
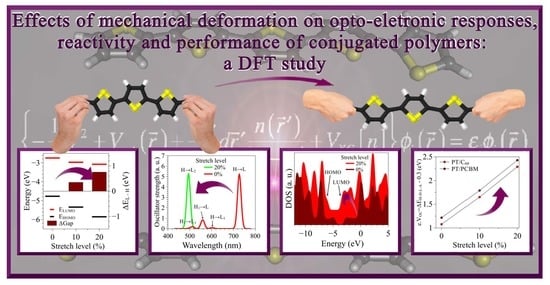
Effects of Mechanical Deformation on the Opto-Electronic Responses, Reactivity, and Performance of Conjugated Polymers: A DFT Study



Abstract
:
1. Introduction
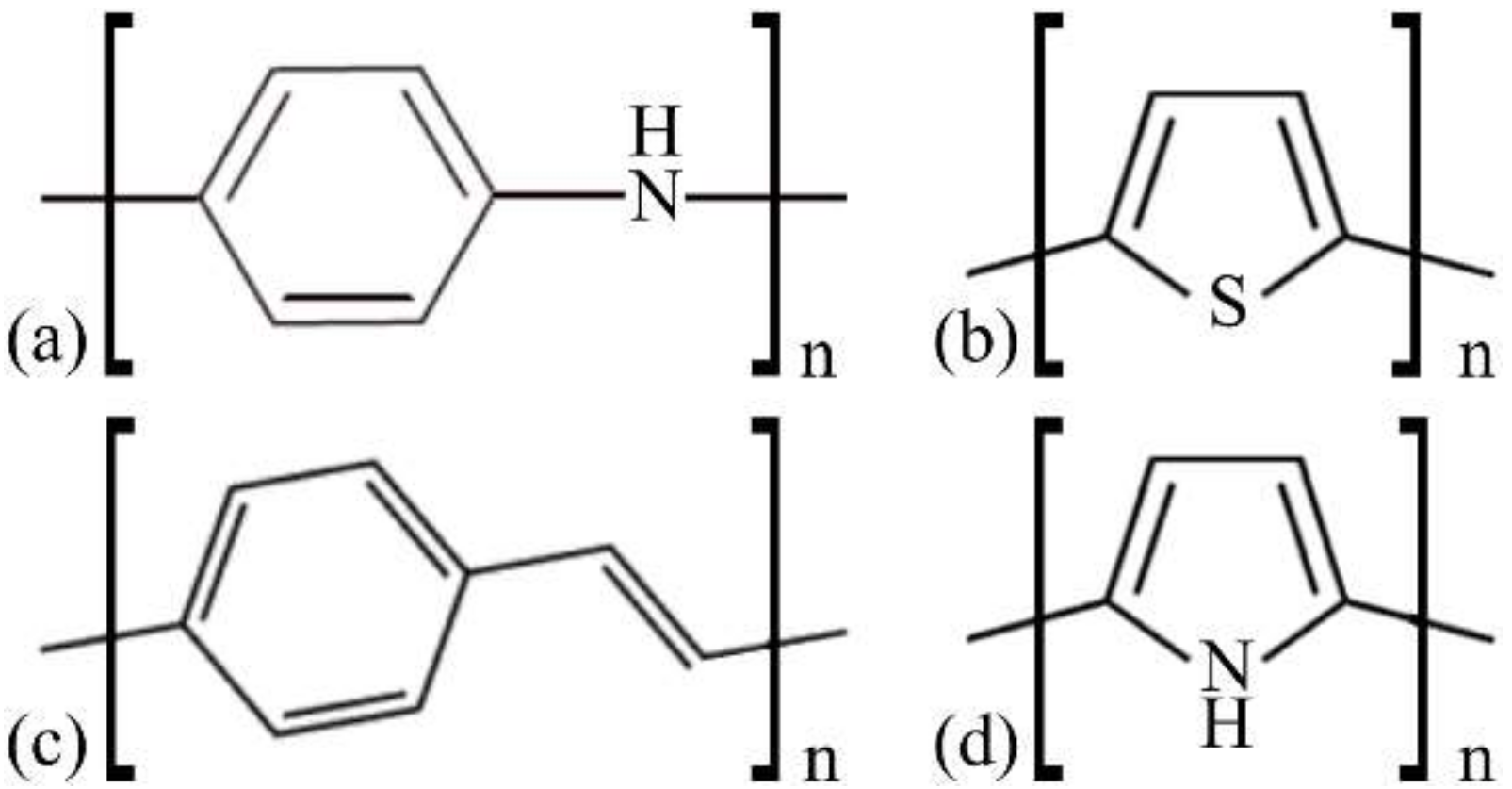
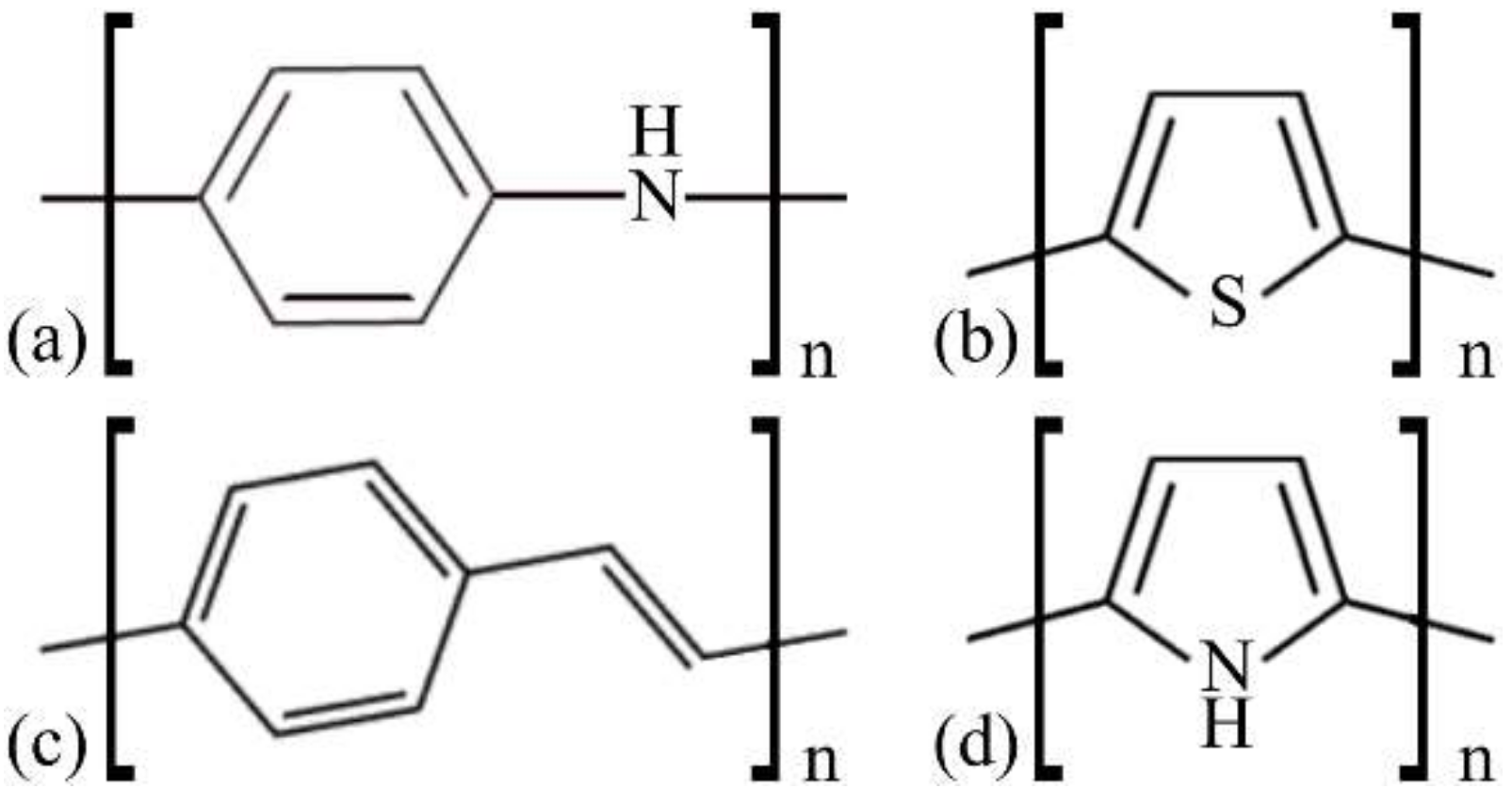
2. Materials and Methods
3. Results and Discussion
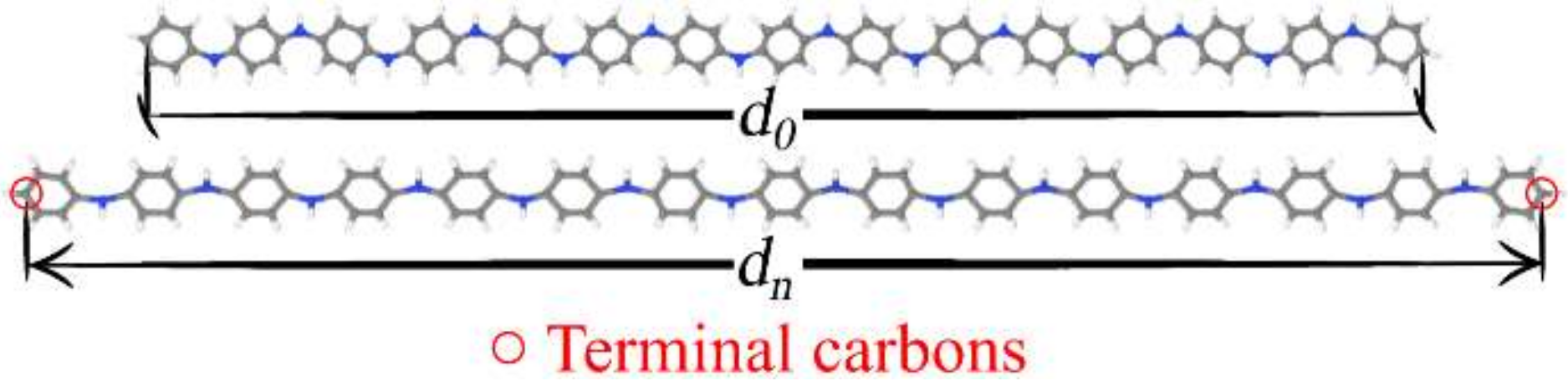
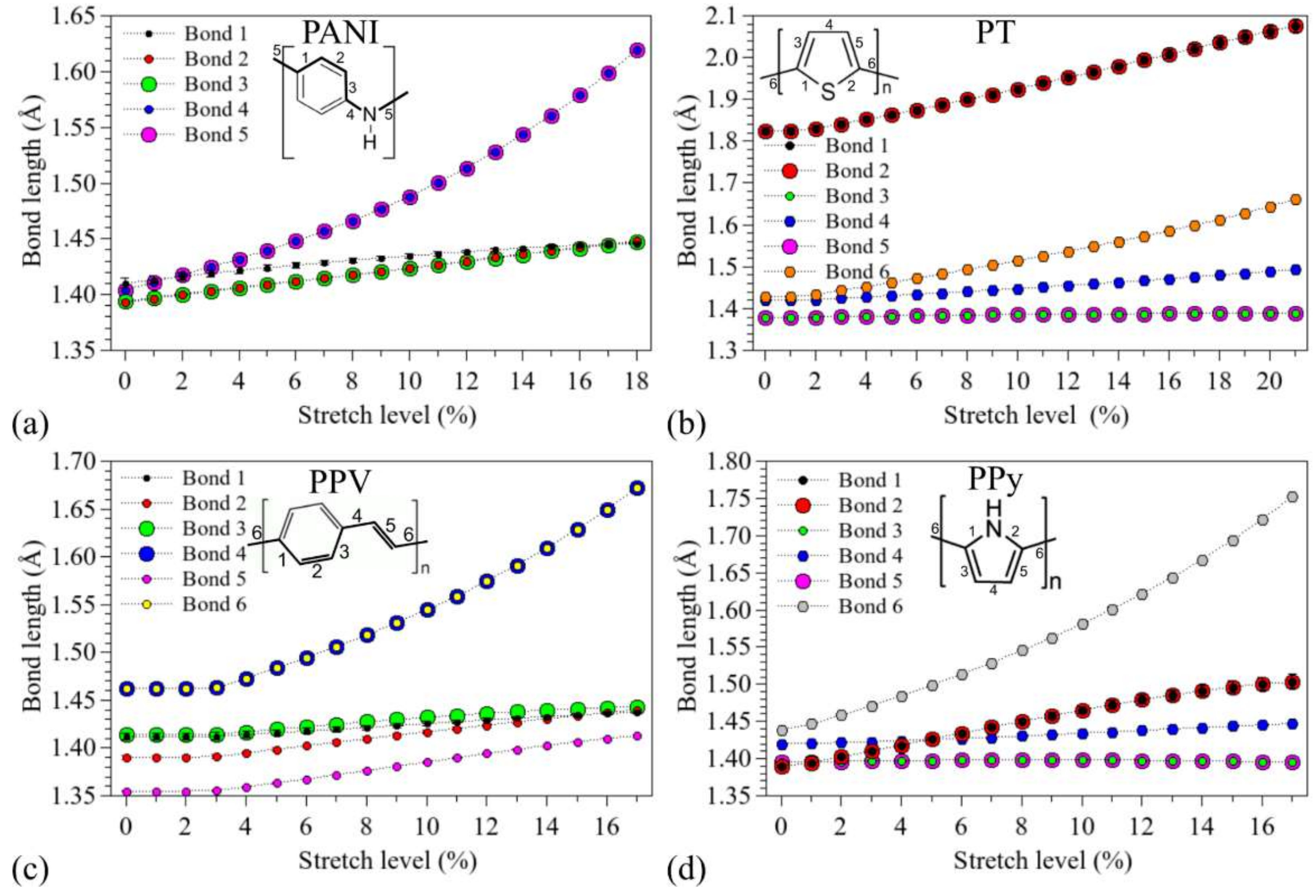
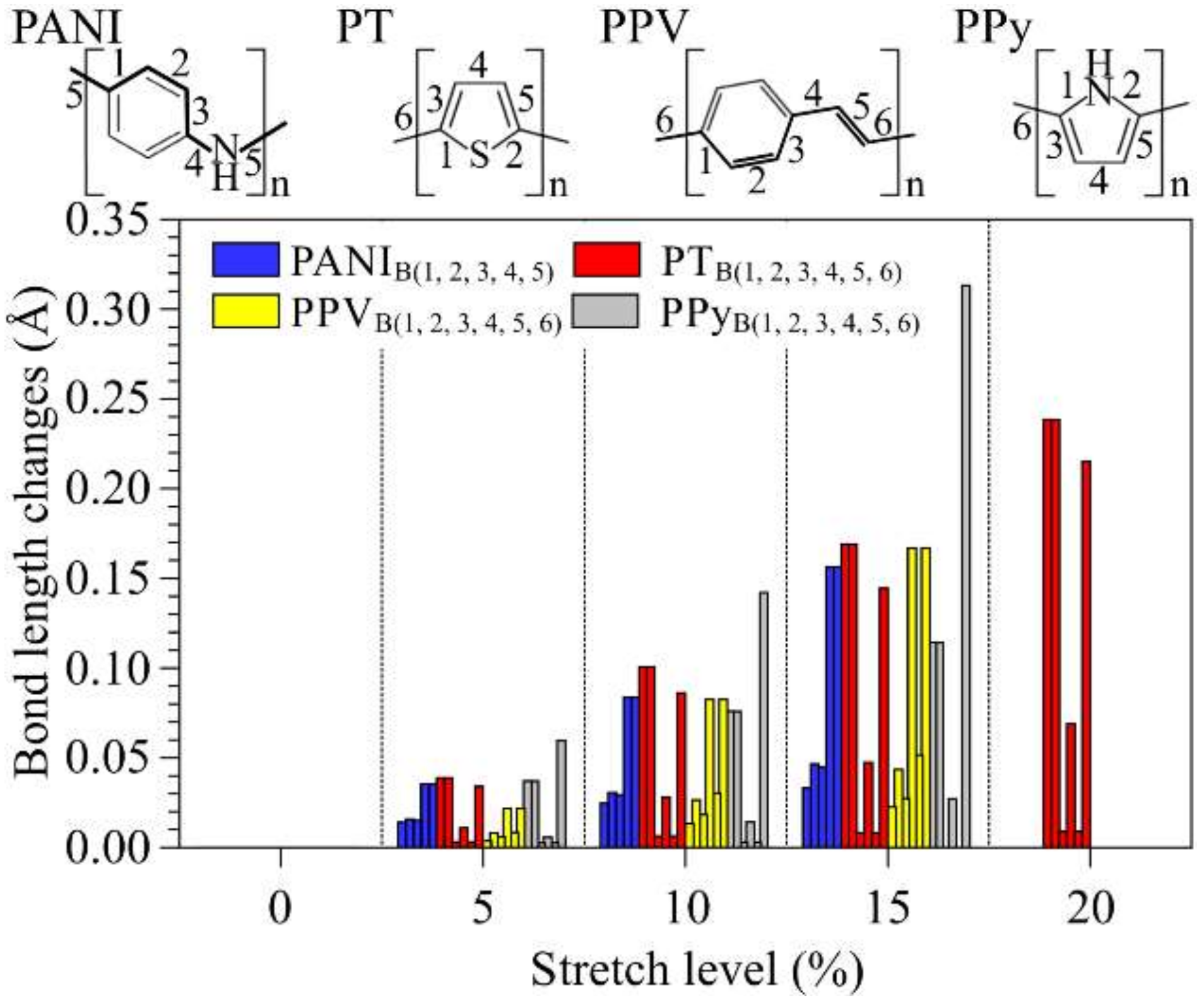
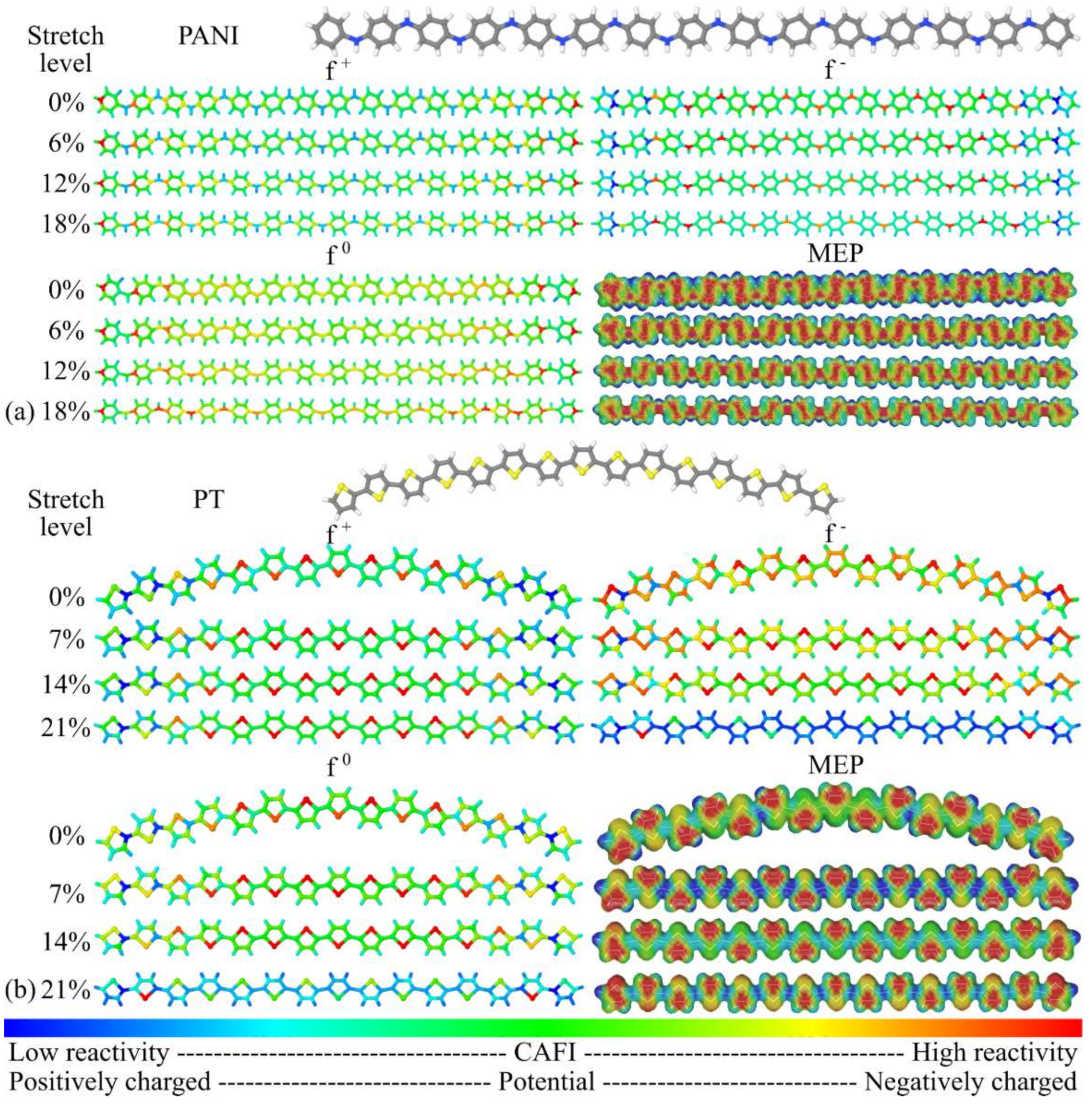
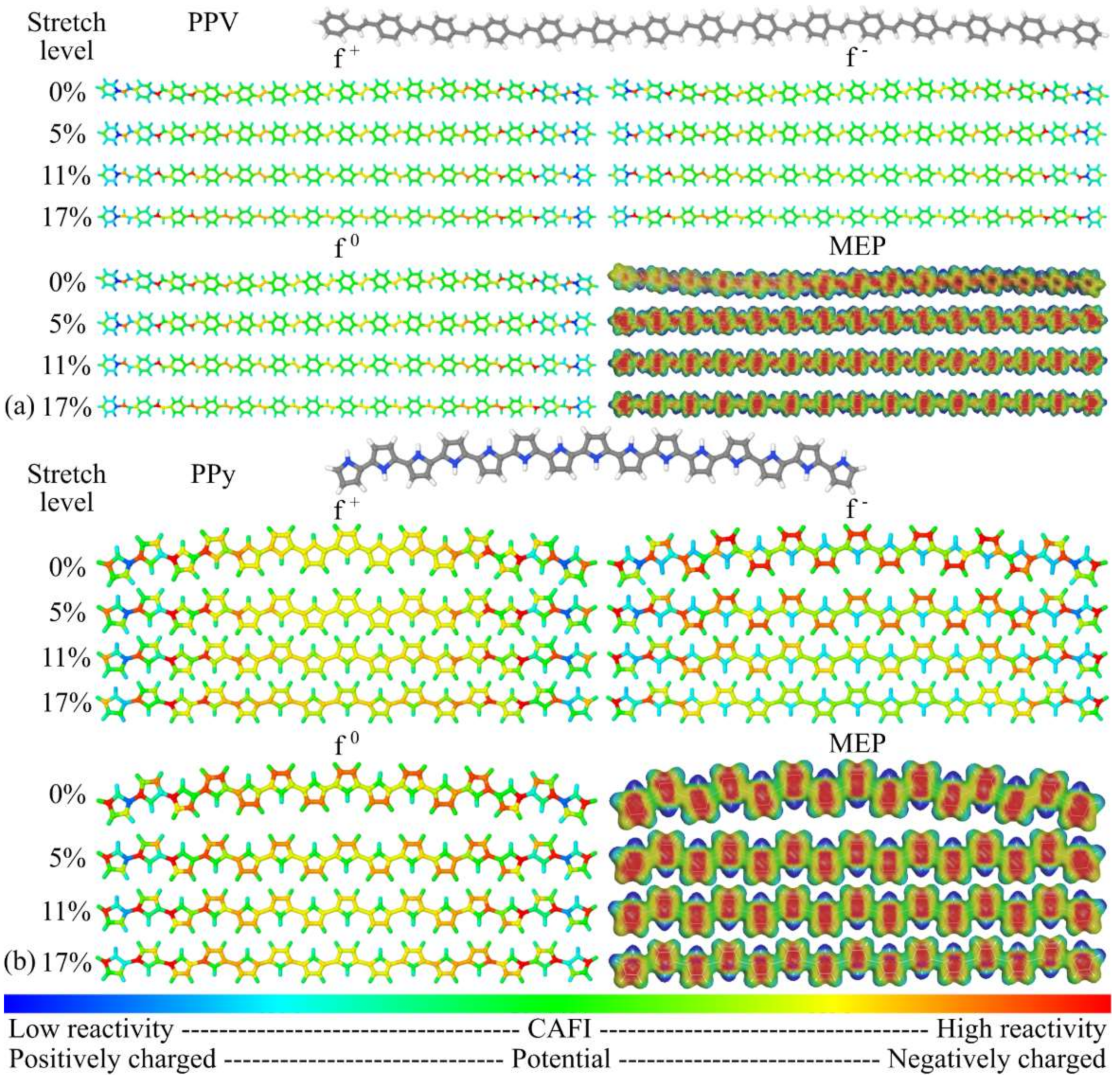
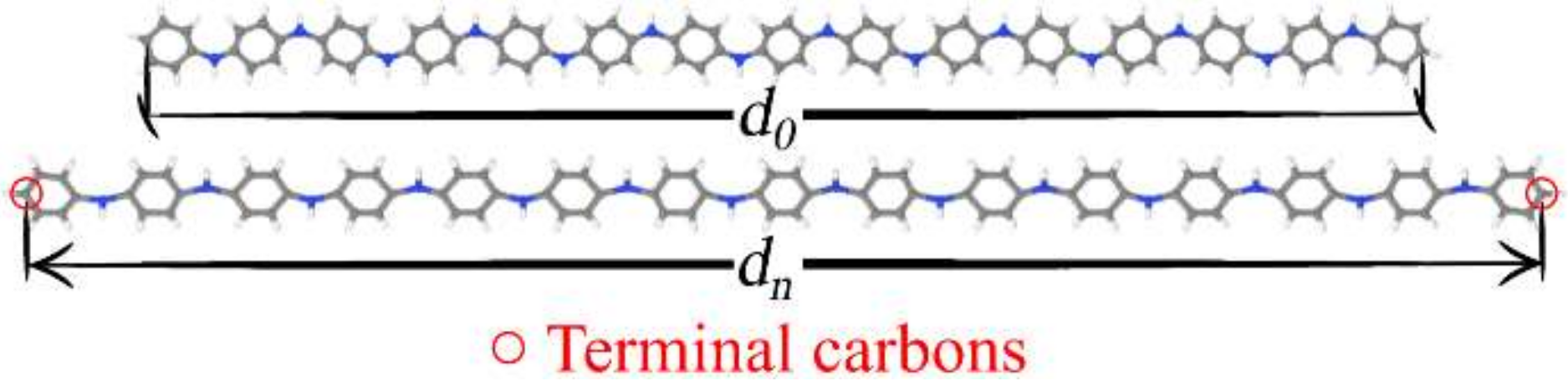
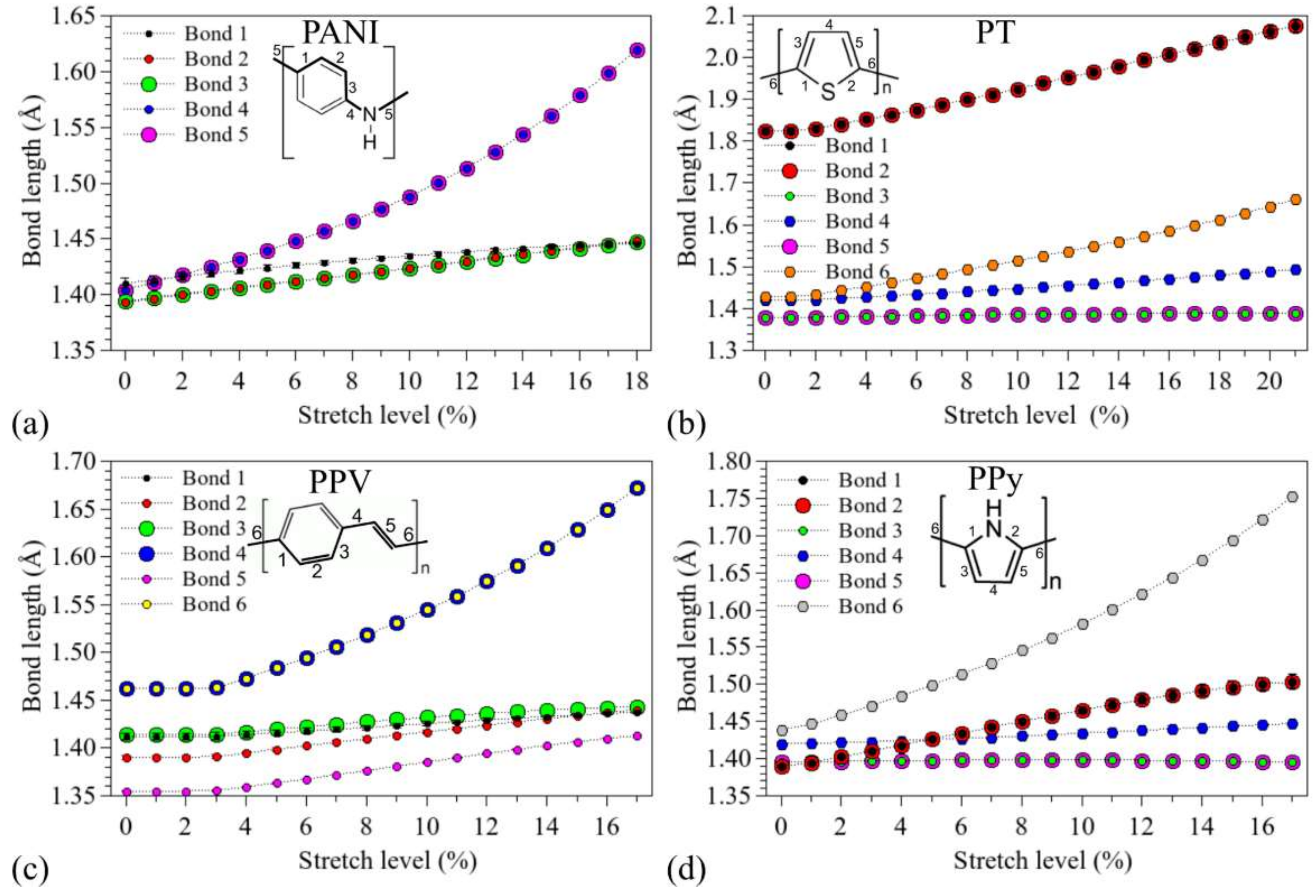
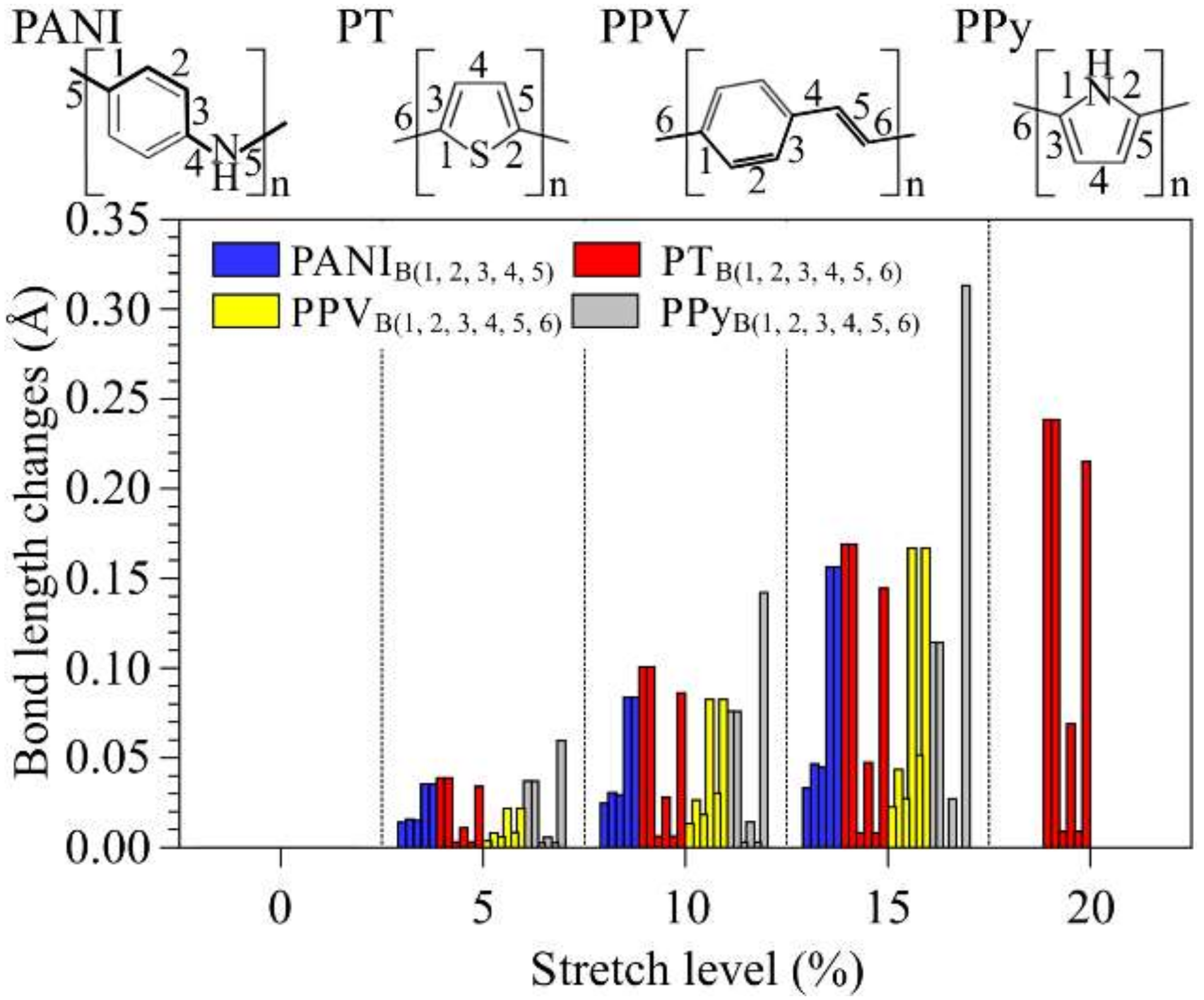
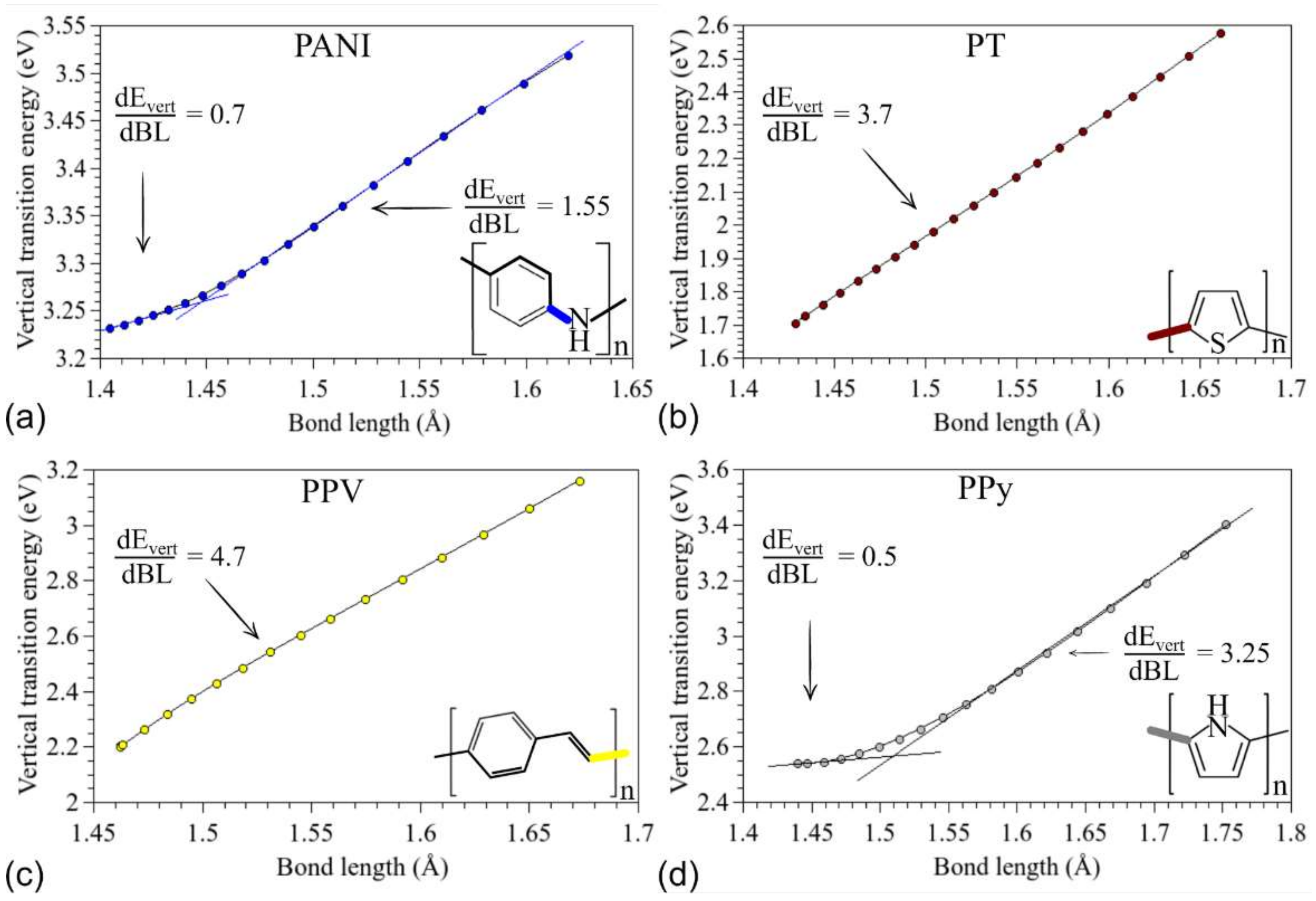
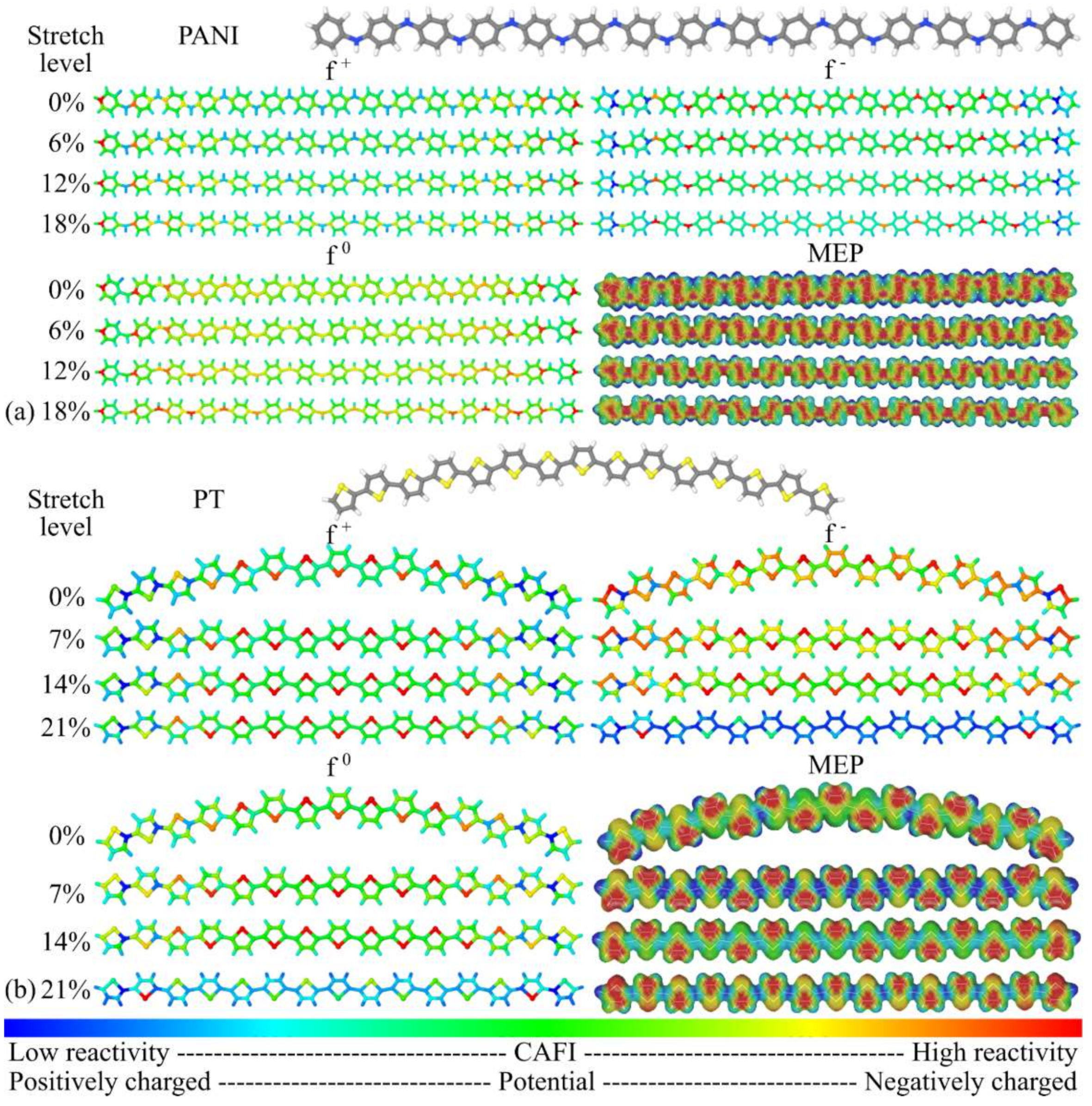
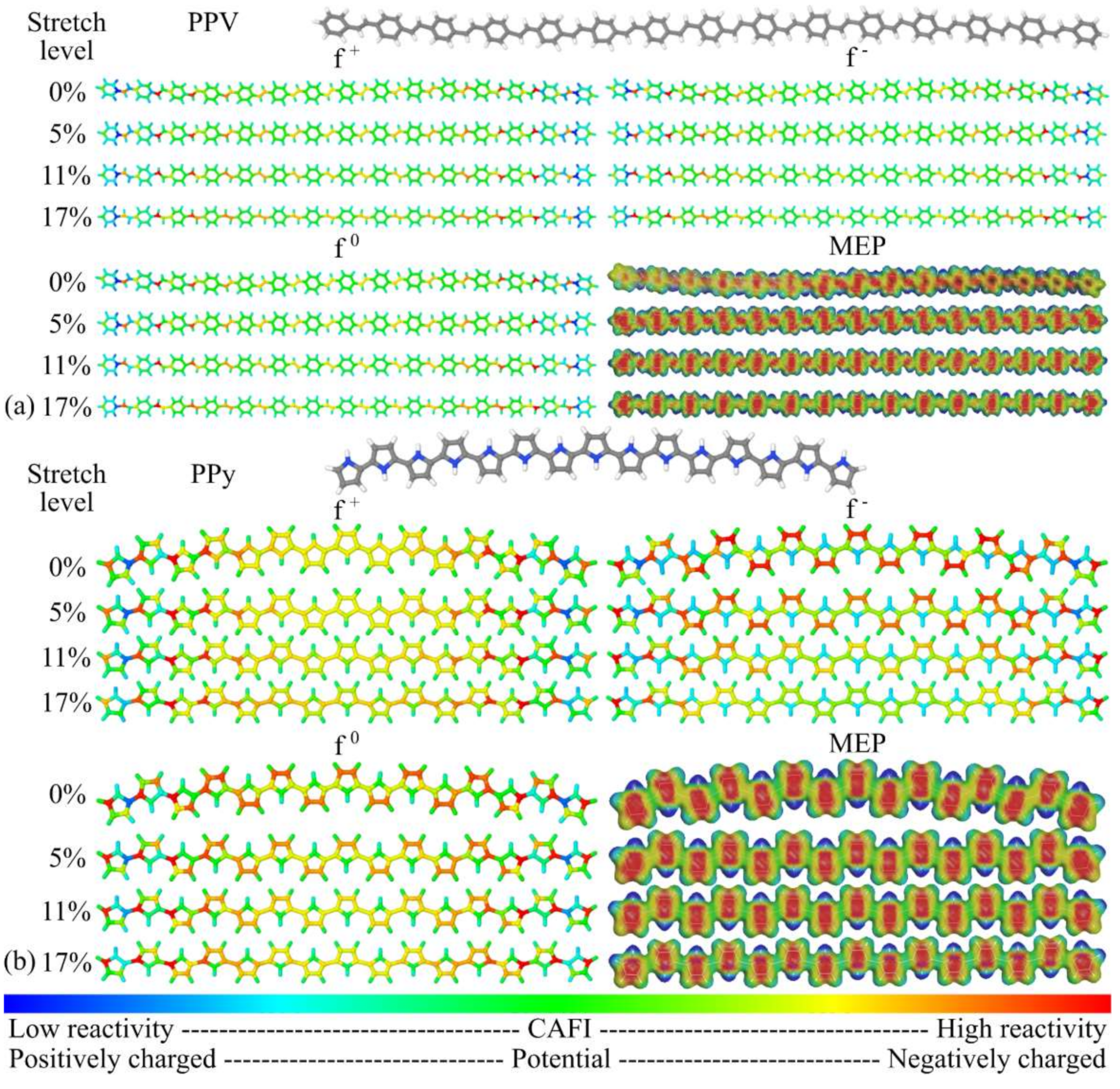
3.1. Structural Changes
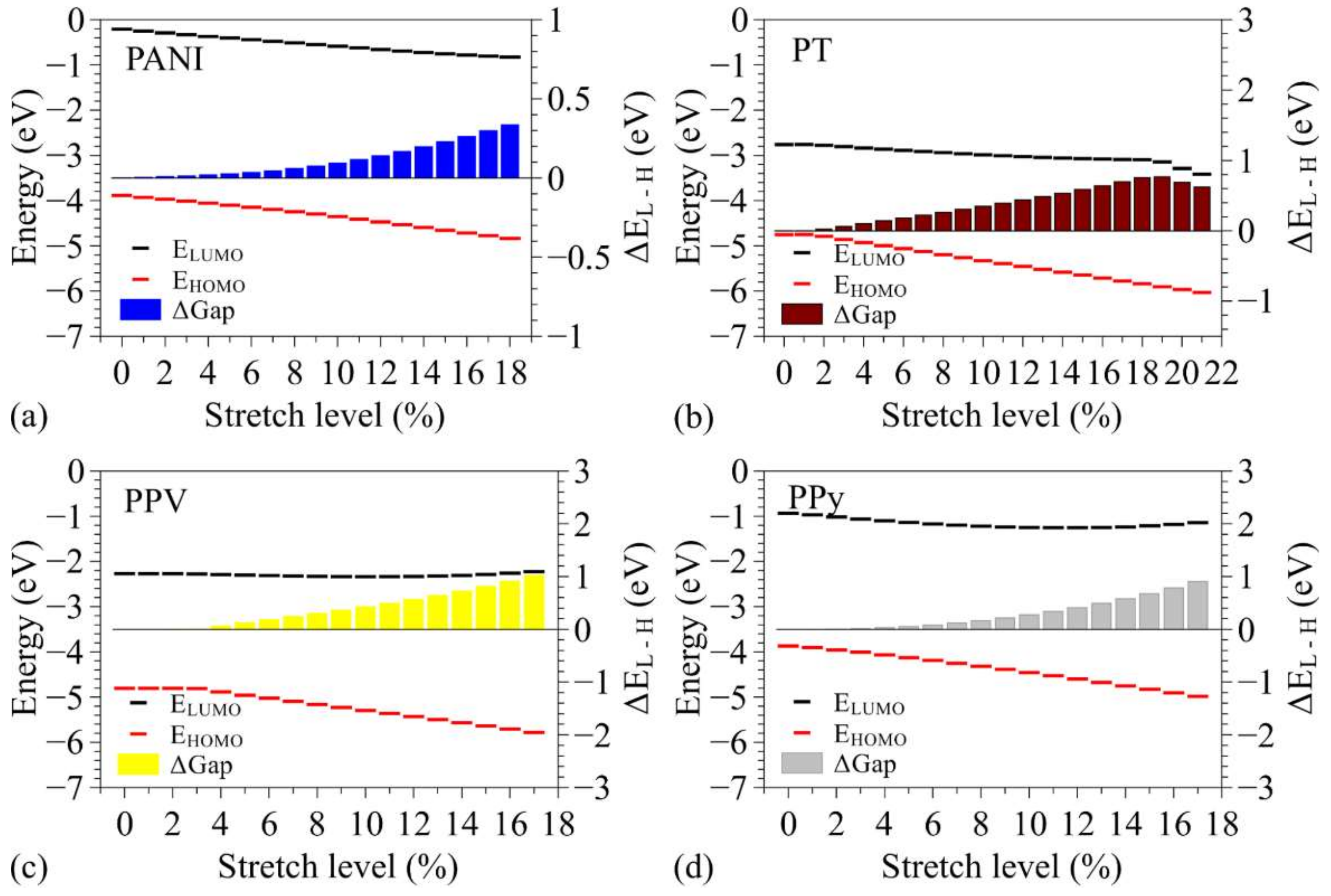
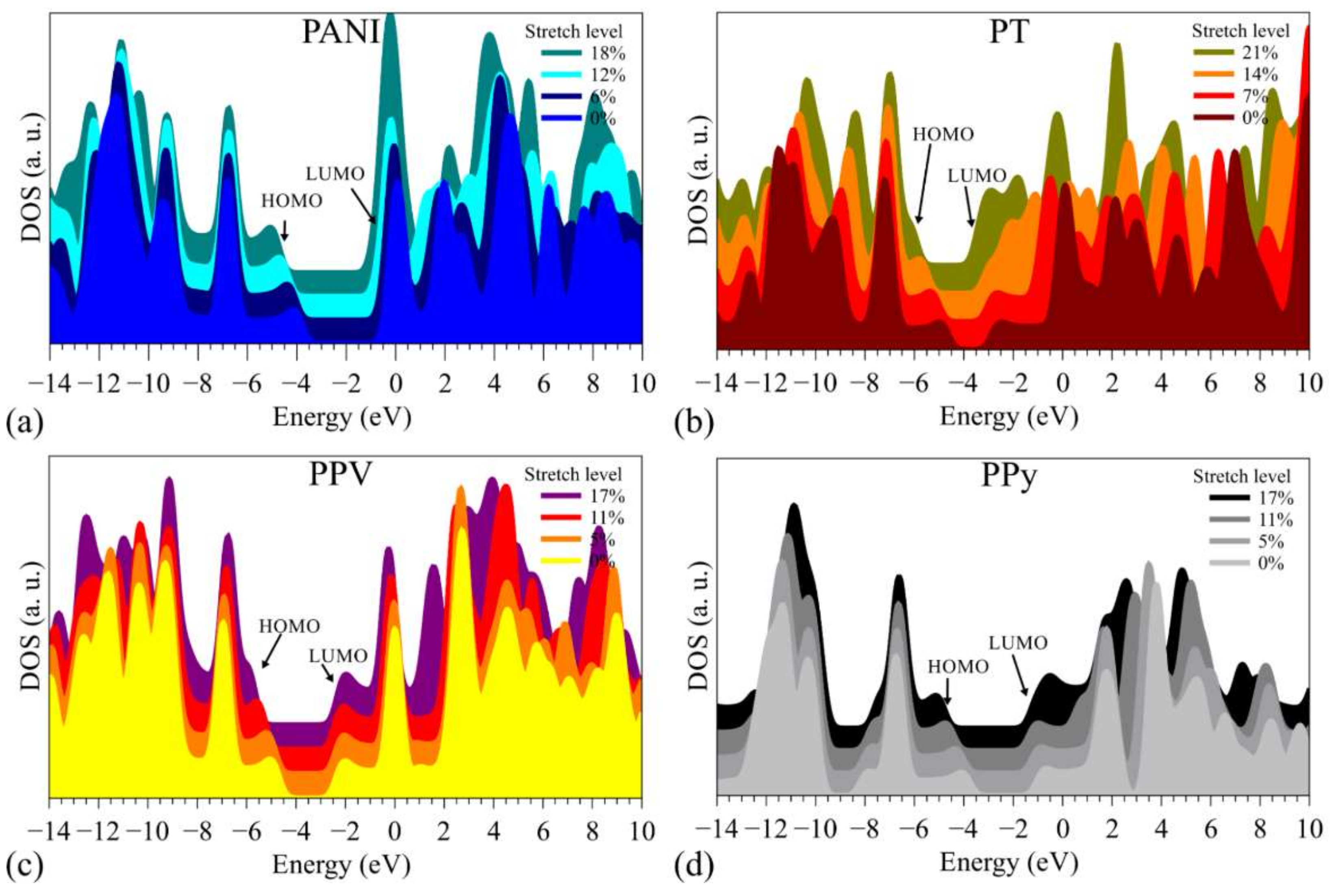
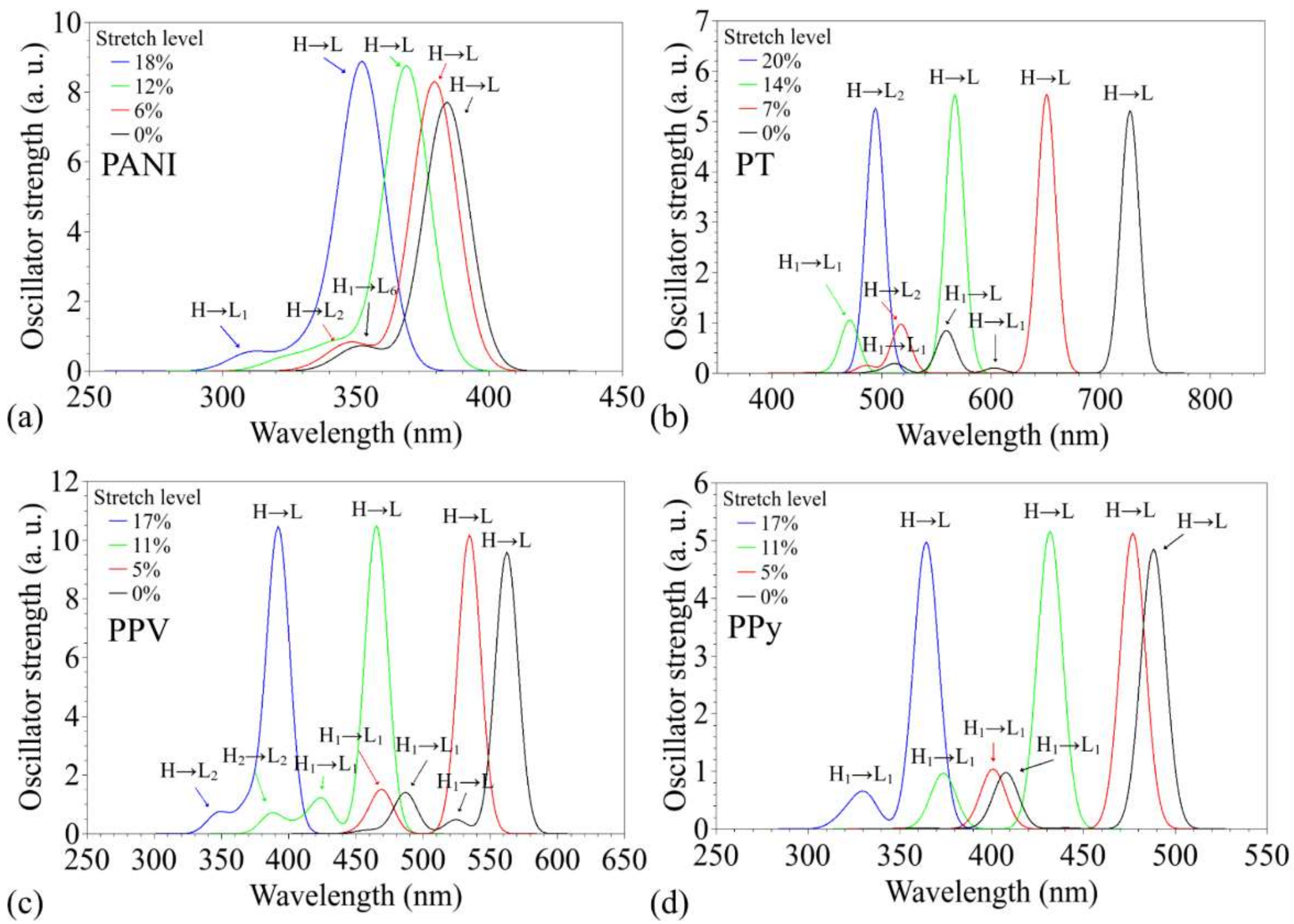
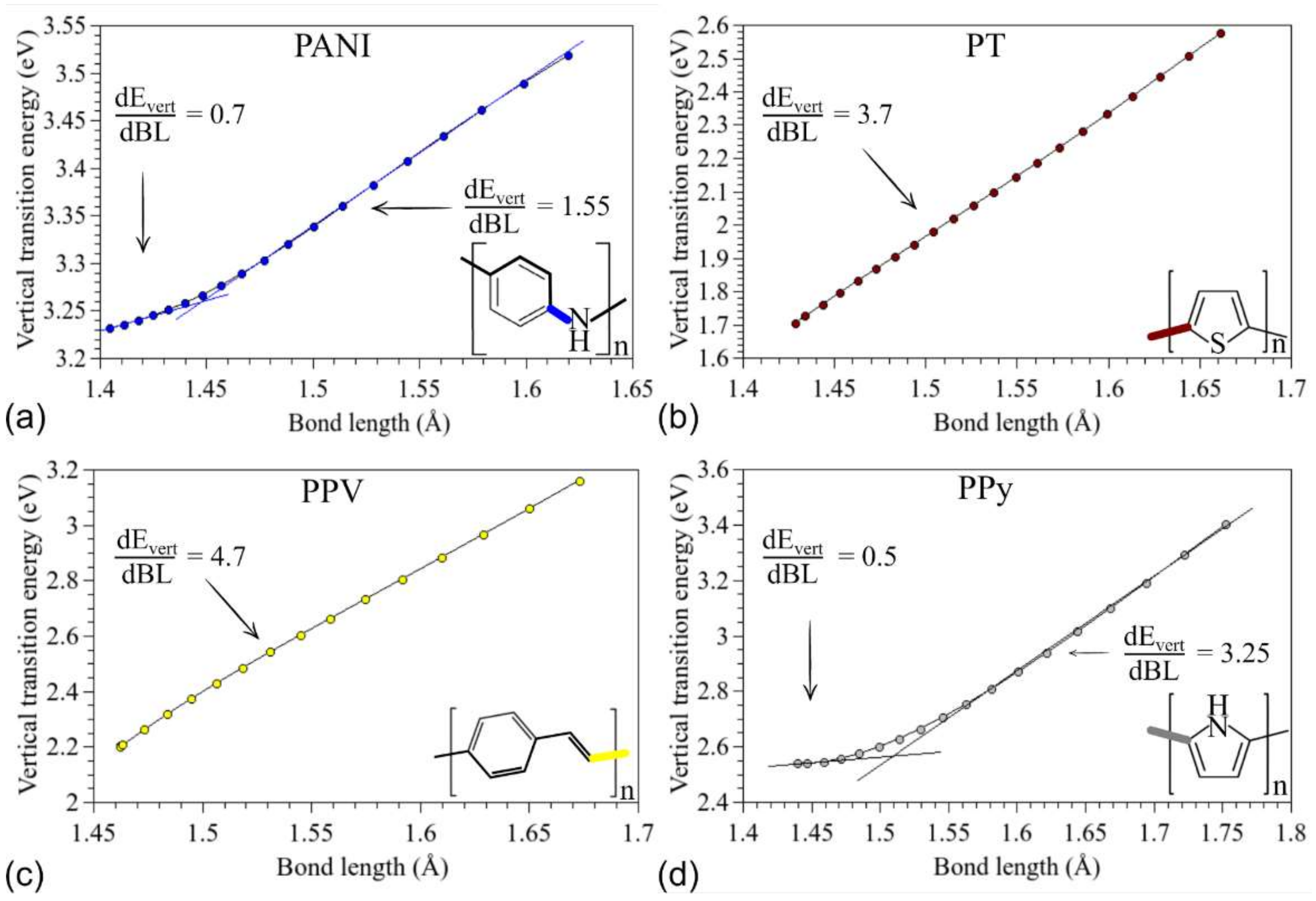
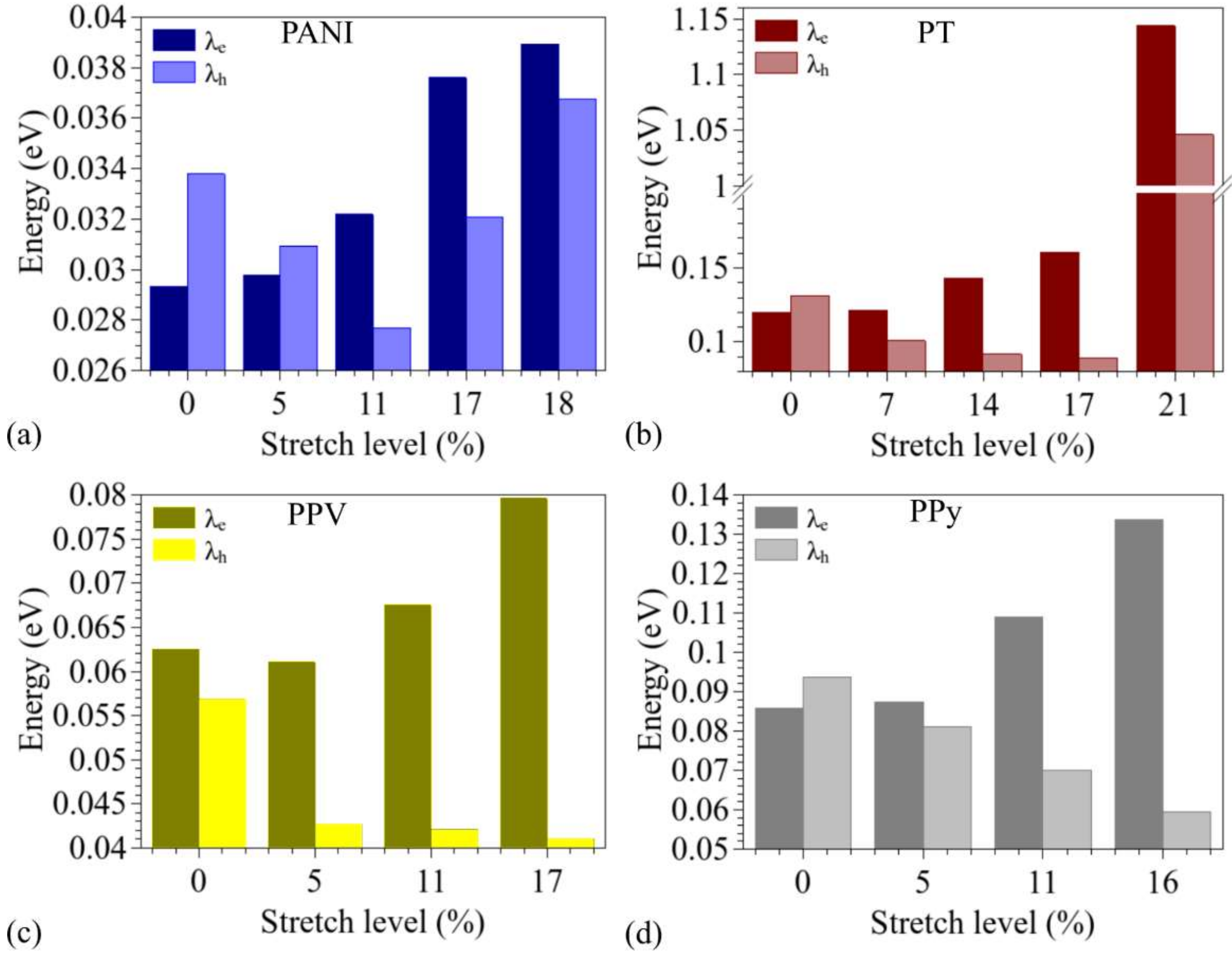
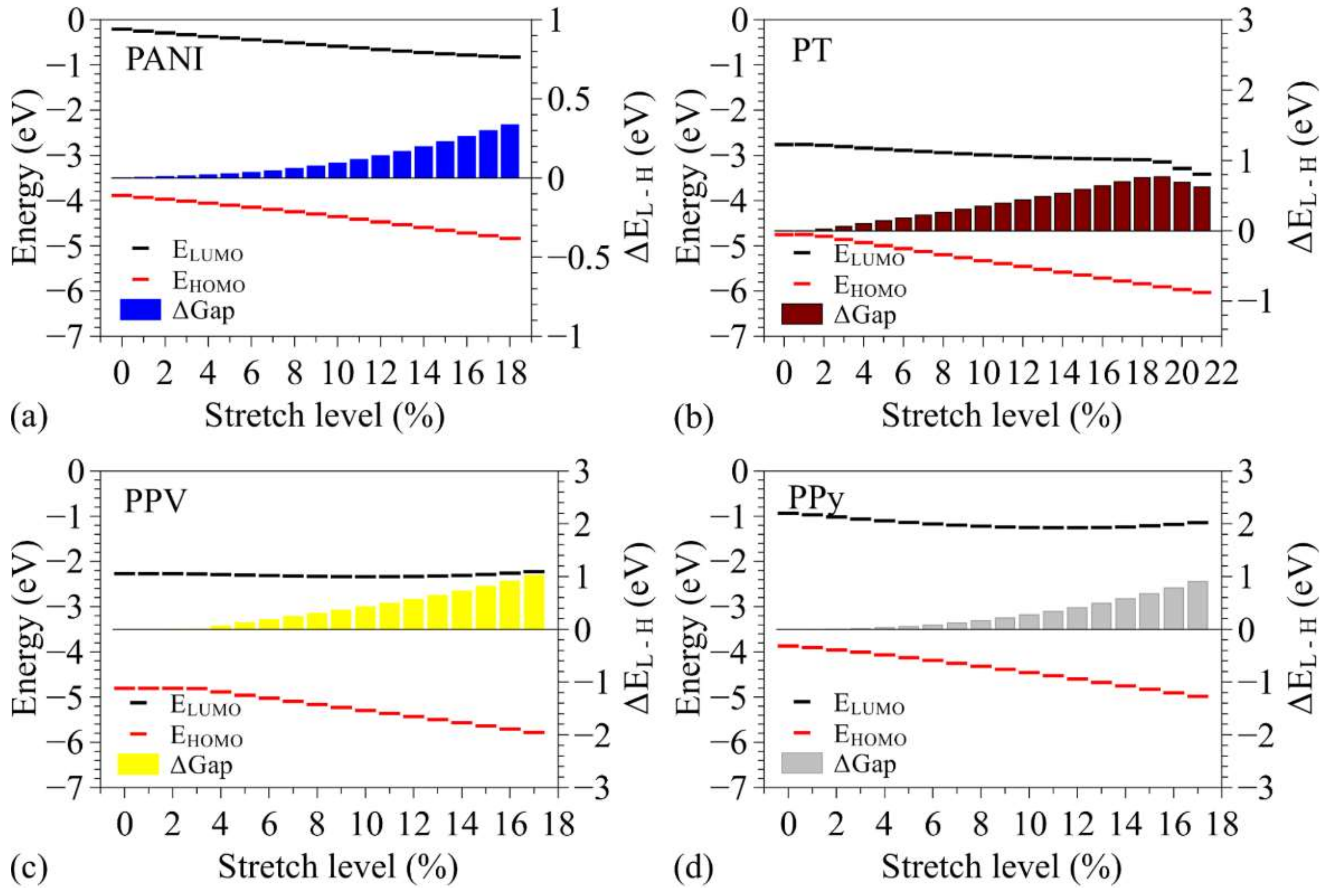
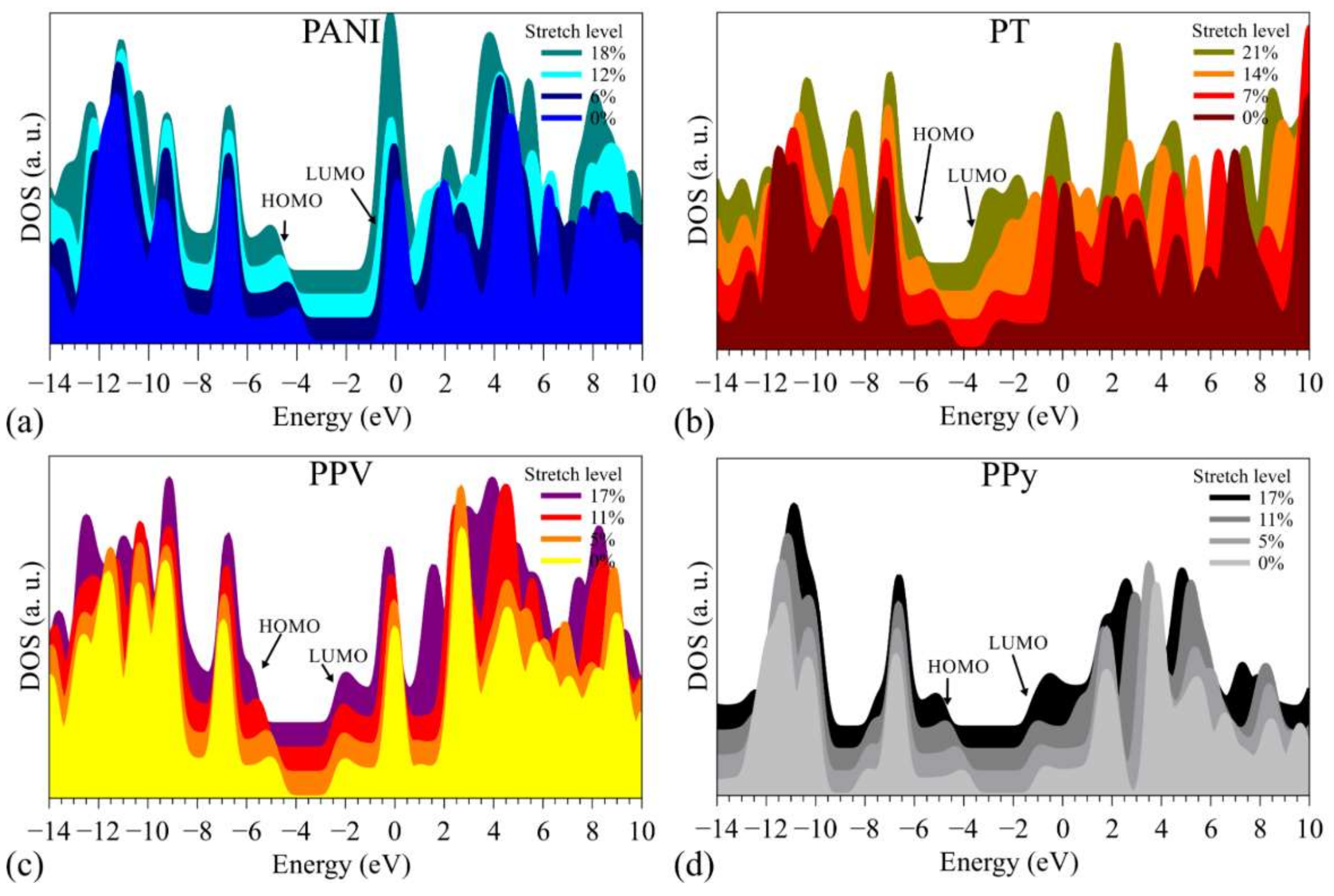
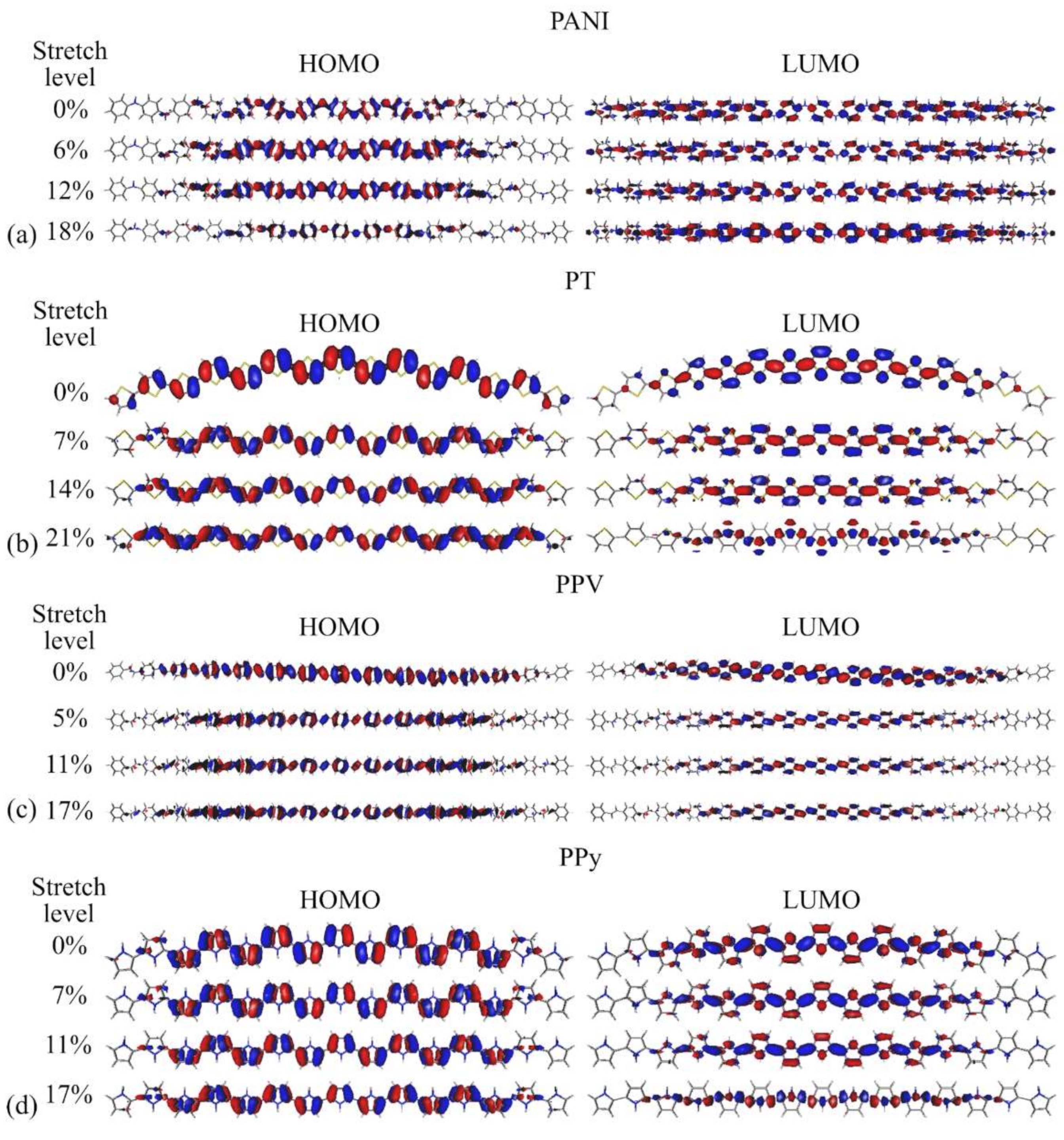
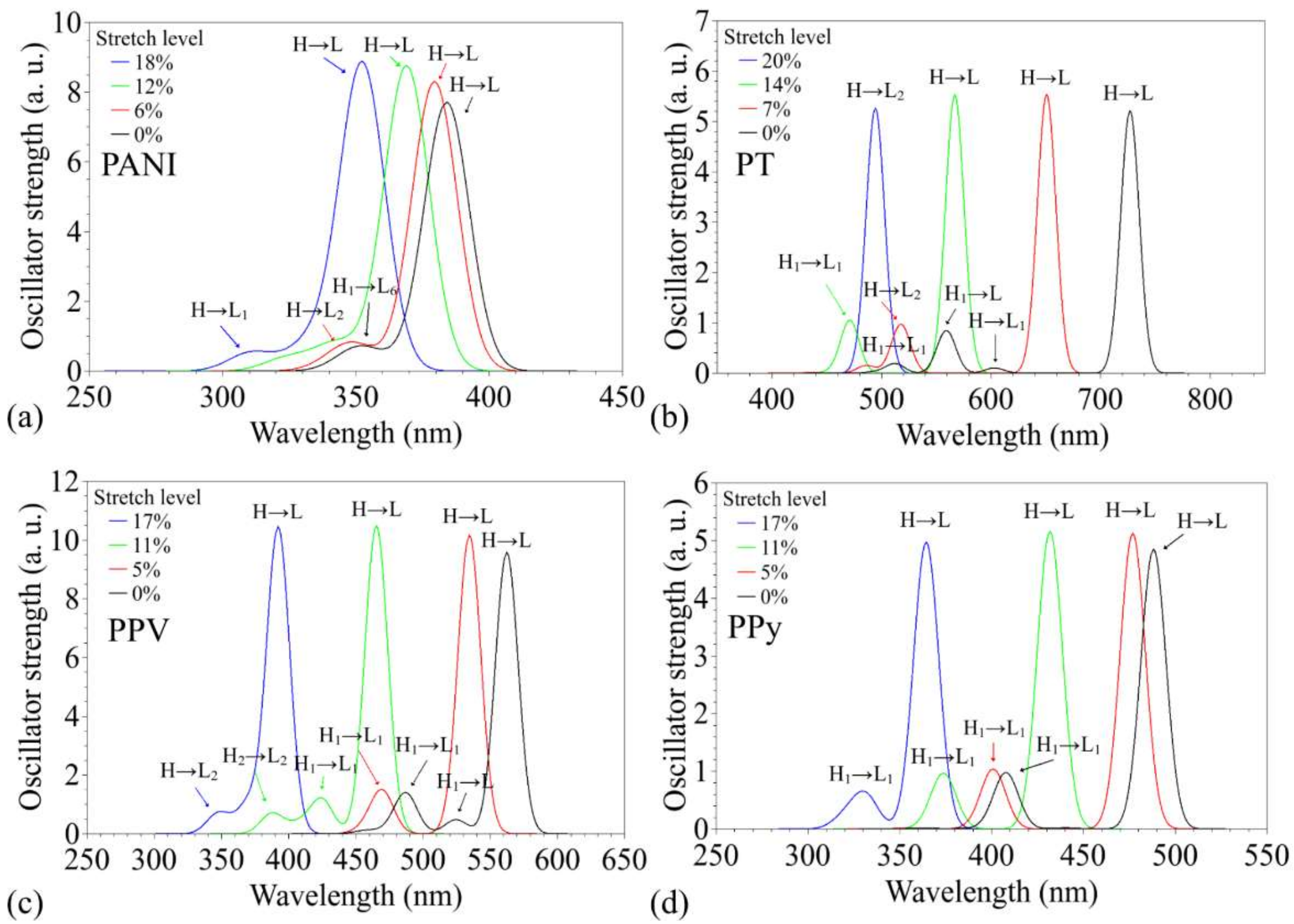
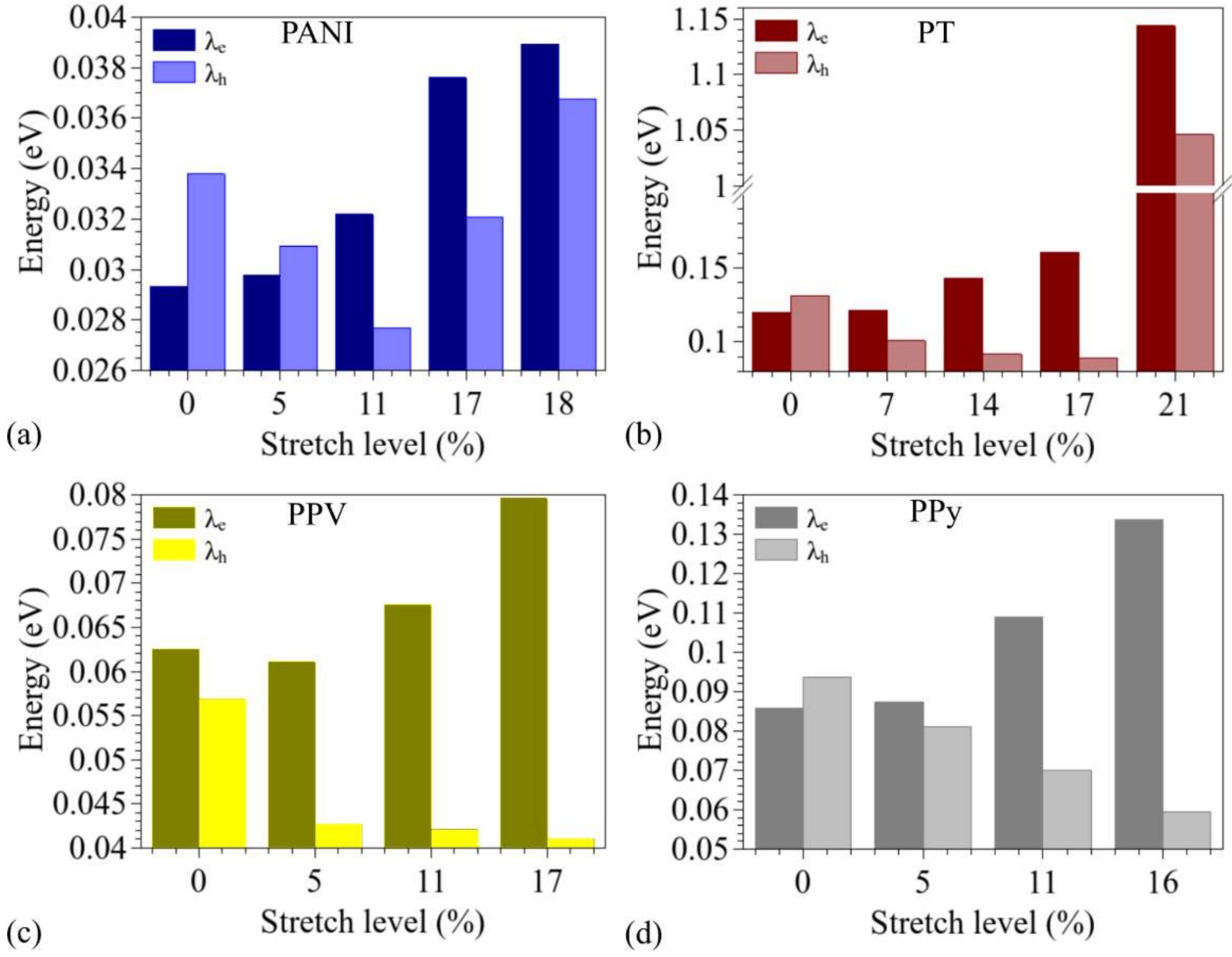
3.2. Changes in Opto-Electronic Properties
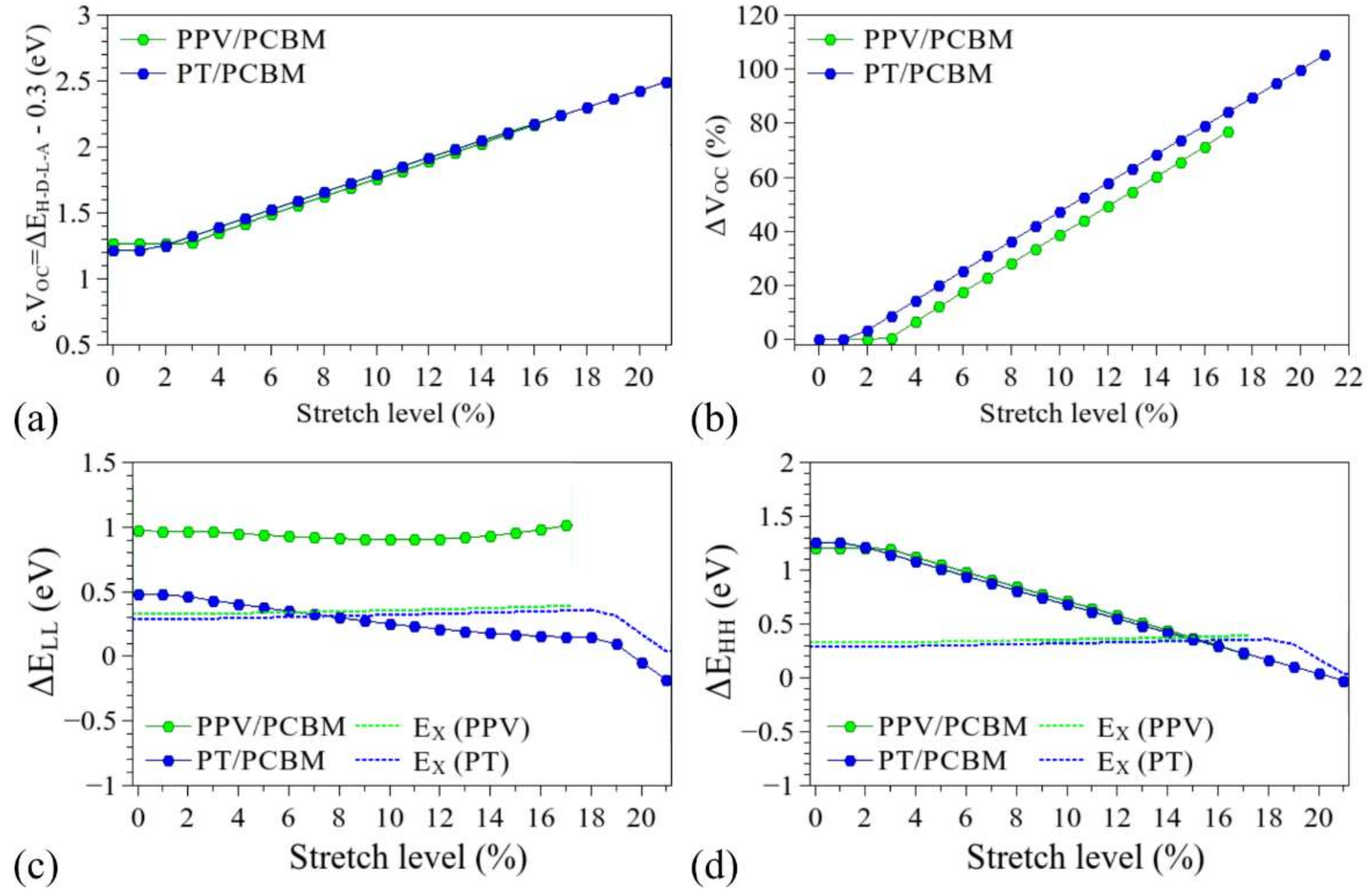
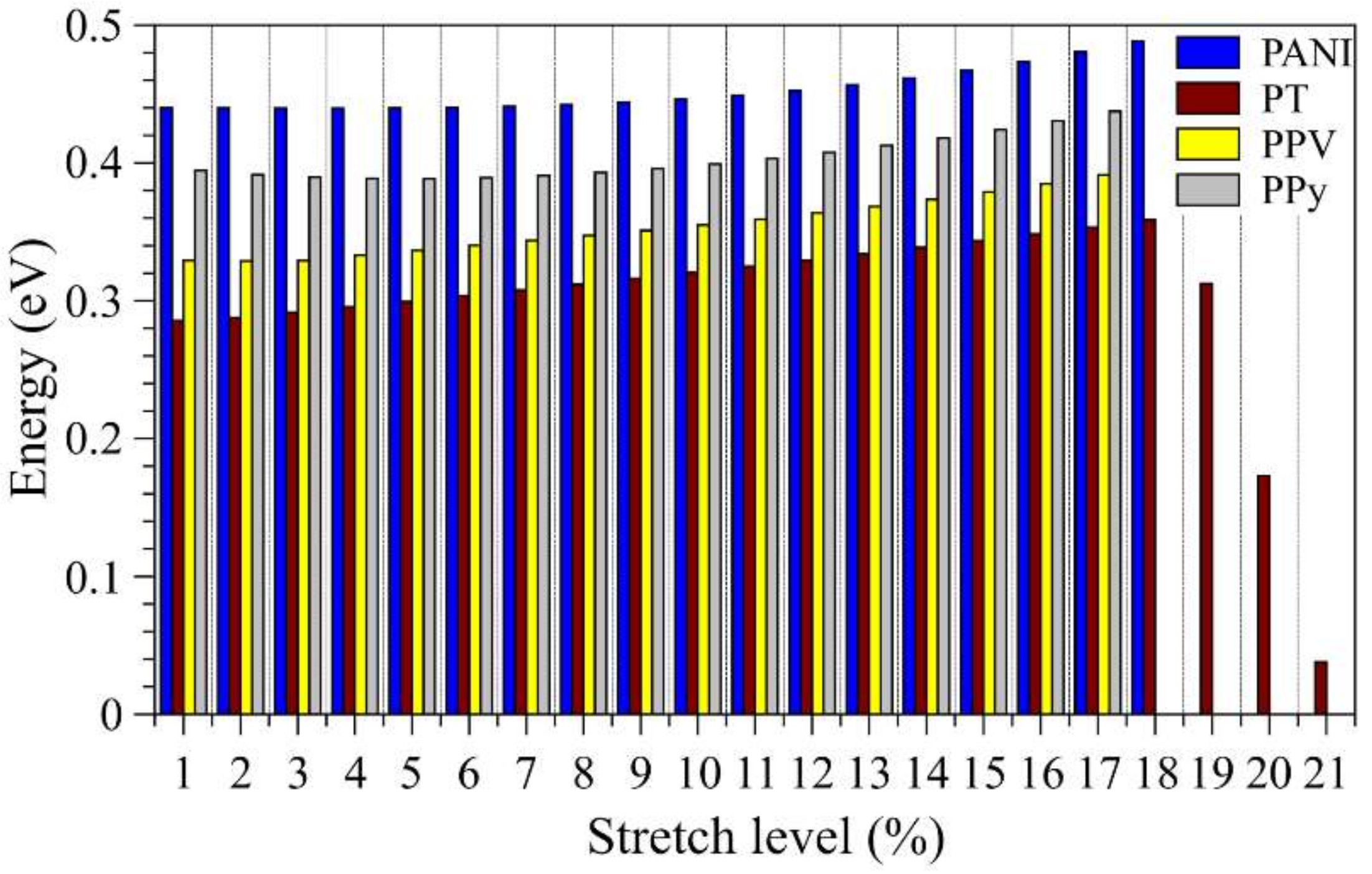
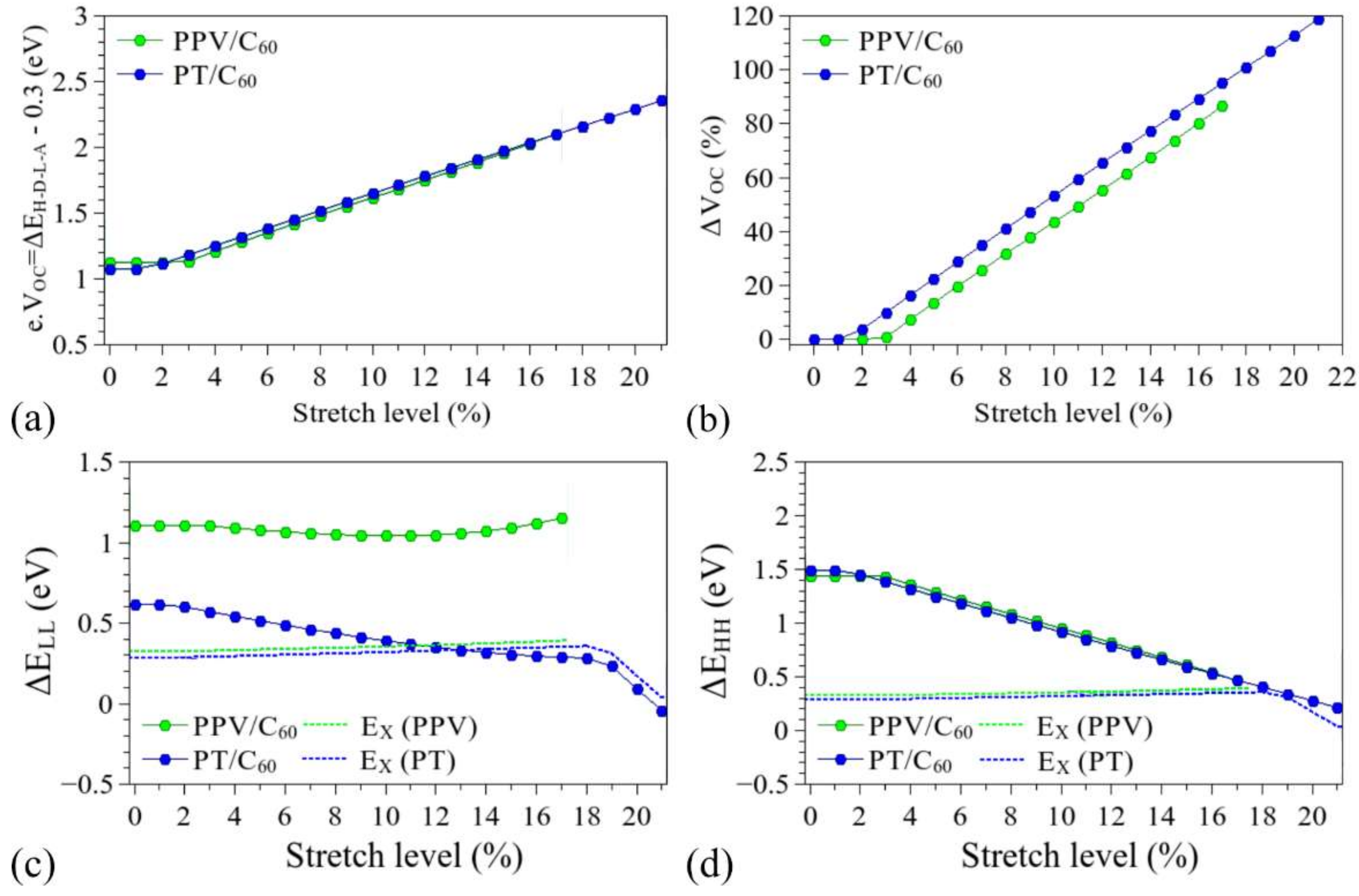
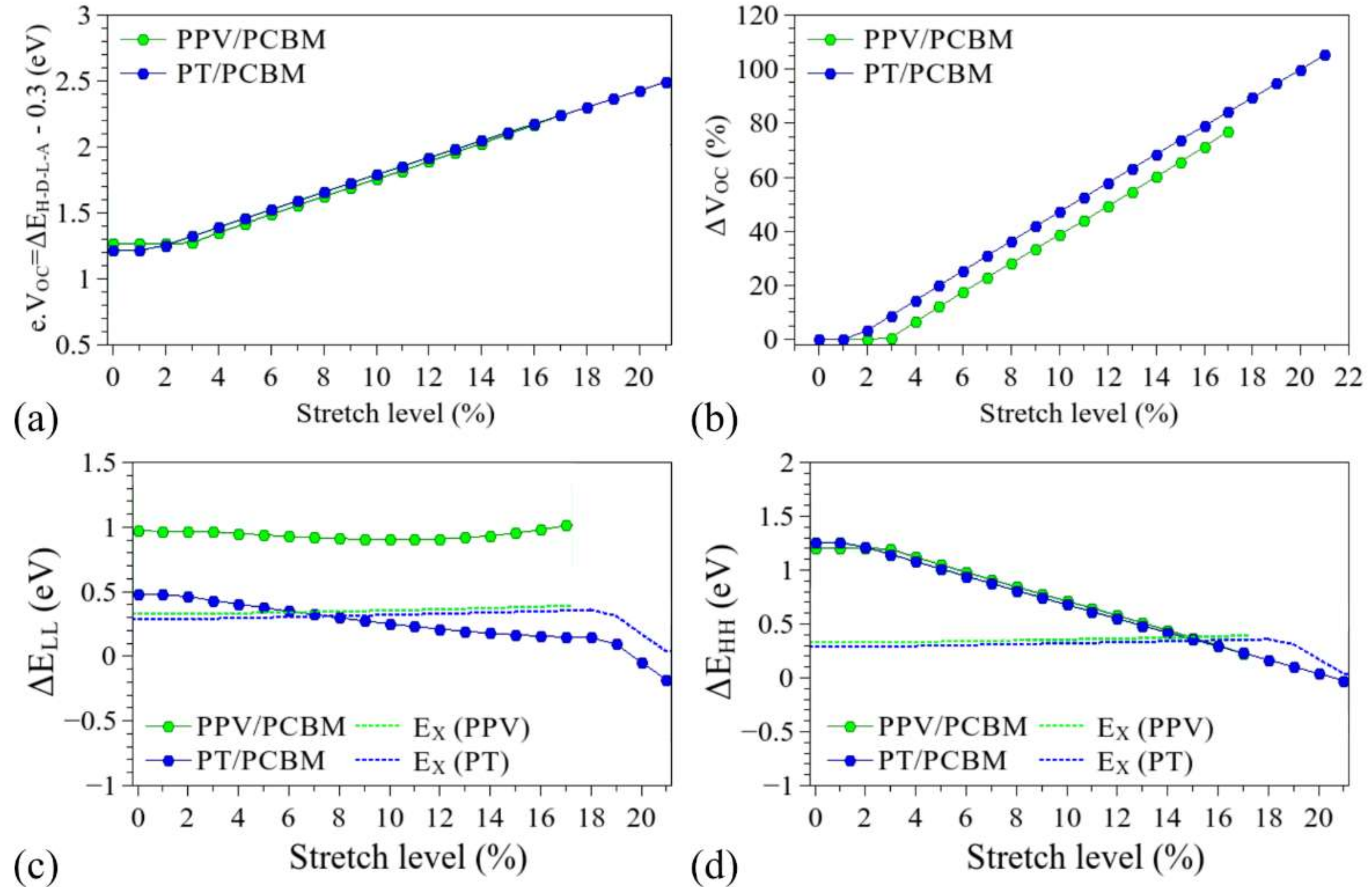
3.3. Considerations Regarding the Effect of Stretching on the Performance of the Compounds in Devices and Identification of Operational Regimes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, J.; Peng, Q.; Peng, X.; Han, L.; Wang, X.; Wang, J.; Zeng, H. Recent Advances in Mechano-Responsive Hydrogels for Biomedical Applications. ACS Appl. Polym. Mater. 2020, 2, 1092–1107. [Google Scholar] [CrossRef]
- Lee, S.; Kim, K.Y.; Jung, S.H.; Lee, J.H.; Yamada, M.; Sethy, R.; Kawai, T.; Jung, J.H. Finely Controlled Circularly Polarized Luminescence of a Mechano-Responsive Supramolecular Polymer. Angew. Chem. 2019, 131, 19054–19058. [Google Scholar] [CrossRef]
- Uǧur, G.; Chang, J.; Xiang, S.; Lin, L.; Lu, J. A Near-Infrared Mechano Responsive Polymer System. Adv. Mater. 2012, 24, 2685–2690. [Google Scholar] [CrossRef]
- Schäfer, C.G.; Gallei, M.; Zahn, J.T.; Engelhardt, J.; Hellmann, G.P.; Rehahn, M. Reversible Light-, Thermo-, and Mechano-Responsive Elastomeric Polymer Opal Films. Chem. Mater. 2013, 25, 2309–2318. [Google Scholar] [CrossRef]
- Willis-Fox, N.; Rognin, E.; Aljohani, T.; Daly, R. Polymer Mechanochemistry: Manufacturing Is Now a Force to Be Reckoned with. Chem 2018, 4, 2499–2537. [Google Scholar] [CrossRef] [Green Version]
- Takacs, L. The historical development of mechanochemistry. Chem. Soc. Rev. 2013, 42, 7649–7659. [Google Scholar] [CrossRef]
- Crawford, D.E.; Casaban, J. Recent Developments in Mechanochemical Materials Synthesis by Extrusion. Adv. Mater. 2016, 28, 5747–5754. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, P.F.M.; Torresi, R.M.; Emmerling, F.; Camargo, P.H.C. Challenges and opportunities in the bottom-up mechanochemical synthesis of noble metal nanoparticles. J. Mater. Chem. A 2020, 8, 16114–16141. [Google Scholar] [CrossRef]
- Szczęśniak, B.; Borysiuk, S.; Choma, J.; Jaroniec, M. Mechanochemical synthesis of highly porous materials. Mater. Horiz. 2020, 7, 1457–1473. [Google Scholar] [CrossRef]
- Boldyreva, E. Mechanochemistry of inorganic and organic systems: What is similar, what is different? Chem. Soc. Rev. 2013, 42, 7719–7738. [Google Scholar] [CrossRef]
- Ma, R.; Feng, J.; Yin, D.; Sun, H.-B. Highly efficient and mechanically robust stretchable polymer solar cells with random buckling. Org. Electron. 2017, 43, 77–81. [Google Scholar] [CrossRef]
- Ostroverkhova, O. Organic Optoelectronic Materials: Mechanisms and Applications. Chem. Rev. 2016, 116, 13279–13412. [Google Scholar] [CrossRef]
