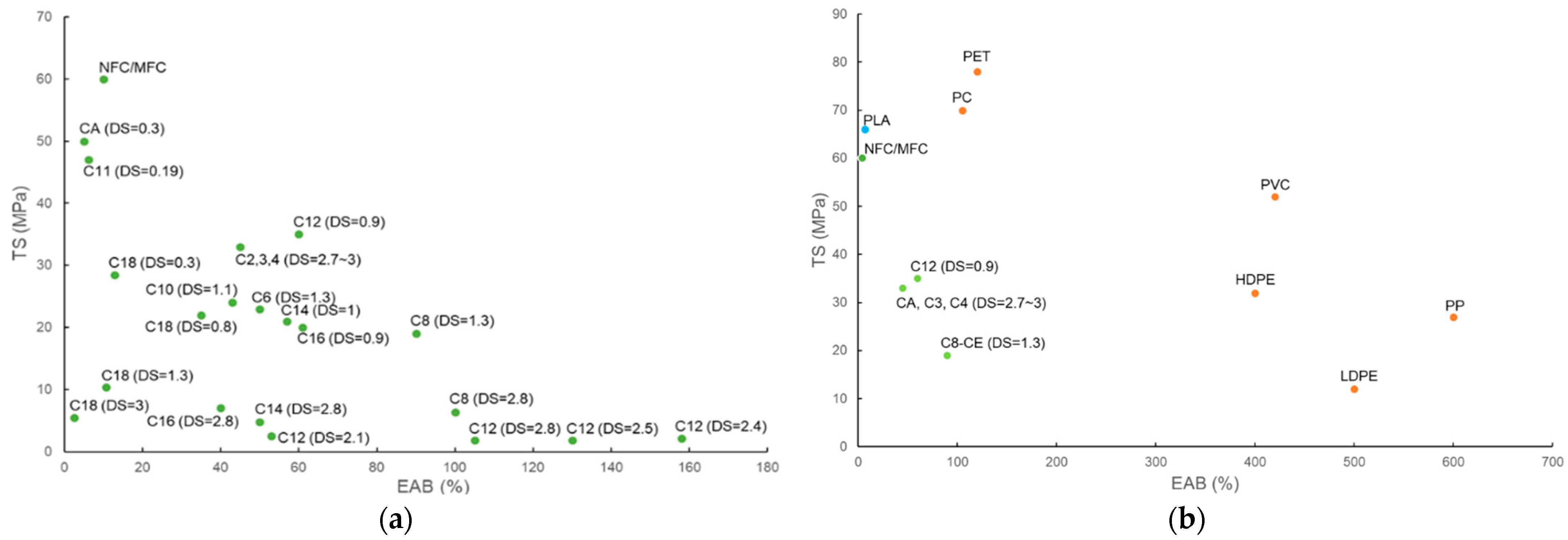
3.2.3. Influence of Substituent Length and DS on the Mechanical Strength of CEMs and CELs
3.2.4. Thermoplasticity of CELs
4. Transesterification
4.1. Transesterification with Vinyl Esters as Acylants
4.2. Transesterification with Plant Oils as Acylants
5. Crosslinking
5.1. Crosslinking on Hydroxyl Groups of Cellulose
5.2. Crosslinking with Functional Groups of Cellulose Derivatives
6. Degradability of Esterified, Cross-Linked Cellulose and Its Derivatives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviation
| Abbr. | Full Name |
| σ | tensile strength |
| ε | elongation at break |
| AGU | β-1,4-linked D-anhydroglucose units |
| BNF | bacterial nanofibrils |
| BTCA | 1,2,3,4-butanetetra-carboxylic acid |
| CA | cellulose acetate |
| CAB | cellulose acetate butyrate |
| CAC | citric acid |
| CB | cellulose butyrate |
| CESs | cellulose esters with short substitution chains (C2–C6) |
| CEMs | cellulose esters with medium substitution chains (C8–C12) |
| CELs | cellulose esters with long substitution chains (> C12) |
| CH | cellulose hexanoate |
| CIN | cinnamaldehyde |
| CMC | cellulose microcrystals |
| CMCS | carboxymethyl cellulose |
| CMF | cellulose microfibrils |
| CNC | cellulose nanocrystals |
| CNF | cellulose nanofibrils |
| CP | cellulose propionate |
| DACNF | dialdehyde CNF |
| DI water | deionized water |
| DMAP | 4-dimethylaminopyridine |
| DMTMM | 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride |
| DS | degree of substitution |
| E | Young’s modulus |
| G-AgNPs | green-synthesized silver nanoparticles |
| G-DACNF | gelatin-crosslinked DACNF |
| GLA | glutaraldehyde |
| HPMC | hydroxy propyl methyl cellulose |
| OP | oxygen permeability |
| OTR | oxygen transmission rate |
| RT | room temperature |
| SA | succinic acid |
| SB | sodium benzoate |
| TCA | tricarballylic acid |
| Td-onset | decomposition onset temperature |
| Tm | melting temperature |
| TOFA | tall oil fatty acid |
| WA | water absorption |
| WCA | water contact angle |
| WS | water solubility |
| WVP | water vapor permeability |
| WVTR | water vapor transmission rate |
References
- Wang, J.; Emmerich, L.; Wu, J.; Vana, P.; Zhang, K. Hydroplastic polymers as eco-friendly hydrosetting plastics. Nat. Sustain. 2021, 4, 877–883. [Google Scholar] [CrossRef]
- Guan, Q.F.; Yang, H.B.; Han, Z.M.; Ling, Z.C.; Yang, K.P.; Yin, C.H.; Yu, S.H. Plant Cellulose Nanofiber-Derived Structural Material with High-Density Reversible Interaction Networks for Plastic Substitute. Nano Lett. 2021, 21, 8999–9004. [Google Scholar] [CrossRef]
- Geyer, R.; Jambeck, J.R.; Law, K.L. Production, use, and fate of all plastics ever made. Sci. Adv. 2017, 3, e1700782. [Google Scholar] [CrossRef] [Green Version]
- Shieh, P.; Zhang, W.; Husted, K.E.L.; Kristufek, S.L.; Xiong, B.; Lundberg, D.J.; Lem, J.; Veysset, D.; Sun, Y.; Nelson, K.A.; et al. Cleavable comonomers enable degradable, recyclable thermoset plastics. Nature 2020, 583, 542–547. [Google Scholar] [CrossRef]
- Bugatti, V.; Viscusi, G.; Gorrasi, G. Formulation of a Bio-Packaging Based on Pure Cellulose Coupled with Cellulose Acetate Treated with Active Coating: Evaluation of Shelf Life of Pasta Ready to Eat. Foods 2020, 9, 1414. [Google Scholar] [CrossRef]
- Liu, C.; Luan, P.; Li, Q.; Cheng, Z.; Sun, X.; Cao, D.; Zhu, H. Biodegradable, Hygienic, and Compostable Tableware from Hybrid Sugarcane and Bamboo Fibers as Plastic Alternative. Matter 2020, 3, 2066–2079. [Google Scholar] [CrossRef]
- Lu, Y.; Weng, L.; Zhang, L. Morphology and Properties of Soy Protein Isolate Thermoplastics Reinforced with Chitin Whiskers. Biomacromolecules 2004, 5, 1046–1051. [Google Scholar] [CrossRef]
- Ma, X.; Chang, P.R.; Yu, J.; Stumborg, M. Properties of biodegradable citric acid-modified granular starch/thermoplastic pea starch composites. Carbohyd. Polym. 2009, 75, 1–8. [Google Scholar] [CrossRef]
- Chen, M.-J.; Li, R.-M.; Zhang, X.-Q.; Feng, J.; Feng, J.; Liu, C.-F.; Shi, Q.-S. Homogeneous Transesterification of Sugar Cane Bagasse toward Sustainable Plastics. ACS Sustain. Chem. Eng. 2016, 5, 360–366. [Google Scholar] [CrossRef]
- He, Y.; Tang, H.; Chen, Y.; Zhang, S. Facile Strategy to Construct Metal–Organic Coordination Thermoplastic Starch with High Hydrophobicity, Glass-Transition Temperature, and Improved Shape Recovery. ACS Sustain. Chem. Eng. 2020, 8, 8655–8663. [Google Scholar] [CrossRef]
- Ortega-Toro, R.; Morey, I.; Talens, P.; Chiralt, A. Active bilayer films of thermoplastic starch and polycaprolactone obtained by compression molding. Carbohyd. Polym. 2015, 127, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Blilid, S.; Kędzierska, M.; Miłowska, K.; Wrońska, N.; El Achaby, M.; Katir, N.; Belamie, E.; Alonso, B.; Lisowska, K.; Lahcini, M.; et al. Phosphorylated Micro- and Nanocellulose-Filled Chitosan Nanocomposites as Fully Sustainable, Biologically Active Bioplastics. ACS Sustain. Chem. Eng. 2020, 8, 18354–18365. [Google Scholar] [CrossRef]
- Guan, Q.F.; Yang, H.B.; Han, Z.M.; Ling, Z.C.; Yu, S.H. An all-natural bioinspired structural material for plastic replacement. Nat. Commun. 2020, 11, 5401. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chen, C.; Brozena, A.H.; Zhu, J.Y.; Xu, L.; Driemeier, C.; Dai, J.; Rojas, O.J.; Isogai, A.; Wagberg, L.; et al. Developing fibrillated cellulose as a sustainable technological material. Nature 2021, 590, 47–56. [Google Scholar] [CrossRef]
- Liu, Y.; Ahmed, S.; Sameen, D.E.; Wang, Y.; Lu, R.; Dai, J.; Li, S.; Qin, W. A review of cellulose and its derivatives in biopolymer-based for food packaging application. Trends Food Sci. Technol. 2021, 112, 532–546. [Google Scholar] [CrossRef]
- Rol, F.; Belgacem, M.N.; Gandini, A.; Bras, J. Recent advances in surface-modified cellulose nanofibrils. Prog. Polym. Sci. 2019, 88, 241–264. [Google Scholar] [CrossRef]
- Saedi, S.; Garcia, C.V.; Kim, J.T.; Shin, G.H. Physical and chemical modifications of cellulose fibers for food packaging applications. Cellulose 2021, 28, 8877–8897. [Google Scholar] [CrossRef]
- Ventura-Cruz, S.; Tecante, A. Nanocellulose and microcrystalline cellulose from agricultural waste: Review on isolation and application as reinforcement in polymeric matrices. Food Hydrocoll. 2021, 118, 106771. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.; Xie, Y.; Zhang, K. Functional nanomaterials through esterification of cellulose: A review of chemistry and application. Cellulose 2018, 25, 3703–3731. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Wang, Z.; Lamm, M.E.; Yuan, L.; Tang, C. Supramolecular Polymer Nanocomposites Derived from Plant Oils and Cellulose Nanocrystals. Macromolecules 2017, 50, 7475–7483. [Google Scholar] [CrossRef]
