
Improved Melt Processabilities of Thermosetting Polyimide Matrix Resins for High Temperature Carbon Fiber Composite Applications


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Polymer Synthesis
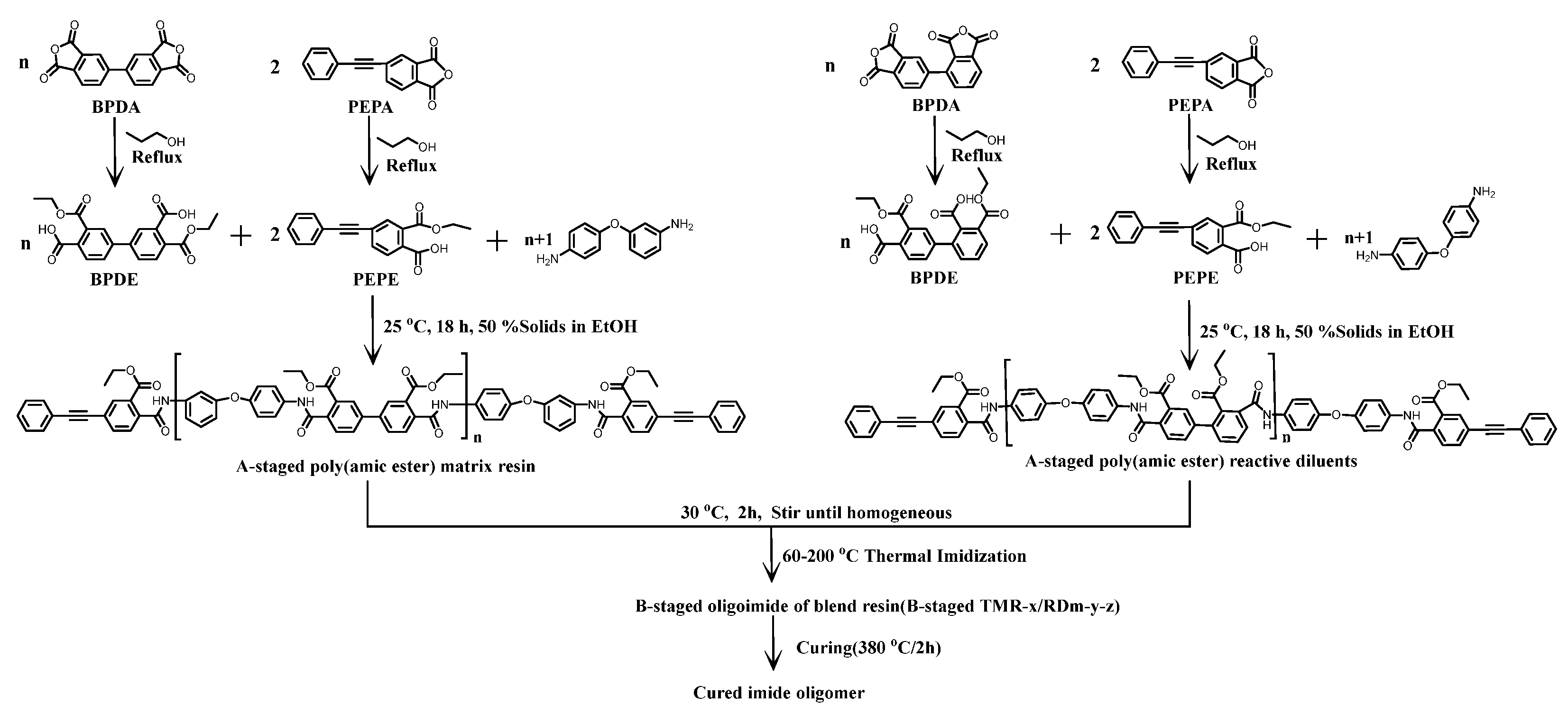
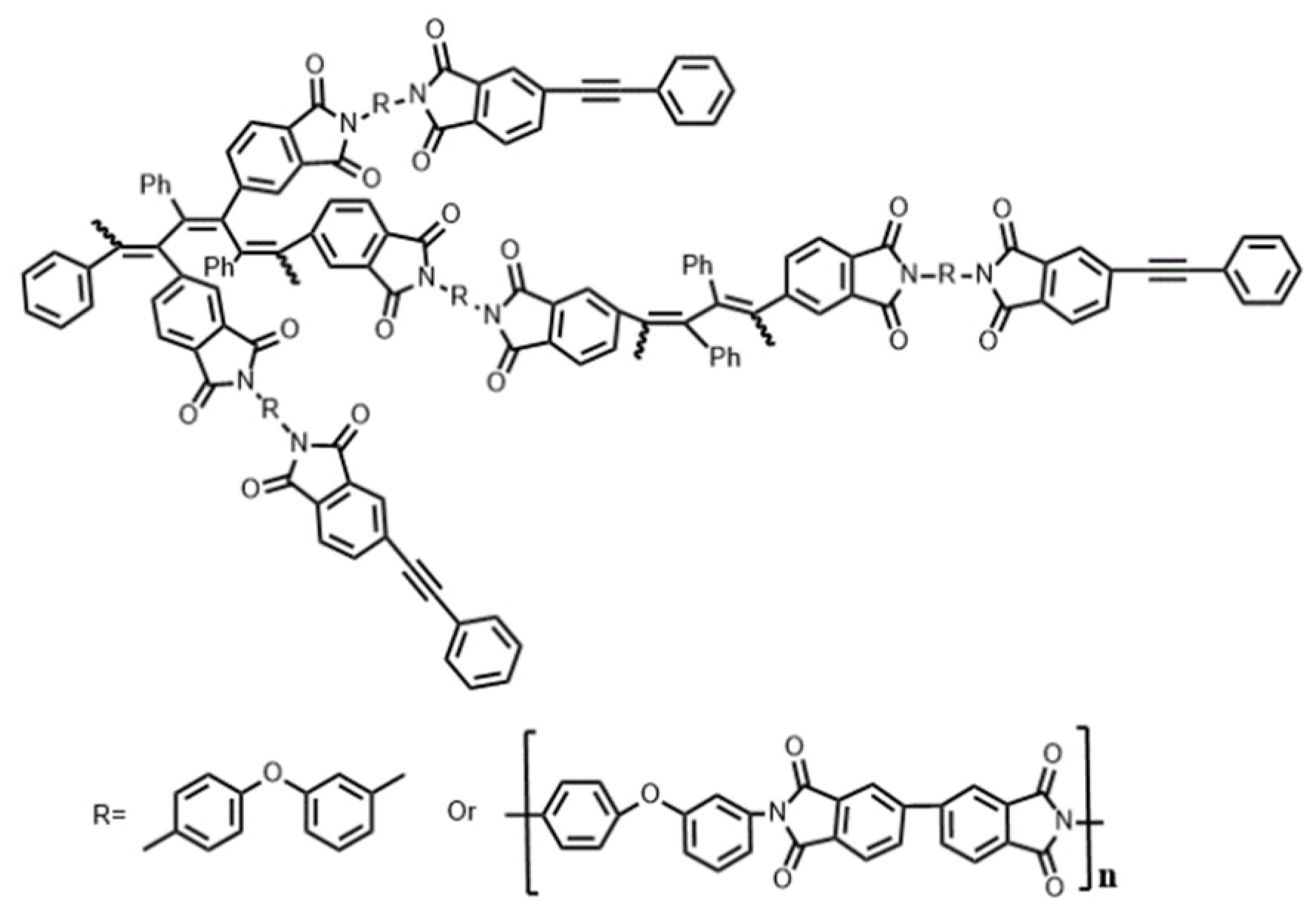
2.2.1. Synthesis of Thermosetting Poly(amic ester) Resins (TMR)
2.2.2. Synthesis of Reactive Diluents (RDm)
2.2.3. Synthesis of Thermosetting Blend Resins (TMR/RDm)
2.2.4. Thermally Cured Blend Neat Resins
2.2.5. Preparation of Carbon Fiber Composites
2.3. Characterization and Measurements
3. Results
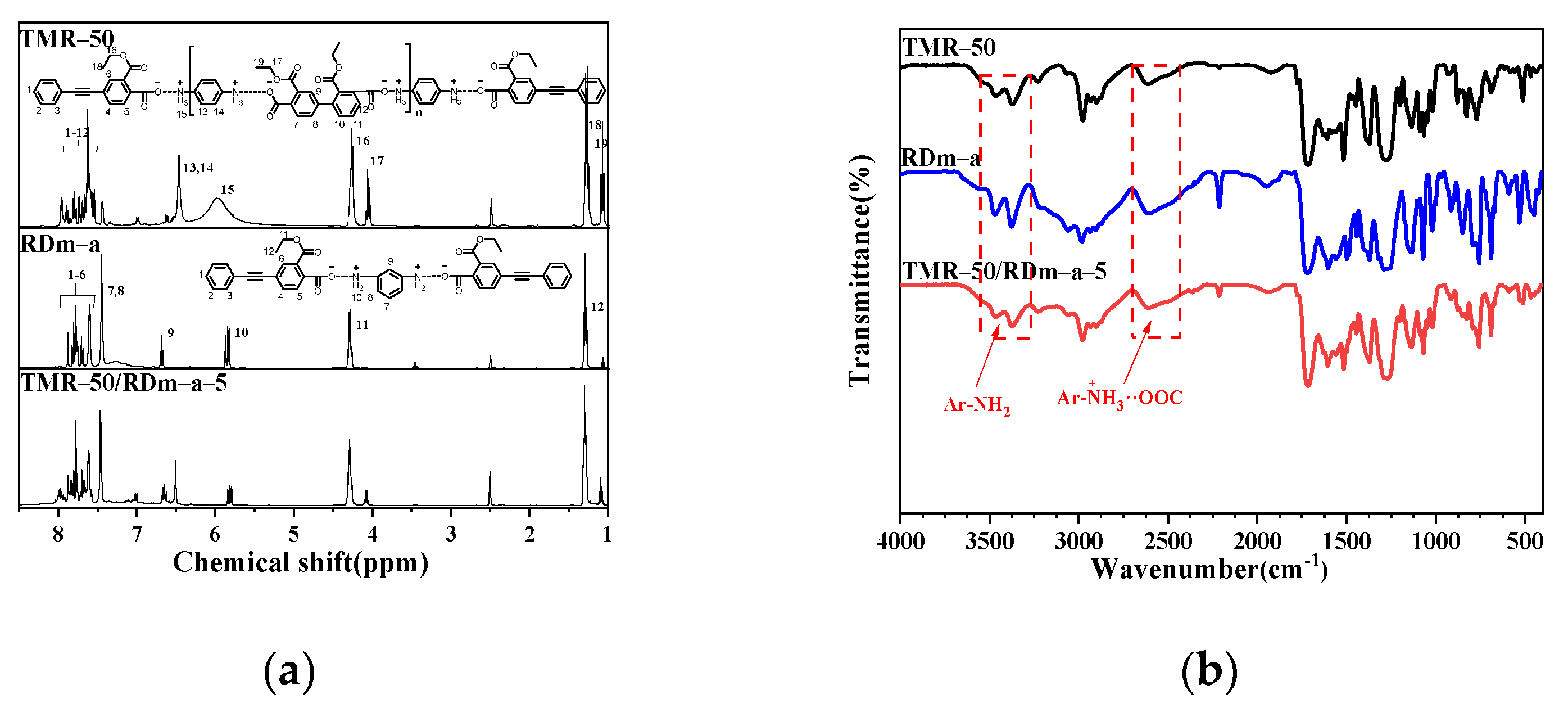
3.1. Characterization of the Thermosetting Blend Resins
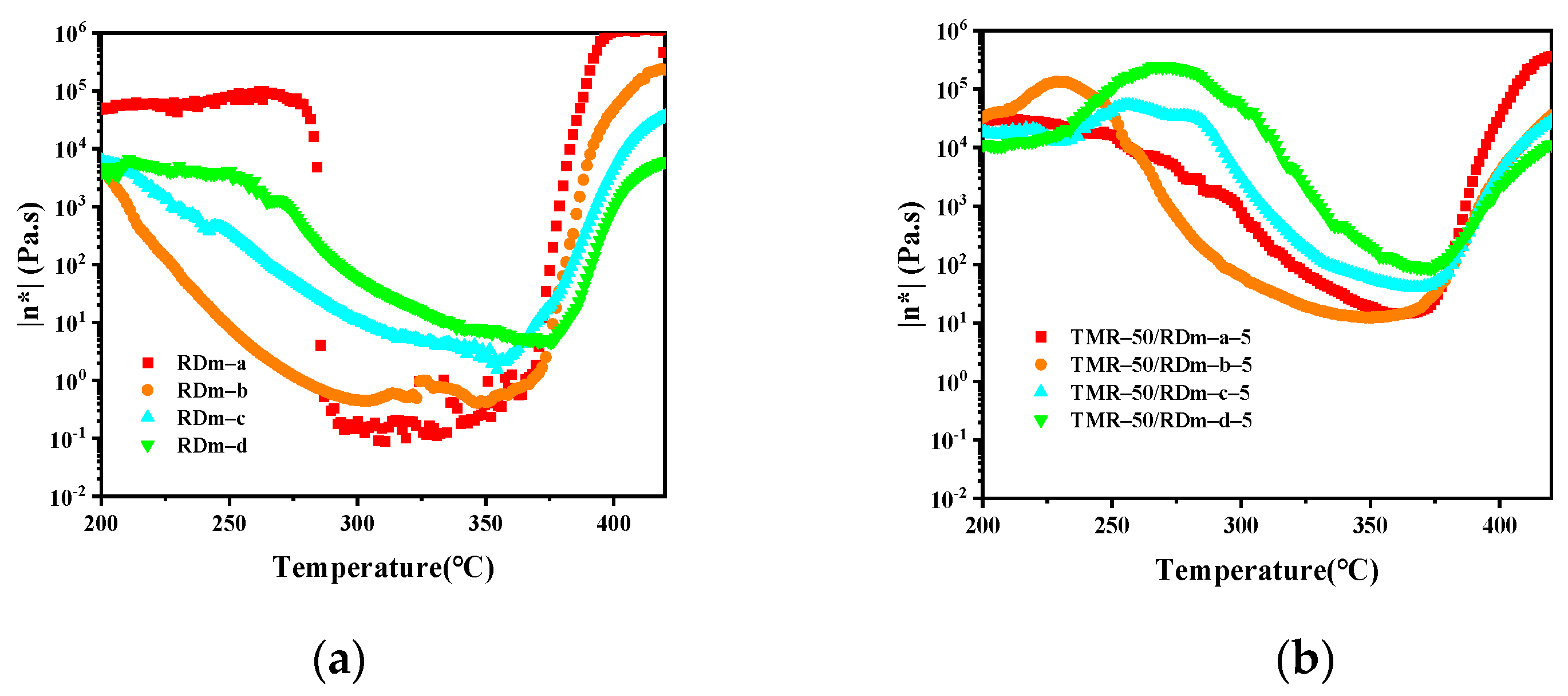
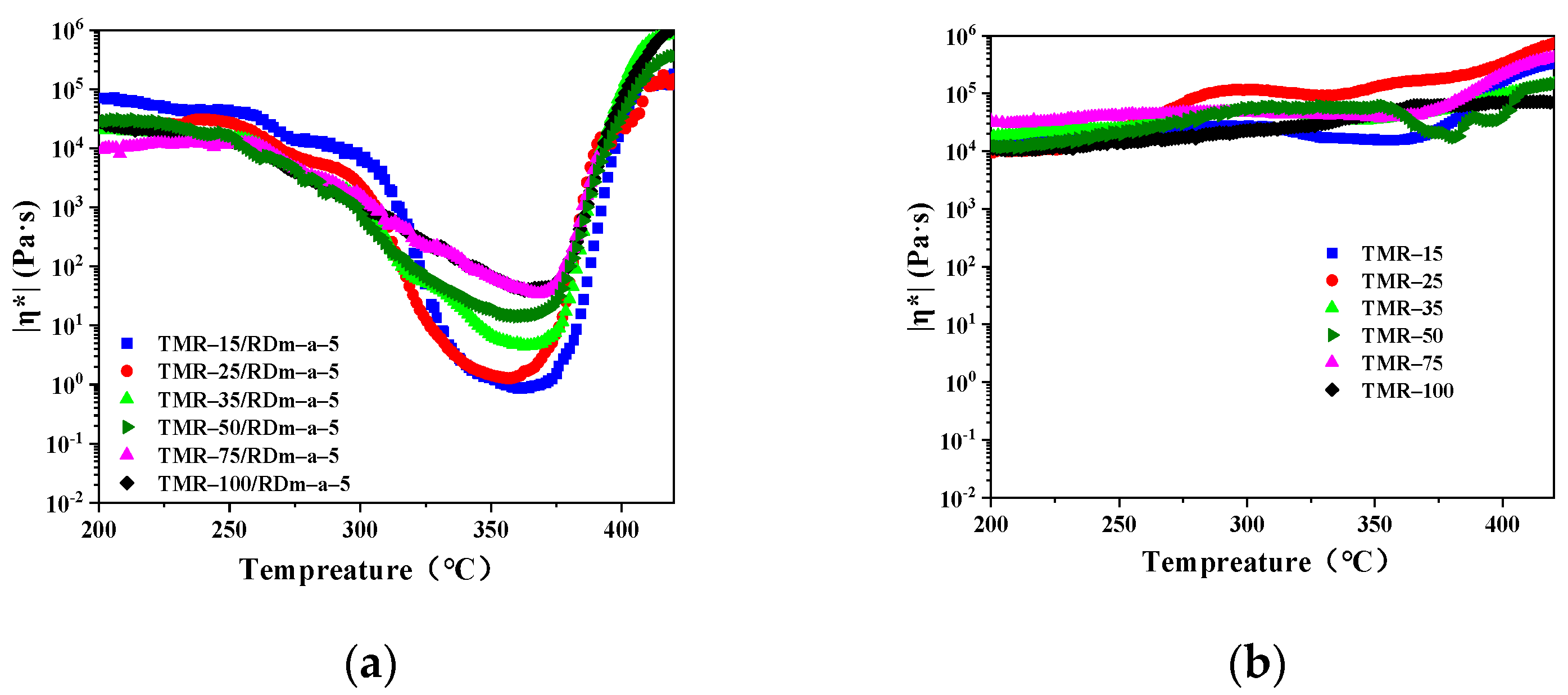
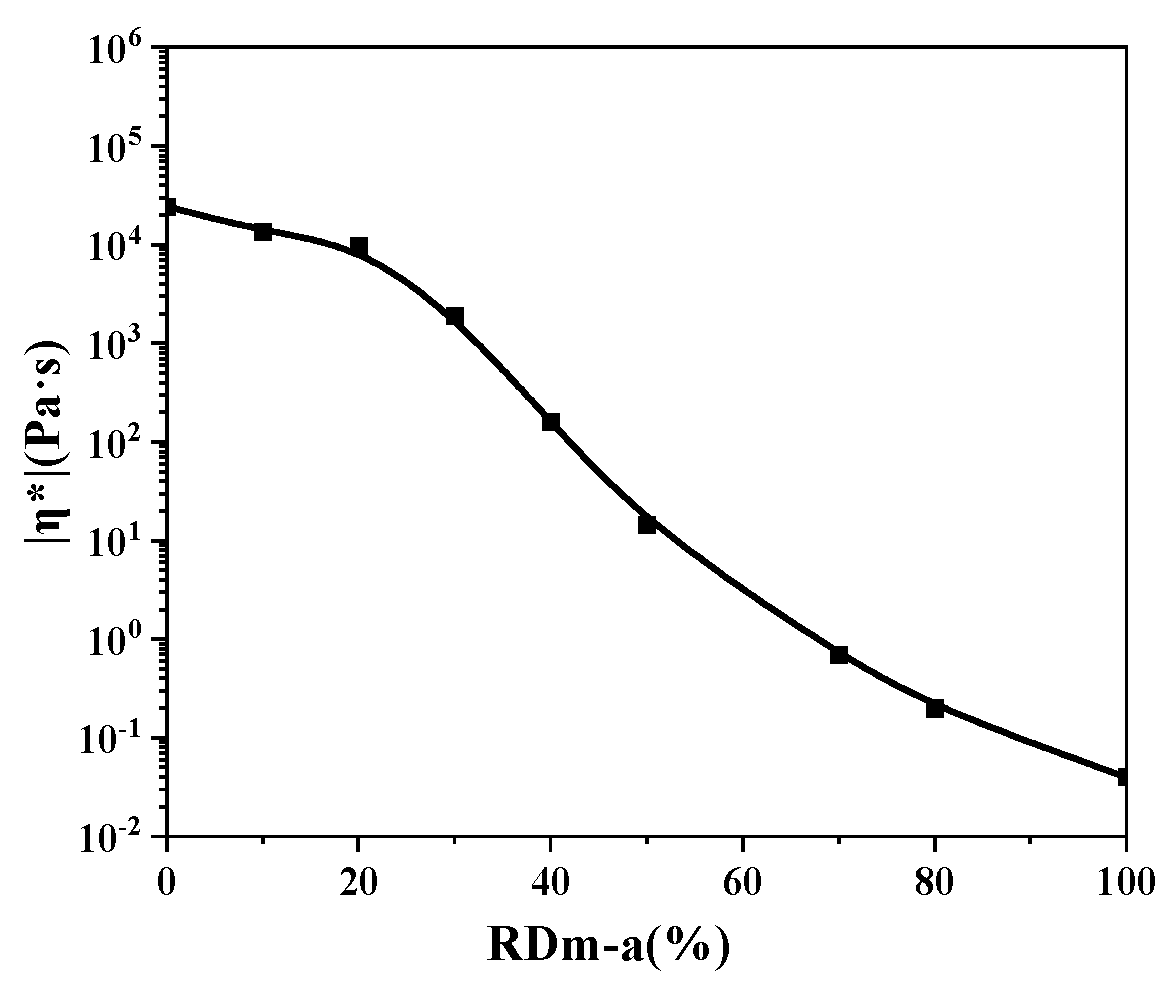
3.2. Melt Processability of the Thermosetting Blend Resins
3.3. Thermal Curing Kinetics of the B-Staged Blend Resins