- Lee, S.-M.; Kwon, J.H.; Kwon, S.; Choi, K.C. A Review of Flexible OLEDs Toward Highly Durable Unusual Displays. IEEE Trans. Electron Devices 2017, 64, 1922–1931. [Google Scholar] [CrossRef]
- Lee, H.B.; Jin, W.-Y.; Ovhal, M.M.; Kumar, N.; Kang, J.-W. Flexible transparent conducting electrodes based on metal meshes for organic optoelectronic device applications: A review. J. Mater. Chem. C 2018, 7, 1087–1110. [Google Scholar] [CrossRef]
- Verboven, I.; Deferme, W. Printing of flexible light emitting devices: A review on different technologies and devices, printing technologies and state-of-the-art applications and future prospects. Prog. Mater. Sci. 2020, 118, 100760. [Google Scholar] [CrossRef]
- Boratto, M.H.; Nozella, N.L.; Ramos, R.A., Jr.; Da Silva, R.A.; Graeff, C.F.O. Flexible conductive blend of natural rubber latex with PEDOT: PSS. APL Mater. 2020, 8, 121107. [Google Scholar] [CrossRef]
- Shen, J.; Fujita, K.; Matsumoto, T.; Hongo, C.; Misaki, M.; Ishida, K.; Mori, A.; Nishino, T. Mechanical, Thermal, and Electrical Properties of Flexible Polythiophene with Disiloxane Side Chains. Macromol. Chem. Phys. 2017, 218, 1700197. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Wang, X.; Chen, F.; Zhang, N.; Ma, M. Extremely strong and tough polythiophene composite for flexible electronics. Chem. Eng. J. 2019, 368, 933–940. [Google Scholar] [CrossRef]
- Etxebarria, I.; Ajuria, J.; Pacios, R. Solution-processable polymeric solar cells: A review on materials, strategies and cell architectures to overcome 10%. Org. Electron. 2015, 19, 34–60. [Google Scholar] [CrossRef]
- Grimsdale, A.C.; Chan, K.L.; Martin, R.E.; Jokisz, P.G.; Holmes, A.B. Synthesis of Light-Emitting Conjugated Polymers for Applications in Electroluminescent Devices. Chem. Rev. 2009, 109, 897–1091. [Google Scholar] [CrossRef]
- Zhang, Z.; Liao, M.; Lou, H.; Hu, Y.; Sun, X.; Peng, H. Conjugated Polymers for Flexible Energy Harvesting and Storage. Adv. Mater. 2018, 30, e1704261. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Kim, J.-H.; Kang, T.E.; Lee, C.; Kang, H.; Shin, M.; Wang, C.; Ma, B.; Jeong, U.; Kim, T.-S.; et al. Flexible, Highly Efficient All-Polymer Solar Cells. Nat. Commun. 2015, 6, 8547. [Google Scholar] [CrossRef] [Green Version]
- Savagatrup, S.; Makaram, A.S.; Burke, D.J.; Lipomi, D.J. Mechanical Properties of Conjugated Polymers and Polymer-Fullerene Composites as a Function of Molecular Structure. Adv. Funct. Mater. 2013, 24, 1169–1181. [Google Scholar] [CrossRef]
- Kaltenbrunner, M.; White, M.; Głowacki, E.D.; Sekitani, T.; Someya, T.; Sariciftci, N.S.; Bauer, S. Ultrathin and lightweight organic solar cells with high flexibility. Nat. Commun. 2012, 3, 770. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Mongare, A.; Plant, A.; Ryu, D. Strain–Microstructure–Optoelectronic Inter-Relationship toward Engineering Mechano-Optoelectronic Conjugated Polymer Thin Films. Polymers 2021, 13, 935. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Yu, K.; Someya, T. The Future of Flexible Organic Solar Cells. Adv. Energy Mater. 2020, 10, 2000765. [Google Scholar] [CrossRef]
- Schwartz, B.J. Conjugated Polymers as Molecular Materials: How Chain Conformation and Film Morphology Influence Energy Transfer and Interchain Interactions. Annu. Rev. Phys. Chem. 2003, 54, 141–172. [Google Scholar] [CrossRef] [Green Version]
- Meier, H.; Stalmach, U.; Kolshorn, H. Effective conjugation length and UV/vis spectra of oligomers. Acta Polym. 1997, 48, 379–384. [Google Scholar] [CrossRef]
- Rissler, J. Effective conjugation length of π-conjugated systems. Chem. Phys. Lett. 2004, 395, 92–96. [Google Scholar] [CrossRef]
- He, J.; Crase, J.L.; Wadumethrige, S.H.; Thakur, K.; Dai, L.; Zou, S.; Rathore, R.; Hartley, C.S. Ortho-Phenylenes: Unusual Conjugated Oligomers with a Surprisingly Long Effective Conjugation Length. J. Am. Chem. Soc. 2010, 132, 13848–13857. [Google Scholar] [CrossRef]