- Farooq, M.; Zou, T.; Riviere, G.; Sipponen, M.H.; Osterberg, M. Strong, Ductile, and Waterproof Cellulose Nanofibril Composite Films with Colloidal Lignin Particles. Biomacromolecules 2019, 20, 693–704. [Google Scholar] [CrossRef]
- Guan, Q.F.; Yang, H.B.; Han, Z.M.; Zhou, L.C.; Yu, S.H. Lightweight, tough, and sustainable cellulose nanofiber-derived bulk structural materials with low thermal expansion coefficient. Sci. Adv. 2020, 6, eaaz1114. [Google Scholar] [CrossRef] [PubMed]
- Fei, Y.; Chen, F.; Fang, W.; Xu, L.; Ruan, S.; Liu, X.; Zhong, M.; Kuang, T. High-strength, flexible and cycling-stable piezo-resistive polymeric foams derived from thermoplastic polyurethane and multi-wall carbon nanotubes. Compos. Part B 2020, 199, 108279. [Google Scholar] [CrossRef]
- Fei, Y.; Chen, F.; Fang, W.; Hejna, A.; Xu, L.; Liu, T.; Zhong, M.; Yang, J.; Kuang, T. Conductive thermoplastic polyurethane nanocomposite foams derived from a cellulose/MWCNTs aerogel framework: Simultaneous enhancement of piezoresistance, strength, and endurance. J. Mater. Chem. C 2021, 9, 13103–13114. [Google Scholar] [CrossRef]
- Cui, X.; Lee, J.J.L.; Chen, W.N. Eco-friendly and biodegradable cellulose hydrogels produced from low cost okara: Towards non-toxic flexible electronics. Sci. Rep. 2019, 9, 18166. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.B.; Liu, Z.X.; Yin, C.H.; Han, Z.M.; Guan, Q.F.; Zhao, Y.X.; Ling, Z.C.; Liu, H.C.; Yang, K.P.; Sun, W.B.; et al. Edible, Ultrastrong, and Microplastic-Free Bacterial Cellulose-Based Straws by Biosynthesis. Adv. Funct. Mater. 2021, 2111713. [Google Scholar] [CrossRef]
- Jiang, B.; Chen, C.; Liang, Z.; He, S.; Kuang, Y.; Song, J.; Mi, R.; Chen, G.; Jiao, M.; Hu, L. Lignin as a Wood-Inspired Binder Enabled Strong, Water Stable, and Biodegradable Paper for Plastic Replacement. Adv. Funct. Mater. 2019, 30, 1906307. [Google Scholar] [CrossRef]
- Shu, L.; Zhang, X.-F.; Wang, Z.; Yao, J. Structure reorganization of cellulose hydrogel by green solvent exchange for potential plastic replacement. Carbohyd. Polym. 2022, 275, 118695. [Google Scholar] [CrossRef]
- Basu, A.; Lindh, J.; Alander, E.; Stromme, M.; Ferraz, N. On the use of ion-crosslinked nanocellulose hydrogels for wound healing solutions: Physicochemical properties and application-oriented biocompatibility studies. Carbohyd. Polym. 2017, 174, 299–308. [Google Scholar] [CrossRef]
- Aditya, T.; Allain, J.P.; Jaramillo, C.; Restrepo, A.M. Surface Modification of Bacterial Cellulose for Biomedical Applications. Int. J. Mol. Sci. 2022, 23, 610. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, D.; Li, Y.; Sun, S.; Zheng, J.; Cui, J.; Wang, G.; Zheng, L.; Wang, Y.; Zhou, H. High-performance green electronic substrate employing flexible and transparent cellulose films. Carbohydr. Polym. 2021, 270, 118359. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, E.S.; Cranston, E.D.; Rezende, C.A. Naturally Hydrophobic Foams from Lignocellulosic Fibers Prepared by Oven-Drying. ACS Sustain. Chem. Eng. 2020, 8, 8267–8278. [Google Scholar] [CrossRef]
- Ajdary, R.; Tardy, B.L.; Mattos, B.D.; Bai, L.; Rojas, O.J. Plant Nanomaterials and Inspiration from Nature: Water Interactions and Hierarchically Structured Hydrogels. Adv. Mater. 2020, 33, 2001085. [Google Scholar] [CrossRef] [PubMed]
- Zaitoon, A.; Lim, L.-T.; Scott-Dupree, C. Activated release of ethyl formate vapor from its precursor encapsulated in ethyl Cellulose/Poly(Ethylene oxide) electrospun nonwovens intended for active packaging of fresh produce. Food Hydrocoll. 2021, 112, 106313. [Google Scholar] [CrossRef]
- Xia, C.; Wang, W.; Wang, L.; Liu, H.; Xiao, J. Multilayer zein/gelatin films with tunable water barrier property and prolonged antioxidant activity. Food Packag. Shelf Life 2019, 19, 76–85. [Google Scholar] [CrossRef]
- Kasaai, M.R.; Moosavi, A. Treatment of Kraft paper with citrus wastes for food packaging applications: Water and oxygen barrier properties improvement. Food Packag. Shelf Life 2017, 12, 59–65. [Google Scholar] [CrossRef]
- Marrez, D.A.; Abdelhamid, A.E.; Darwesh, O.M. Eco-friendly cellulose acetate green synthesized silver nano-composite as antibacterial packaging system for food safety. Food Packag. Shelf Life 2019, 20, 100302. [Google Scholar] [CrossRef]
- Wu, J.; Chen, S.; Ge, S.; Miao, J.; Li, J.; Zhang, Q. Preparation, properties and antioxidant activity of an active film from silver carp (Hypophthalmichthys molitrix) skin gelatin incorporated with green tea extract. Food Hydrocoll. 2013, 32, 42–51. [Google Scholar] [CrossRef]
- Wang, W.; Xiao, J.; Chen, X.; Luo, M.; Liu, H.; Shao, P. Fabrication and characterization of multilayered kafirin/gelatin film with one-way water barrier property. Food Hydrocoll. 2018, 81, 159–168. [Google Scholar] [CrossRef]
- He, X.; Li, M.; Gong, X.; Niu, B.; Li, W. Biodegradable and antimicrobial CSC films containing cinnamon essential oil for preservation applications. Food Packag. Shelf Life 2021, 29, 100697. [Google Scholar] [CrossRef]
- Wan, Z.; Wang, L.; Yang, X.; Guo, J.; Yin, S. Enhanced water resistance properties of bacterial cellulose multilayer films by incorporating interlayers of electrospun zein fibers. Food Hydrocoll. 2016, 61, 269–276. [Google Scholar] [CrossRef]
- Wen, Y.; Liu, J.; Jiang, L.; Zhu, Z.; He, S.; He, S.; Shao, W. Development of intelligent/active food packaging film based on TEMPO-oxidized bacterial cellulose containing thymol and anthocyanin-rich purple potato extract for shelf life extension of shrimp. Food Packag. Shelf Life 2021, 29, 100709. [Google Scholar] [CrossRef]
- Yang, J.; Li, M.; Wang, Y.; Wu, H.; Zhen, T.; Xiong, L.; Sun, Q. Double Cross-Linked Chitosan Composite Films Developed with Oxidized Tannic Acid and Ferric Ions Exhibit High Strength and Excellent Water Resistance. Biomacromolecules 2019, 20, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.W.; Nair, S.S.; Chen, H.; Yan, N.; Farnood, R.; Li, F.Y. Thermally stable, enhanced water barrier, high strength starch bio-composite reinforced with lignin containing cellulose nanofibrils. Carbohydr. Polym. 2020, 230, 115626. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, X.; Jiang, Q.; Yu, D.; Xu, Y.; Wang, B.; Xia, W. Development and properties of bacterial cellulose, curcumin, and chitosan composite biodegradable films for active packaging materials. Carbohydr. Polym. 2021, 260, 117778. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Cheng, R.; Wang, B.; Zeng, J.; Xu, J.; Li, J.; Kang, L.; Cheng, Z.; Gao, W.; Chen, K. Biodegradable sandwich-architectured films derived from pea starch and polylactic acid with enhanced shelf-life for fruit preservation. Carbohydr. Polym. 2021, 251, 117117. [Google Scholar] [CrossRef]