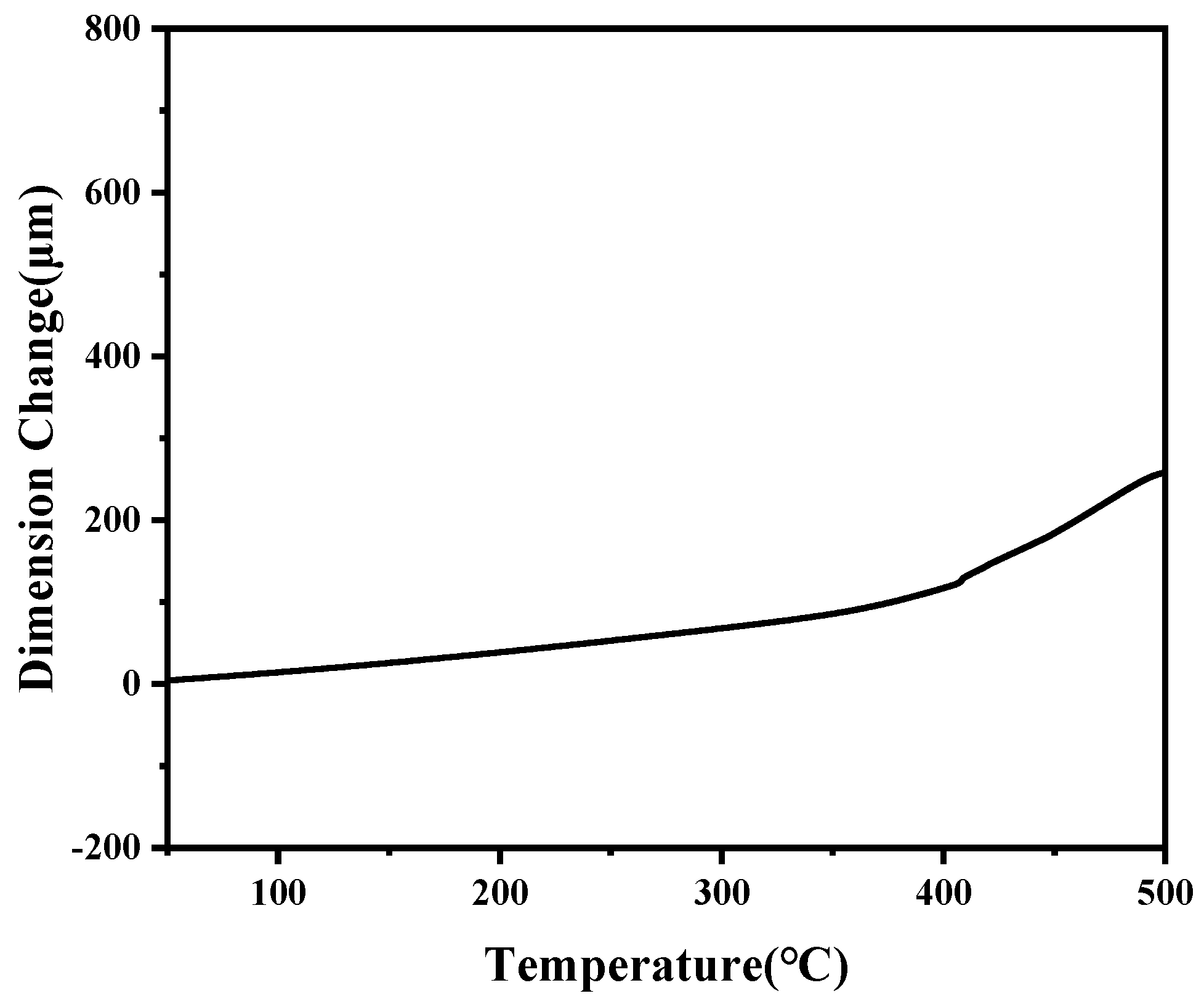
3.4. Thermal Stabilities of the Thermally Cured Blend Resins
3.5. Mechanical Properties of the Thermally Cured Blend Resins
3.6. Mechanical Properties of Polyimide/Carbon Fiber Composites
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wang, T.; Yang, Y.; Song, N.; Zhu, S.; Yao, H.; Zhang, Y.; Guan, S. Thermal crosslinking polymerization of aromatic alkynyl monomers to microporous polyimides in diphenyl sulfone. Microporous Mesoporous Mater. 2021, 328, 111447. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, B.; Liu, C.; Sun, M.; Zhang, X.; Li, J.; Xue, G. Effect on the thermal resistance and thermal decomposition properties of thermally cross-linkable polyimide films obtained from a reactive acetylene. React. Funct. Polym. 2021, 167, 104994. [Google Scholar] [CrossRef]