- Wiebeler, C.; Gopalakrishna Rao, A.; Gärtner, W.; Schapiro, I. The Effective Conjugation Length is Responsible for the Red/Green Spectral Tuning in the Cyanobacteriochrome Slr1393g3. Angew. Chem. Int. Ed. 2019, 58, 1934–1938. [Google Scholar] [CrossRef] [Green Version]
- Kwak, K.; Cho, K.; Kim, S. Stable Bending Performance of Flexible Organic Light-Emitting Diodes Using IZO Anodes. Sci. Rep. 2013, 3, 2787. [Google Scholar] [CrossRef]
- Ma, R.; Chou, S.-Y.; Xie, Y.; Pei, Q. Morphological/nanostructural control toward intrinsically stretchable organic electronics. Chem. Soc. Rev. 2019, 48, 1741–1786. [Google Scholar] [CrossRef]
- Caruso, M.M.; Davis, D.A.; Shen, Q.; Odom, S.A.; Sottos, N.R.; White, S.R.; Moore, J.S. Mechanically-Induced Chemical Changes in Polymeric Materials. Chem. Rev. 2009, 109, 5755–5798. [Google Scholar] [CrossRef]
- Rubner, M.F. Novel optical properties of polyurethane-diacetylene segmented copolymers. Macromolecules 1986, 19, 2129–2138. [Google Scholar] [CrossRef]
- Chen, J.-Y.; Hsieh, H.-C.; Chiu, Y.-C.; Lee, W.-Y.; Hung, C.-C.; Chueh, C.-C.; Chen, W.-C. Electrospinning-induced elastomeric properties of conjugated polymers for extremely stretchable nanofibers and rubbery optoelectronics. J. Mater. Chem. C 2019, 8, 873–882. [Google Scholar] [CrossRef]
- Chow, P.C.Y.; Someya, T. Organic Photodetectors for Next-Generation Wearable Electronics. Adv. Mater. 2019, 32, e1902045. [Google Scholar] [CrossRef]
- Sagara, Y.; Traeger, H.; Li, J.; Okado, Y.; Schrettl, S.; Tamaoki, N.; Weder, C. Mechanically Responsive Luminescent Polymers Based on Supramolecular Cyclophane Mechanophores. J. Am. Chem. Soc. 2021, 143, 5519–5525. [Google Scholar] [CrossRef]
- Onorato, J.; Pakhnyuk, V.; Luscombe, C.K. Structure and design of polymers for durable, stretchable organic electronics. Polym. J. 2016, 49, 41–60. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Baek, P.; Akbarinejad, A.; Barker, D.; Travas-Sejdic, J. Conjugated polymers and composites for stretchable organic electronics. J. Mater. Chem. C 2019, 7, 5534–5552. [Google Scholar] [CrossRef]
- Roldao, J.C.; Batagin-Neto, A.; Lavarda, F.C.; Sato, F. Effects of Mechanical Stretching on the Properties of Conjugated Polymers: Case Study for MEH-PPV and P3HT Oligomers. J. Polym. Sci. Part B Polym. Phys. 2018, 56, 1413–1426. [Google Scholar] [CrossRef]
- Brédas, J.L.; Street, G.B.; Thémans, B.; André, J.M. Organic polymers based on aromatic rings (polyparaphenylene, polypyrrole, polythiophene): Evolution of the electronic properties as a function of the torsion angle between adjacent rings. J. Chem. Phys. 1985, 83, 1323–1329. [Google Scholar] [CrossRef]
- Lee, I.; Rolston, N.; Brunner, P.-L.; Dauskardt, R.H. Hole-Transport Layer Molecular Weight and Doping Effects on Perovskite Solar Cell Efficiency and Mechanical Behavior. ACS Appl. Mater. Interfaces 2019, 11, 23757–23764. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Ma, B.S.; Kim, Y.H.; Kim, T.-S. Mechanical properties of organic semiconductors for flexible electronics. In Organic Flexible Electronics; Elsevier: Amsterdam, The Netherlands, 2021; pp. 199–223. [Google Scholar] [CrossRef]
- Rasmussen, S.C. Conjugated and Conducting Organic Polymers: The First 150 Years. ChemPlusChem 2020, 85, 1412–1429. [Google Scholar] [CrossRef]
- Bhadra, S.; Singha, N.K.; Khastgir, D. Electrochemical synthesis of polyaniline and its comparison with chemically synthesized polyaniline. J. Appl. Polym. Sci. 2007, 104, 1900–1904. [Google Scholar] [CrossRef]
- Mandú, L.O.; Batagin-Neto, A. Chemical sensors based on N-substituted polyaniline derivatives: Reactivity and adsorption studies via electronic structure calculations. J. Mol. Model. 2018, 24, 157. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Liao, M.; Zhang, C.; Yin, M.; Li, D.; Song, Y. The effect of anions on the electrochemical properties of polyaniline for supercapacitors. Phys. Chem. Chem. Phys. 2017, 19, 14030–14041. [Google Scholar] [CrossRef]