- Xia, Q.; Chen, C.; Yao, Y.; Li, J.; He, S.; Zhou, Y.; Li, T.; Pan, X.; Yao, Y.; Hu, L. A strong, biodegradable and recyclable lignocellulosic bioplastic. Nat. Sustain. 2021, 4, 627–635. [Google Scholar] [CrossRef]
- Guan, Q.-F.; Ling, Z.-C.; Han, Z.-M.; Yang, H.-B.; Yu, S.-H. Ultra-Strong, Ultra-Tough, Transparent, and Sustainable Nanocomposite Films for Plastic Substitute. Matter 2020, 3, 1308–1317. [Google Scholar] [CrossRef]
- Shimizu, M.; Saito, T.; Isogai, A. Water-resistant and high oxygen-barrier nanocellulose films with interfibrillar cross-linkages formed through multivalent metal ions. J. Membr. Sci. 2016, 500, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Du, H.; Zhang, F.; Zhang, Y.; Wu, M.; Yu, G.; Liu, C.; Li, B.; Peng, H. Flexible cellulose nanopaper with high wet tensile strength, high toughness and tunable ultraviolet blocking ability fabricated from tobacco stalk via a sustainable method. J. Mater. Chem. A 2018, 6, 13021–13030. [Google Scholar] [CrossRef]
- Zhou, J.; Fang, Z.; Cui, J.; Zhang, X.; Qian, Y.; Liu, W.; Yang, D.; Qiu, X. Wood-inspired strategy to toughen transparent cellulose nanofibril films. Carbohydr. Polym. 2021, 259, 117759. [Google Scholar] [CrossRef] [PubMed]
- Plappert, S.F.; Quraishi, S.; Pircher, N.; Mikkonen, K.S.; Veigel, S.; Klinger, K.M.; Potthast, A.; Rosenau, T.; Liebner, F.W. Transparent, Flexible, and Strong 2,3-Dialdehyde Cellulose Films with High Oxygen Barrier Properties. Biomacromolecules 2018, 19, 2969–2978. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Zhang, Y.; Cha, R.; Yang, J.; Jiang, X. Water-soluble nanocrystalline cellulose films with highly transparent and oxygen barrier properties. Nanoscale 2016, 8, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Isogai, A.; Saito, T.; Fukuzumi, H. TEMPO-oxidized cellulose nanofibers. Nanoscale 2011, 3, 71–85. [Google Scholar] [CrossRef]
- Song, Z.; Xiao, H.; Zhao, Y. Hydrophobic-modified nano-cellulose fiber/PLA biodegradable composites for lowering water vapor transmission rate (WVTR) of paper. Carbohydr. Polym. 2014, 111, 442–448. [Google Scholar] [CrossRef]
- Nguyen, H.L.; Tran, T.H.; Hao, L.T.; Jeon, H.; Koo, J.M.; Shin, G.; Hwang, D.S.; Hwang, S.Y.; Park, J.; Oh, D.X. Biorenewable, transparent, and oxygen/moisture barrier nanocellulose/nanochitin-based coating on polypropylene for food packaging applications. Carbohydr. Polym. 2021, 271, 118421. [Google Scholar] [CrossRef]
- Kriechbaum, K.; Bergstrom, L. Antioxidant and UV-Blocking Leather-Inspired Nanocellulose-Based Films with High Wet Strength. Biomacromolecules 2020, 21, 1720–1728. [Google Scholar] [CrossRef] [Green Version]
- Gaiolas, C.; Belgacem, M.N.; Silva, L.; Thielemans, W.; Costa, A.P.; Nunes, M.; Silva, M.J. Green chemicals and process to graft cellulose fibers. J. Colloid Interface Sci. 2009, 330, 298–302. [Google Scholar] [CrossRef]
- Bayer, I.S.; Fragouli, D.; Attanasio, A.; Sorce, B.; Bertoni, G.; Brescia, R.; Di Corato, R.; Pellegrino, T.; Kalyva, M.; Sabella, S.; et al. Water-repellent cellulose fiber networks with multifunctional properties. ACS Appl. Mater. Interfaces 2011, 3, 4024–4031. [Google Scholar] [CrossRef]
- Eyley, S.; Thielemans, W. Surface modification of cellulose nanocrystals. Nanoscale 2014, 6, 7764–7779. [Google Scholar] [CrossRef] [Green Version]
- Habibi, Y. Key advances in the chemical modification of nanocelluloses. Chem. Soc. Rev. 2014, 43, 1519–1542. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Kaushik, A.; Ahuja, D. Surface functionalization of nanofibrillated cellulose extracted from wheat straw: Effect of process parameters. Carbohyd. Polym. 2016, 150, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Avila Ramirez, J.A.; Gomez Hoyos, C.; Arroyo, S.; Cerrutti, P.; Foresti, M.L. Acetylation of bacterial cellulose catalyzed by citric acid: Use of reaction conditions for tailoring the esterification extent. Carbohyd. Polym. 2016, 153, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.T.; Tajvidi, M. Sustainable Barrier System via Self-Assembly of Colloidal Montmorillonite and Cross-linking Resins on Nanocellulose Interfaces. ACS Appl. Mater. Interfaces 2019, 11, 1604–1615. [Google Scholar]
- Nechyporchuk, O.; Belgacem, M.N.; Bras, J. Production of cellulose nanofibrils: A review of recent advances. Ind. Crops Prod. 2016, 93, 2–25. [Google Scholar] [CrossRef]
- Lavoine, N.; Desloges, I.; Dufresne, A.; Bras, J. Microfibrillated cellulose—Its barrier properties and applications in cellulosic materials: A review. Carbohydr. Polym. 2012, 90, 735–764. [Google Scholar] [CrossRef]
- Abdul Khalil, H.P.S.; Davoudpour, Y.; Saurabh, C.K.; Hossain, M.S.; Adnan, A.S.; Dungani, R.; Paridah, M.T.; Islam Sarker, M.Z.; Fazita, M.R.N.; Syakir, M.I.; et al. A review on nanocellulosic fibres as new material for sustainable packaging: Process and applications. Renew. Sustain. Energy Rev. 2016, 64, 823–836. [Google Scholar] [CrossRef]
- Xiong Chang, X.; Mujawar Mubarak, N.; Ali Mazari, S.; Sattar Jatoi, A.; Ahmad, A.; Khalid, M.; Walvekar, R.; Abdullah, E.C.; Karri, R.R.; Siddiqui, M.T.H.; et al. A review on the properties and applications of chitosan, cellulose and deep eutectic solvent in green chemistry. J. Ind. Eng. Chem. 2021, 104, 362–380. [Google Scholar] [CrossRef]
- Ferrer, A.; Pal, L.; Hubbe, M. Nanocellulose in packaging: Advances in barrier layer technologies. Ind. Crops Prod. 2017, 95, 574–582. [Google Scholar] [CrossRef]
- Liang, L.; Bhagia, S.; Li, M.; Huang, C.; Ragauskas, A.J. Cross-Linked Nanocellulosic Materials and Their Applications. ChemSusChem 2020, 13, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Mascheroni, E.; Piergiovanni, L. The Potential of NanoCellulose in the Packaging Field: A Review. Packag. Technol. Sci. 2015, 28, 475–508. [Google Scholar] [CrossRef]
- Ben Shalom, T.; Belsey, S.; Chasnitsky, M.; Shoseyov, O. Cellulose Nanocrystals and Corn Zein Oxygen and Water Vapor Barrier Biocomposite Films. Nanomaterials 2021, 11, 247. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhou, J.; Wang, Z.; Mu, S.; Wu, R.; Wang, Z. Cellulose nanocrystal/plant oil polymer composites with hydrophobicity, humidity-sensitivity, and high wet strength. Carbohydr. Polym. 2020, 231, 115739. [Google Scholar] [CrossRef]
- Nagalakshmaiah, M.; El Kissi, N.; Dufresne, A. Ionic Compatibilization of Cellulose Nanocrystals with Quaternary Ammonium Salt and Their Melt Extrusion with Polypropylene. ACS Appl. Mater. Interfaces 2016, 8, 8755–8764. [Google Scholar] [CrossRef]