- Ma, S.; Wang, C.; Cong, B.; Zhou, H.; Zhao, X.; Chen, C.; Wang, D.; Liu, C.; Qu, C. Anisotropic all-aromatic polyimide aerogels with robust and high-temperature stable properties for flexible thermal protection. Chem. Eng. J. 2022, 431, 134047. [Google Scholar] [CrossRef]
- Liu, Y.; Xu, X.-Z.; Mo, S.; Lan, B.-W.; Zhu, C.-Z.; Li, C.-H.; Xu, J.; Fan, L. Long-term Thermo-oxidative Degradation Modeling of a Carbon Fiber Reinforced Polyimide Composite: Multistep Degradation Behaviors and Kinetics. Chin. J. Polym. Sci. 2020, 38, 1202–1213. [Google Scholar] [CrossRef]
- Zhang, Y.; Mushtaq, N.; Fang, X.; Chen, G. In situ FTIR analysis for the determination of imidization degree of polyimide precursors. Polymer 2022, 238, 124416. [Google Scholar] [CrossRef]
- Nikolaeva, A.L.; Gofman, I.V.; Yakimansky, A.V.; Ivan’Kova, E.M.; Abalov, I.V.; Baranchikov, A.E.; Ivanov, V.K. Polyimide-Based Nanocomposites with Binary CeO2/Nanocarbon Fillers: Conjointly Enhanced Thermal and Mechanical Properties. Polymers 2020, 12, 1952. [Google Scholar] [CrossRef]