- Hu, Z.; Zu, L.; Jiang, Y.; Lian, H.; Liu, Y.; Li, Z.; Chen, F.; Wang, X.; Cui, X. High Specific Capacitance of Polyaniline/Mesoporous Manganese Dioxide Composite Using KI-H2SO4 Electrolyte. Polymers 2015, 7, 1939–1953. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Lin, J.; Shen, Z.X. Polyaniline (PANi) based electrode materials for energy storage and conversion. J. Sci. Adv. Mater. Devices 2016, 1, 225–255. [Google Scholar] [CrossRef] [Green Version]
- Kaloni, T.P.; Giesbrecht, P.K.; Schreckenbach, G.; Freund, M.S. Polythiophene: From Fundamental Perspectives to Applications. Chem. Mater. 2017, 29, 10248–10283. [Google Scholar] [CrossRef]
- Kanal, I.Y.; Owens, S.G.; Bechtel, J.S.; Hutchison, G. Efficient Computational Screening of Organic Polymer Photovoltaics. J. Phys. Chem. Lett. 2013, 4, 1613–1623. [Google Scholar] [CrossRef]
- Huong, V.T.T.; Nguyen, H.T.; Tai, T.B.; Nguyen, M.T. π-Conjugated Molecules Containing Naphtho[2,3-b]thiophene and Their Derivatives: Theoretical Design for Organic Semiconductors. J. Phys. Chem. C 2013, 117, 10175–10184. [Google Scholar] [CrossRef]
- Nimith, K.; Satyanarayan, M.; Umesh, G. Enhancement in fluorescence quantum yield of MEH-PPV:BT blends for polymer light emitting diode applications. Opt. Mater. 2018, 80, 143–148. [Google Scholar] [CrossRef]
- Farjamtalab, I.; Sabbaghi-Nadooshan, R. Current density of anodes, recombination rate and luminance in MEH-PPV, MDMO-PPV, and P3HT polymers in polymer light-emitting diodes. Polym. Sci. Ser. A 2016, 58, 726–731. [Google Scholar] [CrossRef]
- Cernini, R.; Li, X.-C.; Spencer, G.; Holmes, A.; Moratti, S.; Friend, R. Electrochemical and optical studies of PPV derivatives and poly(aromatic oxadiazoles). Synth. Met. 1997, 84, 359–360. [Google Scholar] [CrossRef]
- Yussuf, A.; Al-Saleh, M.; Al-Enezi, S.; Abraham, G. Synthesis and Characterization of Conductive Polypyrrole: The Influence of the Oxidants and Monomer on the Electrical, Thermal, and Morphological Properties. Int. J. Polym. Sci. 2018, 2018, 1–8. [Google Scholar] [CrossRef]
- Coleone, A.P.; Lascane, L.G.; Batagin-Neto, A. Polypyrrole derivatives for optoelectronic applications: A DFT study on the influence of side groups. Phys. Chem. Chem. Phys. 2019, 21, 17729–17739. [Google Scholar] [CrossRef]
- Bibi, S.; Ullah, H.; Ahmad, S.M.; Ali Shah, A.-U.; Bilal, S.; Tahir, A.A.; Ayub, K. Molecular and Electronic Structure Elucidation of Polypyrrole Gas Sensors. J. Phys. Chem. C 2015, 119, 15994–16003. [Google Scholar] [CrossRef]
- Ghoorchian, A.; Alizadeh, N. Chemiresistor gas sensor based on sulfonated dye-doped modified conducting polypyrrole film for high sensitive detection of 2,4,6-trinitrotoluene in air. Sens. Actuators B Chem. 2018, 255, 826–835. [Google Scholar] [CrossRef]
- Gierschner, J.; Cornil, J.; Egelhaaf, H.-J. Optical Bandgaps of π-Conjugated Organic Materials at the Polymer Limit: Experiment and Theory. Adv. Mater. 2007, 19, 173–191. [Google Scholar] [CrossRef]
- Bronze-Uhle, E.S.; Batagin-Neto, A.; Lavarda, F.C.; Graeff, C. Ionizing radiation induced degradation of poly (2-methoxy-5-(2’-ethyl-hexyloxy) -1,4-phenylene vinylene) in solution. J. Appl. Phys. 2011, 110, 073510. [Google Scholar] [CrossRef] [Green Version]
- Batagin-Neto, A.; Oliveira, E.F.; Graeff, C.; Lavarda, F.C. Modelling polymers with side chains: MEH-PPV and P3HT. Mol. Simul. 2013, 39, 309–321. [Google Scholar] [CrossRef]
- de Oliveira, E.F.; Camilo, A., Jr.; da Silva-Filho, L.C.; Lavarda, F.C. Effect of chemical modifications on the electronic structure of poly(3-hexylthiophene). J. Polym. Sci. Part B Polym. Phys. 2013, 51, 842–846. [Google Scholar] [CrossRef]