- Wang, S.; Jiang, F.; Xu, X.; Kuang, Y.; Fu, K.; Hitz, E.; Hu, L. Super-Strong, Super-Stiff Macrofibers with Aligned, Long Bacterial Cellulose Nanofibers. Adv. Mater. 2017, 29, 1702498. [Google Scholar] [CrossRef] [PubMed]
- Jonoobi, M.; Harun, J.; Mathew, A.P.; Hussein, M.Z.B.; Oksman, K. Preparation of cellulose nanofibers with hydrophobic surface characteristics. Cellulose 2010, 17, 299–307. [Google Scholar] [CrossRef]
- Avila Ramirez, J.A.; Suriano, C.J.; Cerrutti, P.; Foresti, M.L. Surface esterification of cellulose nanofibers by a simple organocatalytic methodology. Carbohydr. Polym. 2014, 114, 416–423. [Google Scholar] [CrossRef]
- Saito, T.; Nishiyama, Y.; Putaux, J.-L.; Vignon, M.; Isogai, A. Homogeneous Suspensions of Individualized Microfibrils from TEMPO-Catalyzed Oxidation of Native Cellulose. Biomacromolecules 2006, 7, 1687–1691. [Google Scholar] [CrossRef]
- Tang, L.; Huang, B.; Lu, Q.; Wang, S.; Ou, W.; Lin, W.; Chen, X. Ultrasonication-assisted manufacture of cellulose nanocrystals esterified with acetic acid. Bioresour. Technol. 2013, 127, 100–105. [Google Scholar] [CrossRef]
- Yoo, Y.; Youngblood, J.P. Green One-Pot Synthesis of Surface Hydrophobized Cellulose Nanocrystals in Aqueous Medium. ACS Sustain. Chem. Eng. 2016, 4, 3927–3938. [Google Scholar] [CrossRef]
- Minelli, M.; Baschetti, M.G.; Doghieri, F.; Ankerfors, M.; Lindström, T.; Siró, I.; Plackett, D. Investigation of mass transport properties of microfibrillated cellulose (MFC) films. J. Membr. Sci. 2010, 358, 67–75. [Google Scholar] [CrossRef]
- Fukuzumi, H.; Saito, T.; Iwata, T.; Kumamoto, Y.; Isogai, A. Transparent and High Gas Barrier Films of Cellulose Nanofibers Prepared by TEMPO-Mediated Oxidation. Biomacromolecules 2009, 10, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Missoum, K.; Belgacem, M.N.; Barnes, J.-P.; Brochier-Salon, M.-C.; Bras, J. Nanofibrillated cellulose surface grafting in ionic liquid. Soft Matter 2012, 8, 8338. [Google Scholar] [CrossRef]
- Uschanov, P.; Johansson, L.-S.; Maunu, S.L.; Laine, J. Heterogeneous modification of various celluloses with fatty acids. Cellulose 2011, 18, 393–404. [Google Scholar] [CrossRef]
- Cunha, A.G.; Freire, C.S.; Silvestre, A.J.; Neto, C.P.; Gandini, A. Reversible hydrophobization and lipophobization of cellulose fibers via trifluoroacetylation. J. Colloid Interface Sci. 2006, 301, 333–336. [Google Scholar] [CrossRef]
- Crepy, L.; Chaveriat, L.; Banoub, J.; Martin, P.; Joly, N. Synthesis of cellulose fatty esters as plastics-influence of the degree of substitution and the fatty chain length on mechanical properties. ChemSusChem 2009, 2, 165–170. [Google Scholar] [CrossRef]
- Sealey, J.E.; Samaranayake, G.; Todd, J.G.; Glasser, W.G. Novel Cellulose Derivatives. Preparation and Thermal Analysis of Waxy Esters of Cellulose. J. Polym. Sci. Part B Polym. Phys. 1996, 34, 1613–1620. [Google Scholar] [CrossRef]
- Cao, X.; Sun, S.; Peng, X.; Zhong, L.; Sun, R.; Jiang, D. Rapid synthesis of cellulose esters by transesterification of cellulose with vinyl esters under the catalysis of NaOH or KOH in DMSO. J. Agric. Food Chem. 2013, 61, 2489–2495. [Google Scholar] [CrossRef]
- Wang, W.; Gu, F.; Deng, Z.; Zhu, Y.; Zhu, J.; Guo, T.; Song, J.; Xiao, H. Multilayer surface construction for enhancing barrier properties of cellulose-based packaging. Carbohydr. Polym. 2021, 255, 117431. [Google Scholar] [CrossRef]
- Swatloski, R.P.; Spear, S.K.; Holbrey, J.D.; Rogers, R.D. Dissolution of Cellose with Ionic Liquids. J. Am. Chem. Soc. 2002, 124, 4974–4975. [Google Scholar] [CrossRef]
- Chen, J.; Xu, J.; Wang, K.; Cao, X.; Sun, R. Cellulose acetate fibers prepared from different raw materials with rapid synthesis method. Carbohydr. Polym. 2016, 137, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Onwukamike, K.N.; Grelier, S.; Grau, E.; Cramail, H.; Meier, M.A.R. Sustainable Transesterification of Cellulose with High Oleic Sunflower Oil in a DBU-CO2 Switchable Solvent. ACS Sustain. Chem. Eng. 2018, 6, 8826–8835. [Google Scholar] [CrossRef]
- Rodionova, G.; Lenes, M.; Eriksen, Ø.; Gregersen, Ø. Surface chemical modification of microfibrillated cellulose: Improvement of barrier properties for packaging applications. Cellulose 2011, 18, 127–134. [Google Scholar] [CrossRef]
- Abraham, E.; Nevo, Y.; Slattegard, R.; Attias, N.; Sharon, S.; Lapidot, S.; Shoseyov, O. Highly Hydrophobic Thermally Stable Liquid Crystalline Cellulosic Nanomaterials. ACS Sustain. Chem. Eng. 2016, 4, 1338–1346. [Google Scholar] [CrossRef]
- Ioelovich, M. Adjustment of Hydrophobic Properties of Cellulose Materials. Polymers 2021, 13, 1241. [Google Scholar] [CrossRef]
- Deng, S.; Huang, R.; Zhou, M.; Chen, F.; Fu, Q. Hydrophobic cellulose films with excellent strength and toughness via ball milling activated acylation of microfibrillated cellulose. Carbohydr. Polym. 2016, 154, 129–138. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, Z.; Chen, S.; Dong, H.; Zhang, X.; Qin, Y.; Yao, C.; Xu, F. High-barrier, strong, and antibacterial paper fabricated by coating acetylated cellulose and cinnamaldehyde for food packaging. Cellulose 2021, 28, 4371–4384. [Google Scholar] [CrossRef]
- Saha, N.R.; Sarkar, G.; Roy, I.; Bhattacharyya, A.; Rana, D.; Dhanarajan, G.; Banerjee, R.; Sen, R.; Mishra, R.; Chattopadhyay, D. Nanocomposite films based on cellulose acetate/polyethylene glycol/modified montmorillonite as nontoxic active packaging material. RSC Adv. 2016, 6, 92569–92578. [Google Scholar] [CrossRef]
- Assis, R.Q.; Rios, P.D.A.; Rios, A.D.O.; Olivera, F.C. Biodegradable packaging of cellulose acetate incorporated with norbixin, lycopene or zeaxanthin. Ind. Crops Prod. 2020, 147, 112212. [Google Scholar] [CrossRef]
- Assis, R.Q.; Pagno, C.H.; Stoll, L.; Rios, P.D.; Rios, A.O.; Olivera, F.C. Active food packaging of cellulose acetate: Storage stability, protective effect on oxidation of riboflavin and release in food simulants. Food Chem. 2021, 349, 129140. [Google Scholar] [CrossRef]
- Mulyadi, A.; Deng, Y. Surface modification of cellulose nanofibrils by maleated styrene block copolymer and their composite reinforcement application. Cellulose 2016, 23, 519–528. [Google Scholar] [CrossRef]
- Kulomaa, T.; Matikainen, J.; Karhunen, P.; Heikkilä, M.; Fiskari, J.; Kilpeläinen, I. Cellulose fatty acid esters as sustainable film materials—Effect of side chain structure on barrier and mechanical properties. RSC Adv. 2015, 5, 80702–80708. [Google Scholar] [CrossRef]
- Lee, K.Y.; Quero, F.; Blaker, J.J.; Hill, C.A.; Eichhorn, S.J.; Bismarck, A. Surface only modification of bacterial cellulose nanofibers with organic acids. Cellulose 2011, 18, 595–605. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Geissler, A.; Standhardt, M.; Mehlhase, S.; Gallei, M.; Chen, L.; Marie Thiele, C. Moisture-responsive films of cellulose stearoyl esters showing reversible shape transitions. Sci. Rep. 2015, 5, 11011. [Google Scholar] [CrossRef]
- Havimo, M.; Jalomäki, J.; Granström, M.; Rissanen, A.; Iivanainen, T.; Kemell, M.; Heikkilä, M.; Sipi, M.; Kilpeläinen, I. Mechanical strength and water resistance of paperboard coated with long chain cellulose esters. Packag. Technol. Sci. 2011, 24, 249–258. [Google Scholar] [CrossRef]
- Willberg-Keyriläinen, P.; Vartiainen, J.; Harlin, A.; Ropponen, J. The effect of side-chain length of cellulose fatty acid esters on their thermal, barrier and mechanical properties. Cellulose 2017, 24, 505–517. [Google Scholar] [CrossRef]
- Bras, J.; Vaca-Garcia, C.; Borredon, M.-E.; Glasser, W. Oxygen and water vapor permeability of fully substituted long chain cellulose esters (LCCE). Cellulose 2007, 14, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Li, W.; Wang, W.; Wang, S.; Qin, C. Reactive superhydrophobic paper from one-step spray-coating of cellulose-based derivative. Appl. Surf. Sci. 2019, 497, 143816. [Google Scholar] [CrossRef]
- Qin, C.; Wang, W.; Li, W.; Zhang, S. Reactive Water Vapor Barrier Coatings Derived from Cellulose Undecenoyl Esters for Paper Packaging. Coatings 2020, 10, 1032. [Google Scholar] [CrossRef]
- Balasubramaniam, S.L.; Patel, A.S.; Nayak, B. Surface modification of cellulose nanofiber film with fatty acids for developing renewable hydrophobic food packaging. Food Packag. Shelf Life 2020, 26, 100587. [Google Scholar] [CrossRef]
- Sun, B.; Wang, W.; Zhang, M.; Sain, M. Biomass-based edible film with enhanced mass barrier capacity and gas permeable selectivity. Cellulose 2018, 25, 5919–5937. [Google Scholar] [CrossRef]
- Crépy, L.; Miri, V.; Joly, N.; Martin, P.; Lefebvre, J.-M. Effect of side chain length on structure and thermomechanical properties of fully substituted cellulose fatty esters. Carbohydr. Polym. 2011, 83, 1812–1820. [Google Scholar] [CrossRef]
- Chauvelon, G.; Gergaud, N.; Saulniera, L.; Lourdin, D.; Buleon, A.; Thibault, J.-F.; Krausz, P. Esterification of cellulose-enriched agricultural by-products and characterization of mechanical properties of cellulosic films. Carbohydr. Polym. 2000, 42, 385–392. [Google Scholar] [CrossRef]
- Sehaqui, H.; Zimmermann, T.; Tingaut, P. Hydrophobic cellulose nanopaper through a mild esterification procedure. Cellulose 2014, 21, 367–382. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wang, S.; Wang, W.; Qin, C.; Wu, M. Facile preparation of reactive hydrophobic cellulose nanofibril film for reducing water vapor permeability (WVP) in packaging applications. Cellulose 2019, 26, 3271–3284. [Google Scholar] [CrossRef]
- Attallah, O.A.; Mojicevic, M.; Garcia, E.L.; Azeem, M.; Chen, Y.; Asmawi, S.; Brenan Fournet, M. Macro and Micro Routes to High Performance Bioplastics: Bioplastic Biodegradability and Mechanical and Barrier Properties. Polymers 2021, 13, 2155. [Google Scholar] [CrossRef] [PubMed]
- Ratanakamnuan, U.; Atong, D.; Aht-Ong, D. Cellulose esters from waste cotton fabric via conventional and microwave heating. Carbohydr. Polym. 2012, 87, 84–94. [Google Scholar] [CrossRef]
- Wen, X.; Wang, H.; Wei, Y.; Wang, X.; Liu, C. Preparation and characterization of cellulose laurate ester by catalyzed transesterification. Carbohydr. Polym. 2017, 168, 247–254. [Google Scholar] [CrossRef]
- Geissler, A.; Bonaccurso, E.; Heim, L.-O.; Heinze, T.; Zhang, K. Temperature-Responsive Thin Films from Cellulose Stearoyl Triester. J. Phys. Chem. C 2014, 118, 2408–2417. [Google Scholar] [CrossRef]
- Brand, J.; Pecastaings, G.; Sebe, G. A versatile method for the surface tailoring of cellulose nanocrystal building blocks by acylation with functional vinyl esters. Carbohydr. Polym. 2017, 169, 189–197. [Google Scholar] [CrossRef]
- Cao, X.; Peng, X.; Zhong, L.; Sun, S.; Yang, D.; Zhang, X.; Sun, R. A novel transesterification system to rapidly synthesize cellulose aliphatic esters. Cellulose 2014, 21, 581–594. [Google Scholar] [CrossRef]
- Dankovich, T.A.; Hsieh, Y.-L. Surface modification of cellulose with plant triglycerides for hydrophobicity. Cellulose 2007, 14, 469–480. [Google Scholar] [CrossRef]
- Dong, X.; Dong, Y.; Jiang, M.; Wang, L.; Tong, J.; Zhou, J. Modification of microcrystalline cellulose by using soybean oil for surface hydrophobization. Ind. Crops Prod. 2013, 46, 301–303. [Google Scholar] [CrossRef]
- Gorade, V.G.; Kotwal, A.; Chaudhary, B.U.; Kale, R.D. Surface modification of microcrystalline cellulose using rice bran oil: A bio-based approach to achieve water repellency. J. Polym. Res. 2019, 26, 217. [Google Scholar] [CrossRef]
- Herrera, M.A.; Mathew, A.P.; Oksman, K. Barrier and mechanical properties of plasticized and cross-linked nanocellulose coatings for paper packaging applications. Cellulose 2017, 24, 3969–3980. [Google Scholar] [CrossRef]
- Coma, V.; Sebti, I.; Pardon, P.; Pichavant, F.H.; Deschamps, A. Film properties from crosslinking of cellulosic derivatives with a polyfunctional carboxylic acid. Carbohydr. Polym. 2003, 51, 265–271. [Google Scholar] [CrossRef]
- Shahbazi, M.; Ahmadi, S.J.; Seif, A.; Rajabzadeh, G. Carboxymethyl cellulose film modification through surface photo-crosslinking and chemical crosslinking for food packaging applications. Food Hydrocoll. 2016, 61, 378–389. [Google Scholar] [CrossRef]
- Beghetto, V.; Gatto, V.; Conca, S.; Bardella, N.; Buranello, C.; Gasparetto, G.; Sole, R. Development of 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methyl-morpholinium chloride cross-linked carboxymethyl cellulose films. Carbohydr. Polym. 2020, 249, 116810. [Google Scholar] [CrossRef]
- Zhou, Y.J.; Luner, P.; Caluwe, P. Mechanism of Crosslinking of Papers with Polyfunctional Carboxylic Acids. Appl. Polym. Sci. 1995, 58, 1523–1534. [Google Scholar] [CrossRef]
- He, X.; Luzi, F.; Yang, W.; Xiao, Z.; Torre, L.; Xie, Y.; Puglia, D. Citric Acid as Green Modifier for Tuned Hydrophilicity of Surface Modified Cellulose and Lignin Nanoparticles. ACS Sustain. Chem. Eng. 2018, 6, 9966–9978. [Google Scholar] [CrossRef]
- Wu, Y.; Luo, X.; Li, W.; Song, R.; Li, J.; Li, Y.; Li, B.; Liu, S. Green and biodegradable composite films with novel antimicrobial performance based on cellulose. Food Chem. 2016, 197, 250–256. [Google Scholar] [CrossRef]
- Andersson, C. New ways to enhance the functionality of paperboard by surface treatment—A review. Packag. Technol. Sci. 2008, 21, 339–373. [Google Scholar] [CrossRef]
- Frank, B.P.; Smith, C.; Caudill, E.R.; Lankone, R.S.; Carlin, K.; Benware, S.; Pedersen, J.A.; Fairbrother, D.H. Biodegradation of Functionalized Nanocellulose. Environ. Sci. Technol. 2021, 55, 10744–10757. [Google Scholar] [CrossRef] [PubMed]