- Oliveira, E.F.; Lavarda, F.C. Reorganization energy for hole and electron transfer of poly(3-hexylthiophene) derivatives. Polymer 2016, 99, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, G.P.; Barboza, B.H.; Batagin-Neto, A. Polyaniline-based gas sensors: DFT study on the effect of side groups. Comput. Theor. Chem. 2021, 1207, 113526. [Google Scholar] [CrossRef]
- Coleone, A.P.; Barboza, B.H.; Batagin-Neto, A. Polypyrrole derivatives for detection of toxic gases: A theoretical study. Polym. Adv. Technol. 2021, 32, 4464–4478. [Google Scholar] [CrossRef]
- Schaftenaar, G.; Noordik, J.H. Molden: A pre- and post-processing program for molecular and electronic structures. J. Comput. Aided Mol. Des. 2000, 14, 123–134. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef] [Green Version]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- McCormick, T.M.; Bridges, C.R.; Carrera, E.I.; DiCarmine, P.M.; Gibson, G.L.; Hollinger, J.; Kozycz, L.M.; Seferos, D.S. Conjugated Polymers: Evaluating DFT Methods for More Accurate Orbital Energy Modeling. Macromolecules 2013, 46, 3879–3886. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Beyer, M.K. The mechanical strength of a covalent bond calculated by density functional theory. J. Chem. Phys. 2000, 112, 7307–7312. [Google Scholar] [CrossRef]
- Alves, G.G.; Oliveira, E.F.; Batagin-Neto, A.; Lavarda, F.C. Molecular modeling of low bandgap diblock co-oligomers with π-bridges for applications in photovoltaics. Comput. Mater. Sci. 2018, 152, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Yi, Y.; Chen, L.; Shuai, Z. Exciton binding energy of electronic polymers: A first principles study. J. Theor. Comput. Chem. 2008, 7, 517–530. [Google Scholar] [CrossRef]
- Organic Electronics. Advances in Polymer Science; Grasser, T., Meller, G., Li, L., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; Volume 223, ISBN 978-3-642-04537-0. [Google Scholar]
- Hutchison, G.R.; Ratner, M.A.; Marks, T.J. Hopping Transport in Conductive Heterocyclic Oligomers: Reorganization Energies and Substituent Effects. J. Am. Chem. Soc. 2005, 127, 2339–2350. [Google Scholar] [CrossRef]
- Yang, W.; Mortier, W.J. The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines. J. Am. Chem. Soc. 1986, 108, 5708–5711. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, A.F.; Langenaeker, W. Conceptual Density Functional Theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Maia, R.A.; Ventorim, G.; Batagin-Neto, A. Reactivity of lignin subunits: The influence of dehydrogenation and formation of dimeric structures. J. Mol. Model. 2019, 25, 1–11. [Google Scholar] [CrossRef]
- Alves, G.G.; Lavarda, F.C.; Graeff, C.F.; Batagin-Neto, A. Reactivity of eumelanin building blocks: A DFT study of monomers and dimers. J. Mol. Graph. Model. 2020, 98, 107609. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- De Proft, F.; Van Alsenoy, C.; Peeters, A.; Langenaeker, W.; Geerlings, P. Atomic charges, dipole moments, and Fukui functions using the Hirshfeld partitioning of the electron density. J. Comput. Chem. 2002, 23, 1198–1209. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.K.; Pal, S.; Hirao, K. On non-negativity of Fukui function indices. J. Chem. Phys. 1999, 110, 8236–8245. [Google Scholar] [CrossRef]
- Chirlian, L.E.; Francl, M. Atomic charges derived from electrostatic potentials: A detailed study. J. Comput. Chem. 1987, 8, 894–905. [Google Scholar] [CrossRef]
- Herráez, A. How to Use Jmol to Study and Present Molecular Structures; Lulu.com: Morrisville, NC, USA, 2007; ISBN 978-1-84799-259-8. [Google Scholar]
- Allouche, A.-R. Gabedit-A graphical user interface for computational chemistry softwares. J. Comput. Chem. 2010, 32, 174–182. [Google Scholar] [CrossRef]
- Wang, Z.; Su, K.; Fan, H.; Hu, L.; Wang, X.; Li, Y.; Wen, Z. Mechanical and electronic properties of C60 under structure distortion studied with density functional theory. Comput. Mater. Sci. 2007, 40, 537–547. [Google Scholar] [CrossRef]