- Rajeswari, A.; Christy, E.J.S.; Swathi, E.; Pius, A. Fabrication of improved cellulose acetate-based biodegradable films for food packaging applications. Environ. Chem. Ecotoxicol. 2020, 2, 107–114. [Google Scholar] [CrossRef]
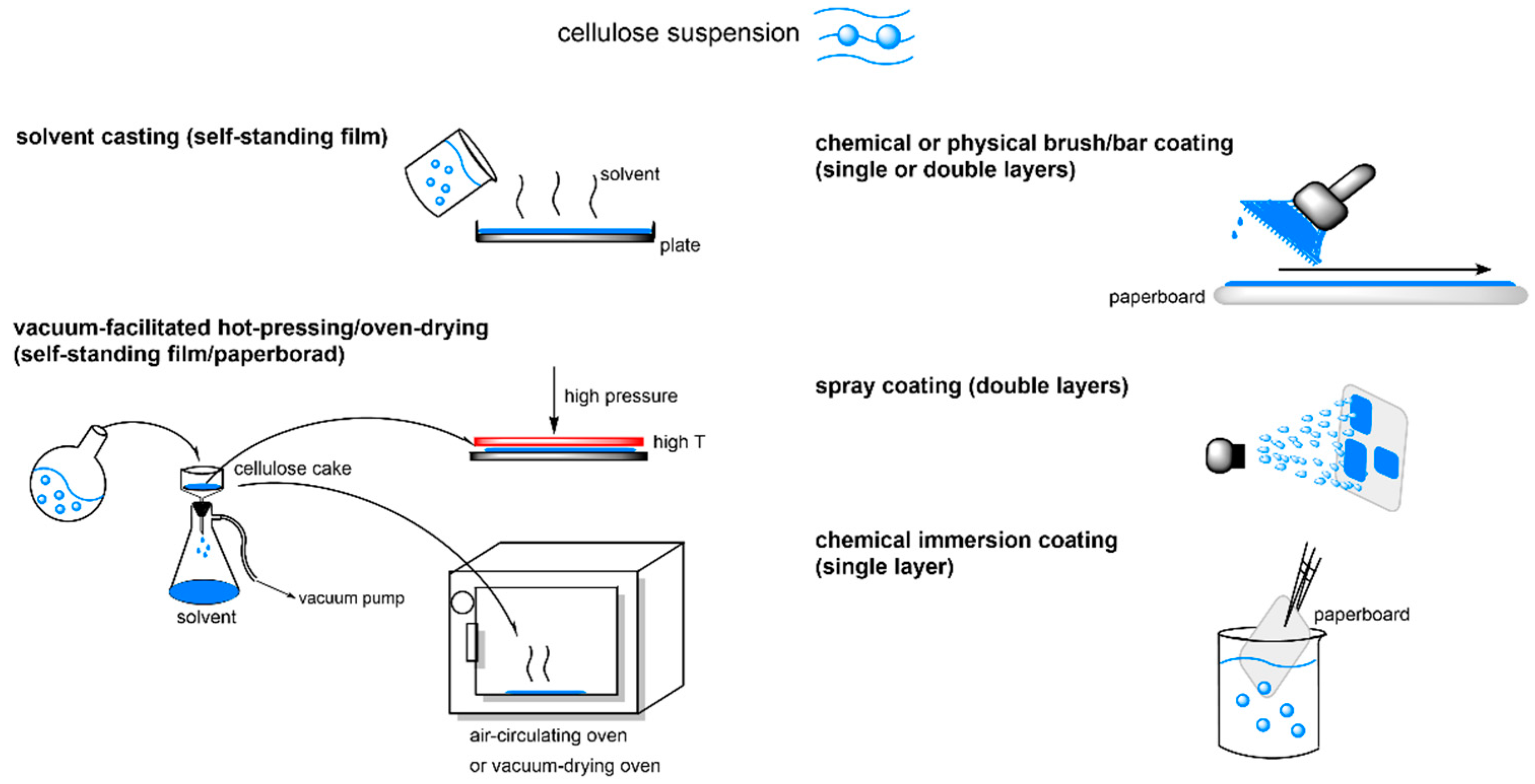

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cellulose Source | Extraction Method | Fiber Size | Crystallinity | Functionality | Reference |
|---|---|---|---|---|---|
| kenaf bast fiber | disintegration, refining, cryo-crushing and cylinder homogenization | CNF, diameter: 10–30 nm | 81% | –OH | [76] |
| Gluconacetobacter xylinus NRRL B-42 | blender homogenization | BNF, fiber size: n.d. a | 91.8% | –OH | [77] |
| tunicin cellulose | TEMPO b-mediated oxidation, blender homogenization, sonication | CMF, diameter: 10–20 nm | n.d. | –OH and sodium carboxylate groups (0.31 mmol/g) | [78] |
| wood pulp filter paper | blender homogenization, refining, freeze-drying | CNC, fiber size: n.d. | 85% | –OH and sulfate groups | [79] |
| commercial never-dried CNC suspension in water | - | CNC, length: 64 nm, width: 7 nm | 72% | –OH and sulfate groups | [80] |
| wheat straw CMF | blender homogenization | CNF, diameter: 10–40 nm | 89% | –OH | [62] |
| softwood pulp dissolved with sulfite | carboxymethylation and cylinder homogenization | CMF, diameter: 5–15 nm | n.d. | –OH and sodium carboxylate groups (586 μ-equiv./g) | [81] |
| softwood and hardwood bleached kraft pulp | TEMPO-mediated oxidation, blender homogenization, sonication | TOCN c, length: several μm, diameter: 3–4 nm | 75% | -OH and sodium carboxylate groups | [82] |
| spruce/pine (w/w = 7/3) bleached softwood pulp | enzymatic treatment and cylinder homogenization | CNF, diameter: 20 nm | n.d. | -OH and sulfate groups | [83] |
| CMC from cotton linters | CNC: acid hydrolysis using sulfuric acid and freeze-dried, regenerated cellulose: treated by N-Methylmorpholine N-oxide under heating | CNC (length: 300 nm, diameter: 10 nm), regenerated cellulose (length: 100 μm) | 80% | CNC: –OH and sulfate groups (70 mmol kg−1) | [84] |
| Cellulose | Acylation Process | Packaging Type and Its Formation | Reaction Degree | Ref. |
|---|---|---|---|---|
| softwood cellulose pulp; CMF | acetic acid or acetic anhydride as acylant with or without sulfuric acid as catalyst, reacted at 60–70 °C for 0.5–4 h with or without toluene as solvent | CA coating or film; CA solution was coated on paper via the hand lay-up technique or solvent-casted in air | DS = 0.21–0.32 for C2-CMF CES | [93,97] |
| CMF | oxalic acid as esterifying agent, reacted at 90 °C for 0.25–4 h in DI water | C2-CES coating; C2-CES aqueous suspension was deposited on filter paper through vacuum filtration, then oven-dried | carboxyl group content of 0.21–0.43 mmol/g fibrils | [89] |
| CNC extracted from bamboo waste pulp | butyric anhydride as acylant and iodine as catalyst, reacted at 105–110 °C for 30 min without solvent | C4-CES film or coating; C4-CES solution was solvent-casted or coated on mung bean seeds | DS = 2.1 | [94] |
| cotton cellulose | acetic, propionic or butyric anhydride as acylant with trifluoroacetic acid as solvent/catalyst, reacted at 50 °C for 1 h | C2, C3, C4 (or their mixture)-CES films or coatings; CES solution was solvent-casted or coated on white paper with a bar coater | DS = 2.7–3 | [95] |
| CMF from wood pulp | hexanoyl chloride as acylant and activated by mechanical ball milling, reacted at RT for 1–12 h in DMF | C6-CES film; C6-CES solution was solvent-casted, dried in an air-circulating oven at 60 °C and treated at 60 °C under vacuum for another 8 h | DS = 0.25–0.45 (3 h of milling time, 1–4 mL of acylant) | [96] |
| Cellulose Packaging | Additives | Packaging Properties | Food Packaging Application | Ref. | ||
|---|---|---|---|---|---|---|
| Barrier Properties | Mechanical Properties | Other Properties | ||||
| kraft paper (CA as coating) | 2–8 mL of CIN (v/v in CA solution) | oil resistance: kit number 12; WA, WVP and OTR of the CIN-CA-coated paper markedly decreased by 96.2%, 76.8%, and million times, respectively | dry and wet σ was increased from 55.8 and 2.3 MPa to 88.2 and 12.9 MPa, respectively, compared with kraft paper | good cytocompatibility, high antioxidation with 8% CIN, excellent antibacterial performance with 6% CIN | extend beef’s shelf-life by 4–5 d at 4 °C | [97] |
| CA-coupled cellulose cardboard (food-grade resin as coating) | 10% (w/w to resin) LDH-HB | n.d. a | n.d. | good cytocompatibility in the release test, excellent antibacterial performance | preserved cooked pasta for up to 30 days at 4 °C | [5] |
| CA film | 10–50 wt% PEG, 1–5 wt% MMTCTAB-MMT or 0.05 wt% G- AgNPs | 3 wt% CTAB-MMT-incorporated CA film showed the lowest WVTR of 5.84 g/m2 24 h; 0.05% G-AgNP-incorporated CA film showed an increased degree of water swelling from 0.28 to 0.44–0.62 | ε of 20 wt% PEG-incorporated CA film (CAP20) was increased from 3.8% to 31.0%, while σ decreased from 43.3 MPa to 32.6 MPa compared with CA film | CAP20 film incorporated with CTAB-MMT showed slightly increased thermal stability, good antimicrobial properties, and no cytotoxicity; G-AgNP-incorporated CA film showed strong antibacterial activity and no cytotoxicity | n.d. | [37,98] |
| CA film | 0.1–1% of carotenoids (lycopene, norbixin and zeaxanthin) (w/w to CA) | 0.1 wt% carotenoids: WVP = 0.035 g × mm m−2 h−1 kPa−1 for norbixin, 0.023 g × m m−2 h−1 kPa−1 for zeaxanthin and 0.022 g × mm m−2 h−1 kPa−1 for lycopene | 0.1 wt% lycopene or zeaxanthin: ε increased from 3.9% to 15%, while ε of 0.1 wt% norbixin-CA film remained unchanged; σ increased from 65.3 MPa to 84, 104 and 86 MPa, respectively, for norbixin-, lycopene- and zeaxanthin-loaded films | films with norbixin or lycopene displayed better light protection for sunflower oil; films with norbixin showed the best UV protection for vitamin B2; film with 0.1% zeaxanthin showed 50 °C lower Td-onset (200 °C) compared with CA film and the films with other carotenoids | n.d. | [99,100] |
| Cellulose | Acylation Process | Packaging Type and Its Formation | Reaction Degree | Ref. |
|---|---|---|---|---|
| unbleached eucalyptus CNF; wheat bran and maize bran residue cellulose; cellulose sheet; softwood cellulose; CMC; α-cellulose | C6–C20 acyl chlorides as acylants, pyridine, sulfuric acid or DMAP as catalyst, reacted at 50–130 °C in DMAc, DMAc/LiCl or cosolvent of toluene and pyridine for hours | C6–C20 films; solvent casting or vacuum dried | DS = 0.19–3 (DS of cellulose esters decreased with increasing substituent chain length from C6 to C18) | [87,102,104,106,107,112,113,115] |
| BNF, α-cellulose | C2–C12 carboxylic acids or C8–C18 acyl chlorides as acylants, reacted at 50–130 °C for 2 h in pyridine or pyridine/tosyl chloride | C2–C12 cellulose ester papers or C8–C18 cellulose ester films; wet cellulose cakes or films were hot-pressed at 90–110 °C | DS = 0.64–3 (DS of cellulose esters decreased with increasing substituent chain length from C2 to C12) | [103,107] |
| α-cellulose, bleached bagasse pulp, CMC | C6–C16 acyl chlorides as acylants and pyridine as catalyst, reacted in DMAc, DMAc/LiCl or pyridine at RT–100 °C for hours or days | C6–C16 cellulose ester-coated paper; cellulose ester suspension or solution was spray- or bar-coated on paperboard via air brush or bar coater | DS = 0.62–2.9 | [105,108,109] |
| premade CNF film | lauroyl, palmitoyl or stearoyl chloride as acylant, reacted in pyridine at 100 °C | C12-, C16- and C18-cellulose ester films; one-sided acylation using a brush and reacted at 100 °C; immersion acylation at 100 °C for 90 min | immersion modification resulted in a higher DS (0.91–1.8) than one-sided modification (0.37–0.55) | [110] |
| oat straw CNF | acetic, butyric, hexanoic or 2-dodecen-1-yl-succinnic anhydride as acylant, reacted at 80 °C in an oven for 2 h with a 10 kg weight on top | C2–C16 cellulose ester nanopapers; CNF wet cake was immersed in acylant liquid for 2 d, then put in an oven at 80 °C for 2 h under hot pressing | DS of cellulose esters decreased from 0.38 to 0.1 with increasing substituent chain length from C2 to C16 | [114] |
| Cellulose Packaging | Packaging Properties | Ref. | ||
|---|---|---|---|---|
| Barrier Properties | Mechanical Properties | Other Properties | ||
| C11-CEM film (DS = 0.19) | higher WCA (101°) and lower WA (6%) than original CNF film (54°, 95%), lower WVP at 3.4 × 10−9 g⋅m−1 s−1 Pa−1 than pristine CNF film (9.0 × 10−9 g⋅m−1 s−1 Pa−1) | decreased σ and E (47 MPa, 2075 MPa) compared with that of neat CNF film (57, 3847 MPa), while the ε was slightly increased (6.2% vs. 4.5%) | Td-onset was increased slightly to 350 °C compared with that of CNF (343 °C) | [115] |
| C18-CEL film (DS = 2.53–2.86) | WVTRs of isostearic-, oleic- and modified TOFA- cellulose ester films were markedly reduced to 21.7, 22.4 and 43.4 g/m2 24 h, poor oxygen resistance (too high for the sensor) | isostearic CEL film showed the highest ε of 101% (twice that of the original cellulose film), while oleic CEL film and modified TOFA CEL film had lower εs (57% and 45%) | Td-onset of C18-CELs (327~340 °C) was 7~22 °C higher than that of unmodified cellulose (320 °C) | [102] |
| C18-CEL film (DS = 0.3–3) | C18-CEL (DS = 0.3) film absorbed less water (13.9%) than pristine CMC film (28.9%), while C18-CEL (DS = 3) did not show significant WA, C18-CEL3 film had higher WCA (110°) than C18-CEL0.3 film (102°), WVPs of C18-CEL3 and C18-CEL0.3 films were decreased to 5 × 10−12 and 75 × 10−12 g m−1 s−1 Pa−1, respectively, compared with 158 × 10−12 g m−1 s−1 Pa−1 for neat CMC film | C18-CEL0.3 film had higher mechanical strength (σ = 28.5 MPa, E = 1118 MPa, ε = 12.7%) than C18-CEL3 film (σ = 5.5 MPa, E = 286 MPa, ε = 2.5%), but both substituted films had decreased mechanical strength compared with unmodified CMC film (σ = 169.2 MPa, E = 7230 MPa, ε = 15.9%) | C18-CEL0.3 film exhibited moisture-responsiveness, while C18-CEL3 film showed thermal responsiveness, the C18-CEL3 film displayed a distinct Tm at 55 °C, while no Tm was found for the C18-CEL0.3 film, reversible changes in the C18-CEL3 film volumes were observed when varying the temperature | [104] |
| C6-, C8-, C10-, C12-, C14- and C18-cellulose ester films (DS = 0.8–1.3) | WCA was increased from 66° to 90°, WVP was decreased from 6 to 1.6 cc × mm m−2 d−1 kPa−1 for C6 to C18, which was markedly lower than 20–25 cc × mm m−2 d−1 kPa−1 for pristine CNF film, C6–C18 cellulose ester films all exhibited poor oxygen resistance | C6-CES, C10-CEM and C12-CEM films had higher E (550–600 MPa) compared with other cellulose ester films, C8-CEM (DS = 1.3) film had the highest ε (90%), C12-CEM (DS = 0.9) film had the highest σ (35 MPa); the other cellulose ester films had lower σ (20–24 MPa) | cellulose ester films were transparent, flexible and heat-sealable, melted at 170–225 °C, and were able to be squeezed through a 2-mm rod die | [106] |
| C11-CEM-coated paper (DS = 2.75) | WCA decreased from 117° to 101° with coating grammage increasing from 0.97 to 6.25 g m−2, WVTR decreased from 441 to 192 g/m2 24 h with increasing coating grammage, compared with 622 g/m2 24 h of uncoated paper | slightly increased ε an nonsignificant change in tensile index | PHGH and MPA were attached to C11-CEM-coated paper, giving the paper desirable antimicrobial performance | [109] |
| C6- and C16-cellulose ester-coated paper (DS = 1.6–2.9) | WCA was enhanced to 95–123°, WVTR of C16-CEL-coated paper (100–300 g/m2 24 h) was considerably lower than that of C6-CES-coated paper (400–1020 g/m2 24 h) | cellulose ester-coated paper showed slightly enhanced σ (12.4–12.7 MPa) compared with uncoated paper (11.8 MPa) | C16-CEL-coated paper showed higher Td-onset (350 °C) than C6-CES-coated paper (320 °C). C6-CES and C16-CEL powder became liquid at 160 °C and 220 °C, respectively, in an oven | [105] |
| C12-, C16- and C18-cellulose ester films, DS (immersion method) = 0.9–1.8, DS (one-sided method) = 0.37–0.55 | WCA of one-sided acylated film: 105–121°, WCA of immersion-acylated film: 113°, WVP of one-sided acylated film did not show any decrease compared with that of unmodified CNF films (WVP = 0.057 ng s−1 m−1 Pa−1), WVP of immersion-acylated film decreased to 0.006–0.021 ng s−1 m−1 Pa−1 | mechanical strength of one-side-acylated film was comparable to that of the original CNF film, immersion-acylated film showed much weaker mechanical strength | thermal stability was increased for immersion-acylated films, whereas that of one-side-acylated films was similar to pristine CNF film (Td-onset = 245 °C) | [110] |
| C2-, C4-, C6- and C16-cellulose ester nanopapers (DS = 0.1–0.38) | WCA was enhanced from 32° to 119° from C2 to C16 and was 24° for the neat CNF nanopaper, C16-CEL paper floated on the water surface for several weeks, while pristine CNF paper sank | C2-CES nanopaper had the highest E, and C16-CEL nanopaper had the lowest E, C16-CEL had the highest wet strength, which was 7- fold greater than that of the reference CNF nanopaper | n.d. a | [114] |
| Technology | Smoothness | WCA a | WVP | TS | Efficiency | Ref. |
|---|---|---|---|---|---|---|
| self-standing film | ‘4–5’ b | 84–107°, ‘2–3’ | reduced 62–90%, ‘4–5’ | decreased 18%, ‘4’ | ‘2’ | [103,106,115] |