- Salaneck, W.R.; Clark, D.T.; Samuelsen, E.J. Science and Applications of Conducting Polymers: Papers from the Sixth European Industrial Workshop; CRC Press: Trondheim, Norway, 2019; ISBN 978-1-00-011224-5. [Google Scholar]
- Chen, X.; Liang, Q.H.; Jiang, J.; Wong, C.K.Y.; Leung, S.Y.; Ye, H.; Yang, D.G.; Ren, T.L. Functionalization-induced changes in the structural and physical properties of amorphous polyaniline: A first-principles and molecular dynamics study. Sci. Rep. 2016, 6, 20621. [Google Scholar] [CrossRef]
- Salzner, U.; Lagowski, J.; Pickup, P.; Poirier, R. Comparison of geometries and electronic structures of polyacetylene, polyborole, polycyclopentadiene, polypyrrole, polyfuran, polysilole, polyphosphole, polythiophene, polyselenophene and polytellurophene. Synth. Met. 1998, 96, 177–189. [Google Scholar] [CrossRef]
- Fu, Y.; Shen, W.; Li, M. Theoretical analysis on the electronic structures and properties of PPV fused with electron-withdrawing unit: Monomer, oligomer and polymer. Polymer 2008, 49, 2614–2620. [Google Scholar] [CrossRef]
- Cumper, C.W.N. The structures of some heterocyclic molecules. Trans. Faraday Soc. 1958, 54, 1266–1270. [Google Scholar] [CrossRef]
- Roöhrig, U.F.; Frank, I. First-principles molecular dynamics study of a polymer under tensile stress. J. Chem. Phys. 2001, 115, 8670–8674. [Google Scholar] [CrossRef]
- Garnier, L.; Gauthier-Manuel, B.; Van Der Vegte, E.W.; Snijders, J.; Hadziioannou, G. Covalent bond force profile and cleavage in a single polymer chain. J. Chem. Phys. 2000, 113, 2497–2503. [Google Scholar] [CrossRef] [Green Version]
- Grandbois, M.; Beyer, M.; Rief, M.; Clausen-Schaumann, H.; Gaub, H.E. How Strong Is a Covalent Bond? Science 1999, 283, 1727–1730. [Google Scholar] [CrossRef]
- Smalø, H.S.; Rybkin, V.V.; Klopper, W.; Helgaker, T.; Uggerud, E. Mechanochemistry: The Effect of Dynamics. J. Phys. Chem. A 2014, 118, 7683–7694. [Google Scholar] [CrossRef]
- Ruini, A.; Caldas, M.J.; Bussi, G.; Molinari, E. Solid State Effects on Exciton States and Optical Properties of PPV. Phys. Rev. Lett. 2002, 88, 206403. [Google Scholar] [CrossRef] [Green Version]
- Brédas, J.-L.; Beljonne, D.; Coropceanu, V.; Cornil, J. Charge-Transfer and Energy-Transfer Processes in π-Conjugated Oligomers and Polymers: A Molecular Picture. Chem. Rev. 2004, 104, 4971–5004. [Google Scholar] [CrossRef]
- Nayak, P.K. Exciton binding energy in small organic conjugated molecule. Synth. Met. 2013, 174, 42–45. [Google Scholar] [CrossRef] [Green Version]
- Melin, J.; Aparicio, F.; Subramanian, V.; Galván, M.; Chattaraj, P.K. Is the Fukui Function a Right Descriptor of Hard−Hard Interactions? J. Phys. Chem. A 2004, 108, 2487–2491. [Google Scholar] [CrossRef]
- Duan, L.; Uddin, A. Progress in Stability of Organic Solar Cells. Adv. Sci. 2020, 7, 1903259. [Google Scholar] [CrossRef] [Green Version]
- Tyler, D.R. Mechanistic Aspects of the Effects of Stress on the Rates of Photochemical Degradation Reactions in Polymers. J. Macromol. Sci. Part C Polym. Rev. 2004, 44, 351–388. [Google Scholar] [CrossRef]
- Li, Z.; Ye, B.; Hu, X.; Ma, X.; Zhang, X.; Deng, Y. Facile electropolymerized-PANI as counter electrode for low cost dye-sensitized solar cell. Electrochem. Commun. 2009, 11, 1768–1771. [Google Scholar] [CrossRef]
- Malhotra, B.; Dhand, C.; Lakshminarayanan, R.; Dwivedi, N.; Mishra, S.; Solanki, P.; Venkatesh, M.; Beuerman, R.W.; Ramakrishna, S.; Mayandi, V. Polyaniline-based biosensors. Nanobiosensors Dis. Diagn. 2015, 4, 25–46. [Google Scholar] [CrossRef] [Green Version]
- Jayasundara, W.S.R.; Schreckenbach, G. Theoretical Study of p- and n-Doping of Polythiophene- and Polypyrrole-Based Conjugated Polymers. J. Phys. Chem. C 2020, 124, 17528–17537. [Google Scholar] [CrossRef]
- Kaloni, T.P.; Schreckenbach, G.; Freund, M.S. Structural and Electronic Properties of Pristine and Doped Polythiophene: Periodic versus Molecular Calculations. J. Phys. Chem. C 2015, 119, 3979–3989. [Google Scholar] [CrossRef] [Green Version]