| physical bar coating on paperboard | ‘3–4’ | 100–124°, ‘3–4’ | reduced 29~69%, ‘2–4’ | no change, ‘5’ | ‘1’ | [105,109] |
| chemical brush coating on film | ‘4’ | 109°, ‘3’ | no change, ‘1’ | decreased 32%, ‘3’ | ‘4’ | [110] |
| spray coating on paperboard | ‘1’ | 152°, ‘5’ | no change, ‘1’ | n.d. c | ‘3’ | [108,109] |
| chemical immersion coating on film | ‘3’ | 114°, ‘3’ | reduced 63~90%, ‘4–5’ | decreased 68~80%, ‘1–2’ | ‘5’ | [110] |
| Parameters | Crosslinker | Water Solubility (WS)/WA | WVTR/WVP | σ, ε | Ref. | |
|---|---|---|---|---|---|---|
| Cellulose | ||||||
| sorbitol plasticized nanocellulose-coated filter paper | CAC | n.d. a | WVP: reduced by 88% | no change in σ; ε: 3.7% vs. 2.5% | [125] | |
| HPMC | CAC | WS: reduced by 74% | WVP: reduced by 43% | n.d. a | [126] | |
| CMCS | UV irradiation | WS: reduced by 21% | WVTR: reduced by 99% | σ: 46.8 MPa vs. 14.2 MPa; ε: 9.1% vs. 19.9% | [127] | |
| DACNF | gelatin | WA: 44% vs. 167% | WVP: reduced by 99.9% | wet σ: 15.4 MPa vs. 9.9 MPa; wet ε: 23.2% vs. 16.5% | [57] | |
| CMCS | DMTMM | WS: 18% vs. 100% | WVP: reduced by 40% | σ: 55 MPa vs. 32 MPa; ε: 25% vs. 30% | [128] | |
| Cellulose | Crosslinking Process | Packaging Type and Its Formation | Reaction Degree | Ref. |
|---|---|---|---|---|
| PEG400 plasticized HPMC | CAC as crosslinker and NaH2PO4 as catalyst, reacted in a mixture of DI water and ethanol with homogenization for 15 min, dried at 60 °C for 60 min and cured at 190 °C for 15 min | CAC-HPMC film; cured at high temperature | crosslinking rate ranged between 0% and 65% with a CAC content of 0–15% (w/w to HPMC) | [126] |
| CMCS | photo-crosslinking via UV irradiation at RT for 30–180 min with SB as photo-initiator or chemical crosslinking with saturated GLA vapor plus gelatin as synergistic crosslinkers at 80 °C for 30–180 min | CMCS/SB/UV or CMCS/GLA/gelatin film; films were prepared by the casting method at 45 °C for 18 h | photo-crosslinked film treated with 20 wt% SB and irradiated for 180 min, or chemically-crosslinked film modified with 0.2 g gelatin and exposed to GLA vapor for 90 min were found to have optimized crosslinking degrees | [127] |
| DACNF | gelatin as crosslinker, reacted in DI water at 60 °C for 3 h | G-DACNF film; vacuum filtration and solvent casting | crosslinking degree = 57% | [57] |
| gelatin plasticized CMCS | DMTMM as crosslinker for CMCS, reacted in DI water at RT for 2 h | DMTMM-CMCS film; films were prepared by the casting method at 40 °C overnight | optimum crosslinking degree was achieved in the presence of 5 wt% DMTMM and 50 wt% glycerol | [128] |
| Cellulose Packaging | Additives | Packaging Properties | Ref. | ||
|---|---|---|---|---|---|
| Barrier Properties | Mechanical Properties | Other Properties | |||
| BTCA, TCA, SA crosslinked paper | - | n.d. a | wet tensile index of BTCA crosslinked (27 mN/g) and TCA crosslinked paper (16.5 mN/g) were markedly enhanced compared with 1.3 mN/g of pristine paper, while papers treated with SA showed little wet strength enhancement | n.d. | [129] |
| CAC-CNC-coated filter paper (three layers of coating) | sorbitol | modified paper showed increased WA (37%) compared with uncoated filter paper (29%), WVP and OP of modified paper was considerably decreased to 0.5 g mm kPa−1 m−2 day−1 and 2 mL µm m−2 day−1 kPa−1 compared with 4 g mm kPa−1 m−2 day−1 and 197 mL µm m−2 day−1 kPa−1 of uncoated filter paper | modified paper had a nearly unchanged σ, a reduced E (from 570 to 310 MPa) and increased ε (from 2.5% to 3.7%) compared with the control filter paper | Td-onset decreased from 311 °C to 288 °C | [125] |
| CAC-HPMC film | PEG400 | increasing CAC content decreased WS of films, with an optimum CAC content of approximately 14%; 5 wt% CAC (w/w to HPMC) reduced WS of CAC-HPMC films by 74%; 15% CAC loading resulted in the highest reduction of WVTR by 47% for CAC-HPMC film (168 g/m2 24 h) compared with neat HPMC film (316 g/m2 24 h) | n.d. | CAC-HPMC films were transparent | [126] |
| CMCS/SB/UV or CMCS/GLA/gelatin film | - | WS of CMCS/SB/UV and CMCS/GLA/gelatin films was reduced to 57.1% and 50.1%, respectively, compared with 78.2% for pristine CMCS film, WVP of CMCS/SB/UV film was markedly decreased to 9 × 10−7 g m−1 s−1 Pa−1 compared with 8.19 × 10−5 g m−1 s−1 Pa−1 for neat CMCS film, while CMCS/GLA/gelatin showed a smaller WVP decrease (to 50 × 10−7 g m−1 s−1 Pa−1) | σ of CMCS/SB/UV and CMCS/GLA/gelatin films were considerably enhanced to 46.8 MPa and 33.7 MPa, respectively, compared with 14.2 MPa of unmodified CMCS film. However, the ε of the CMCS films decreased from 19.9% to 9.1% and 13.8% after crosslinking with SB/UV or GLA/gelatin, respectively | both crosslinked films were noncytotoxic | [127] |
| G-DACNF film | - | G-DACNF displayed greatly decreased WA when immersed in DI water for 1 h, resulting in a weight increase of only 44%, while the neat CNF film weight increased by 167% | G-DACNF film exhibited a much higher wet mechanical strength (ε: 23.2%, wet σ: 15.4 MPa, wet E: 94 MPa) compared with unmodified CNF film (6.7%, 0.9 MPa, 26 MPa) | G-DACNF films were transparent | [57] |
| DMTMM- CMCS film | gelatin (added or not) | 5 wt% DMTMM-CMCS showed the lowest WS (18.1%); while CMCS completely dissolved in water within 4 h, the 10 wt% DMTMM-CMCS film showed a decreased WVP and oil absorption (0.68 × 10−7 g m−1 h−1 Pa−1 and 0.29%); however, the addition of glycerol increased the water sensitivity of the DMTMM-CMCS films | σ was enhanced from 32 MPa to 54.9 MPa, while the ε decreased from 30.1% to 25.4% with the addition of 5 wt% DMTMM compared with neat CMCS film. With the addition of glycerol, the optimum σ and ε values (52.3 MPa, 37.3%) were achieved in the presence of 5 wt% DMTMM and 50 wt% glycerol | DMTMM-CMCS films were transparent with optical transmittance of 80–90% | [128] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Z.; Ngai, T. Recent Advances in Chemically Modified Cellulose and Its Derivatives for Food Packaging Applications: A Review. Polymers 2022, 14, 1533. https://doi.org/10.3390/polym14081533
Jiang Z, Ngai T. Recent Advances in Chemically Modified Cellulose and Its Derivatives for Food Packaging Applications: A Review. Polymers. 2022; 14(8):1533. https://doi.org/10.3390/polym14081533
Chicago/Turabian StyleJiang, Zhuolun, and To Ngai. 2022. "Recent Advances in Chemically Modified Cellulose and Its Derivatives for Food Packaging Applications: A Review" Polymers 14, no. 8: 1533. https://doi.org/10.3390/polym14081533
APA StyleJiang, Z., & Ngai, T. (2022). Recent Advances in Chemically Modified Cellulose and Its Derivatives for Food Packaging Applications: A Review. Polymers, 14(8), 1533. https://doi.org/10.3390/polym14081533