- Sezen, M.; Plank, H.; Fisslthaler, E.; Chernev, B.; Zankel, A.; Tchernychova, E.; Blümel, A.; List, E.J.W.; Grogger, W.; Pölt, P. An investigation on focused electron/ion beam induced degradation mechanisms of conjugated polymers. Phys. Chem. Chem. Phys. 2011, 13, 20235–20240. [Google Scholar] [CrossRef]
- Chambon, S.; Rivaton, A.; Gardette, J.-L.; Firon, M. Photo- and thermal degradation of MDMO-PPV:PCBM blends. Sol. Energy Mater. Sol. Cells 2007, 91, 394–398. [Google Scholar] [CrossRef]
- Chambon, S.; Rivaton, A.; Gardette, J.-L.; Firon, M. Durability of MDMO-PPV and MDMO-PPV: PCBM blends under illumination in the absence of oxygen. Sol. Energy Mater. Sol. Cells 2008, 92, 785–792. [Google Scholar] [CrossRef]
- Le, T.-H.; Kim, Y.; Yoon, H. Electrical and Electrochemical Properties of Conducting Polymers. Polymers 2017, 9, 150. [Google Scholar] [CrossRef]
- Ohtsuka, T. Corrosion Protection of Steels by Conducting Polymer Coating. Int. J. Corros. 2012, 2012, 1–7. [Google Scholar] [CrossRef]
- Li, Y. Molecular Design of Photovoltaic Materials for Polymer Solar Cells: Toward Suitable Electronic Energy Levels and Broad Absorption. Acc. Chem. Res. 2012, 45, 723–733. [Google Scholar] [CrossRef]
- Brédas, J.-L.; Norton, J.E.; Cornil, J.; Coropceanu, V. Molecular Understanding of Organic Solar Cells: The Challenges. Acc. Chem. Res. 2009, 42, 1691–1699. [Google Scholar] [CrossRef]
- Dang, M.T.; Hirsch, L.; Wantz, G.; Wuest, J.D. Controlling the Morphology and Performance of Bulk Heterojunctions in Solar Cells. Lessons Learned from the Benchmark Poly(3-hexylthiophene):[6,6]-Phenyl-C61-butyric Acid Methyl Ester System. Chem. Rev. 2013, 113, 3734–3765. [Google Scholar] [CrossRef]
- Proctor, C.; Kuik, M.; Nguyen, T.-Q. Charge carrier recombination in organic solar cells. Prog. Polym. Sci. 2013, 38, 1941–1960. [Google Scholar] [CrossRef]
- Kim, J.-H.; Noh, J.; Choi, H.; Lee, J.-Y.; Kim, T.-S. Mechanical Properties of Polymer–Fullerene Bulk Heterojunction Films: Role of Nanomorphology of Composite Films. Chem. Mater. 2017, 29, 3954–3961. [Google Scholar] [CrossRef]
- Kim, W.; Choi, J.; Kim, J.-H.; Kim, T.; Lee, C.; Lee, S.; Kim, M.; Kim, B.J.; Kim, T.-S. Comparative Study of the Mechanical Properties of All-Polymer and Fullerene–Polymer Solar Cells: The Importance of Polymer Acceptors for High Fracture Resistance. Chem. Mater. 2018, 30, 2102–2111. [Google Scholar] [CrossRef]
- Cachaneski-Lopes, J.P.; Batagin-Neto, A. Effects of Mechanical Deformation on the Optoelectronic Properties of Fullerenes: A DFT Study. J. Nanostructure Chem. 2021, 1–17. [Google Scholar] [CrossRef]
- Barboza, B.H.; Gomes, O.P.; Batagin-Neto, A. Polythiophene derivatives as chemical sensors: A DFT study on the influence of side groups. J. Mol. Model. 2021, 27, 1–13. [Google Scholar] [CrossRef]
- Lascane, L.G.; Oliveira, E.F.; Galvão, D.S.; Batagin-Neto, A. Polyfuran-based chemical sensors: Identification of promising derivatives via DFT calculations and fully atomistic reactive molecular dynamics. Eur. Polym. J. 2020, 141, 110085. [Google Scholar] [CrossRef]
- Savagatrup, S.; Printz, A.D.; O’Connor, T.F.; Zaretski, A.V.; Rodriquez, D.; Sawyer, E.J.; Rajan, K.M.; Acosta, R.I.; Root, S.E.; Lipomi, D.J. Mechanical degradation and stability of organic solar cells: Molecular and microstructural determinants. Energy Environ. Sci. 2014, 8, 55–80. [Google Scholar] [CrossRef]
- Lipomi, D.J.; Tee, B.C.-K.; Vosgueritchian, M.; Bao, Z. Stretchable Organic Solar Cells. Adv. Mater. 2011, 23, 1771–1775. [Google Scholar] [CrossRef]
- Geniès, E.M.; Boyle, A.; Lapkowski, M.; Tsintavis, C. Polyaniline: A Historical Survey. Synth. Met. 1990, 36, 139–182. [Google Scholar] [CrossRef]
- Ullah, H. Inter-Molecular Interaction in Polypyrrole/TiO2: A DFT Study. J. Alloys Compd. 2017, 692, 140–148. [Google Scholar] [CrossRef]
- Greenham, N.C.; Friend, R.H. Semiconductor Device Physics of Conjugated Polymers. In Solid State Physics; Elsevier: Armsterdam, The Netherlands, 1996; Volume 49, pp. 1–149. ISBN 978-0-12-607749-0. [Google Scholar]
- McCall, R.P.; Ginder, J.M.; Leng, J.M.; Ye, H.J.; Manohar, S.K.; Masters, J.G.; Asturias, G.E.; MacDiarmid, A.G.; Epstein, A.J. Spectroscopy and Defect States in Polyaniline. Phys. Rev. B 1990, 41, 5202–5213. [Google Scholar] [CrossRef] [Green Version]
- Arjmandi, M.; Arjmandi, A.; Peyravi, M.; Pirzaman, A.K. First-Principles Study of Adsorption of XCN (X = F, Cl, and Br) on Surfaces of Polyaniline. Russ. J. Phys. Chem. 2020, 94, 2148–2154. [Google Scholar] [CrossRef]
- Hosseini, H.; Mousavi, S.M. Density Functional Theory Simulation for Cr(VI) Removal from Wastewater Using Bacterial Cellulose/Polyaniline. Int. J. Biol. Macromol. 2020, 165, 883–901. [Google Scholar] [CrossRef] [PubMed]
- Ullah, H.; Shah, A.-H.A.; Bilal, S.; Ayub, K. DFT Study of Polyaniline NH3, CO2, and CO Gas Sensors: Comparison with Recent Experimental Data. J. Phys. Chem. C 2013, 117, 23701–23711. [Google Scholar] [CrossRef]
- Furukawa, Y. Electronic Absorption and Vibrational Spectroscopies of Conjugated Conducting Polymers. J. Phys. Chem. 1996, 100, 15644–15653. [Google Scholar] [CrossRef]
- Zhang, L.; Colella, N.S.; Cherniawski, B.P.; Mannsfeld, S.C.B.; Briseno, A.L. Oligothiophene Semiconductors: Synthesis, Characterization, and Applications for Organic Devices. ACS Appl. Mater. Interfaces 2014, 6, 5327–5343. [Google Scholar] [CrossRef]
- Kaneto, K.; Yoshino, K.; Inuishi, Y. Electrical and Optical Properties of Polythiophene Prepared by Electrochemical Polymerization. Solid State Commun. 1983, 46, 389–391. [Google Scholar] [CrossRef]
- Bredas, J.L.; Silbey, R.; Boudreaux, D.S.; Chance, R.R. Chain-Length Dependence of Electronic and Electrochemical Properties of Conjugated Systems: Polyacetylene, Polyphenylene, Polythiophene, and Polypyrrole. J. Am. Chem. Soc. 1983, 105, 6555–6559. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | d0 (Å) | du (Å) | Δdmax (%) |
|---|---|---|---|
| PANI | 77.447 | 92.162 | 18.0 |
| PT | 57.192 | 69.774 | 21.0 |
| PPV | 94.046 | 110.974 | 17.0 |
| PPy | 52.588 | 62.054 | 17.0 |
| System | Δdn (%) | |ΔEtotal| (Joule × 10−19) | Force (nN) | Elastic Constant (nN/nm) |
|---|---|---|---|---|
| PANI | 0% | --- | --- | ~5.82 |
| 6% | 0.818 | 3.32 | ||
| 12% | 2.995 | 6.01 | ||
| 17% | 6.325 | 7.86 | ||
| PT | 0% | --- | --- | k1 = ~10.41 * k2 = ~5.66 * kmed = ~8.04 |
| 7% | 0.582 | 3.59 | ||
| 14% | 2.700 | 4.53 | ||
| 20% | 5.801 | 8.37 | ||
| PPV | 0% | --- | --- | ~5.49 |
| 5% | 0.146 | 1.44 | ||
| 11% | 1.937 | 4.79 | ||
| 16% | 5.379 | 6.84 | ||
| PPy | 0% | --- | --- | k1 = ~13.87 * k2 = ~7.46 * kmed = ~10.67 |
| 5% | 0.462 | 3.64 | ||
| 11% | 2.219 | 4.31 | ||
| 16% | 4.808 | 8.76 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cachaneski-Lopes, J.P.; Batagin-Neto, A. Effects of Mechanical Deformation on the Opto-Electronic Responses, Reactivity, and Performance of Conjugated Polymers: A DFT Study. Polymers 2022, 14, 1354. https://doi.org/10.3390/polym14071354
Cachaneski-Lopes JP, Batagin-Neto A. Effects of Mechanical Deformation on the Opto-Electronic Responses, Reactivity, and Performance of Conjugated Polymers: A DFT Study. Polymers. 2022; 14(7):1354. https://doi.org/10.3390/polym14071354
Chicago/Turabian StyleCachaneski-Lopes, João P., and Augusto Batagin-Neto. 2022. "Effects of Mechanical Deformation on the Opto-Electronic Responses, Reactivity, and Performance of Conjugated Polymers: A DFT Study" Polymers 14, no. 7: 1354. https://doi.org/10.3390/polym14071354
APA StyleCachaneski-Lopes, J. P., & Batagin-Neto, A. (2022). Effects of Mechanical Deformation on the Opto-Electronic Responses, Reactivity, and Performance of Conjugated Polymers: A DFT Study. Polymers, 14(7), 1354. https://doi.org/10.3390/polym14071354