- Li, Y.; Morgan, R.J. Thermal cure of phenylethynyl-terminated AFR-PEPA-4 imide oligomer and a model compound. J. Appl. Polym. Sci. 2006, 101, 4446–4453. [Google Scholar] [CrossRef]
- Meng, X.; Yan, J.; Fan, W.; Liu, J.; Wang, Z.; Li, G. Thermosetting polyimides and composites based on highly soluble phenylethynyl-terminated isoimide oligomers. RSC Adv. 2014, 4, 37458–37469. [Google Scholar] [CrossRef]
- Delmdahl, R.; Brune, P. Large-area laser-lift-off processing in microelectronics. Phys. Procedia 2013, 41, 241–248. [Google Scholar] [CrossRef] [Green Version]
- Qian, G.; Hu, M.; Zhang, S.; Wang, M.; Chen, C.; Yao, J. Synthesis of Superheat-Resistant Polyimides with Enhanced Dielectric Constant by Introduction of Cu(IotaIota)-Coordination. Polymers 2020, 12, 442. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Yang, J.; Chen, G. Synthesis and characterization of a carborane-containing monofunctional imide monomer as a modifier for imide oligomer. High Perform. Polym. 2017, 30, 812–820. [Google Scholar] [CrossRef]
- Choi, J.-Y.; Jin, S.-W.; Kim, D.-M.; Song, I.-H.; Nam, K.-N.; Park, H.-J.; Chung, C.-M. Enhancement of the Mechanical Properties of Polyimide Film by Microwave Irradiation. Polymers 2019, 11, 477. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.; Kim, Y.H.; Chang, S. Polymers for flexible displays: From material selection to device applications. Prog. Polym. Sci. 2008, 33, 581–630. [Google Scholar] [CrossRef]
- Yamaguchi, H. 11.7-inch flexible AMOLED display driven by a-IGZO TFTs on planstic substrate. SID Dig. 2012, 43, 1002–1005. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, W.; Gao, X.; Cui, P.; Zou, T.; Hu, G.; Tao, L.; Zhai, L. Preparation and Characterization of Semi-Alicyclic Polyimides Containing Trifluoromethyl Groups for Optoelectronic Application. Polymers 2020, 12, 1532. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, G.; Zhan, M.; Yang, J.P. High heat resistant carbon fiber/polyimide composites with neutron shielding performance. Prog. Org. Coat. 2019, 132, 184–190. [Google Scholar] [CrossRef]
- Magato, J.; Klosterman, D. Development of a methodology for characterizing reaction kinetics, rheology, and in situ compaction of polyimide prepregs during cure. J. Compos. Mater. 2019, 54, 835–843. [Google Scholar] [CrossRef]
- Yue, J.; Li, Y.; Li, H.; Zhao, Y.; Zhao, C.; Wang, X. Thermal curing of novel carborane-containing phenylethynyl terminated imide oligomers. RSC Adv. 2015, 5, 98010–98019. [Google Scholar] [CrossRef]
- Meador, M.A.; Johnston, J.C.; Cavano, P.J. Elucidation of the Cross-Link Structure of Nadic-End-Capped Polyimides Using NMR of 13C-Labeled Polymers. Macromolecules 1997, 30, 515–519. [Google Scholar] [CrossRef]
- Nakamura, K.; Ando, S.; Takeichi, T. Thermal analysis and solid-state 13C NMR study of crosslink in polyimides containing acetylene groups in the main chain. Polymer 2001, 42, 4045–4054. [Google Scholar] [CrossRef]
- Holland, T.V.; Glass, T.E.; McGrath, J.E. Investigation of the thermal curing chemistry of the phenylethynyl group using a model aryl ether imide. Polymer 2000, 41, 4965–4990. [Google Scholar] [CrossRef]
- Russell, J.D.; Kardos, J.L. Crosslinking characterization of a polyimide: AFR700B. Polym. Compos. 2004, 18, 595–612. [Google Scholar] [CrossRef]
- Zhang, Y.; Jain, A.; Grunenfelder, L.K.; Miyauchi, M.; Nutt, S. Process development for phenylethynyl-terminated PMDA-type asymmetric polyimide composites. High Perform. Polym. 2017, 30, 731–741. [Google Scholar] [CrossRef]
- Hu, Y.B.; Li, H.G.; Cai, L.; Zhu, J.P.; Pan, L.; Xu, J.; Tao, J. Preparation and properties of Fibre-Metal Laminates based on carbon fibre reinforced PMR polyimide. Compos. Part B Eng. 2015, 69, 587–591. [Google Scholar] [CrossRef]
- Qu, X.; Ji, M.; Fan, L.; Yang, S. Thermoset polyimide matrix resins with improved toughness and high Tg for high temperature carbon fiber composites. High Perform. Polym. 2011, 23, 281–289. [Google Scholar] [CrossRef]
- Serafini, T.T.; Delvigs, P.; Lightsey, G.R. Thermally stable polyimides from solutions of monomeric reactants. J. Appl. Polym. Sci. 1972, 16, 905–915. [Google Scholar] [CrossRef] [Green Version]
- Rao, X.; Dang, G.; Zhou, H.; Yang, W.; Cui, G.; Chen, C.; Yokota, R. Design and synthesis of a tribranched phenylethynyl-terminated aryl ether compound and its use as a reactive diluent for PETI-5. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 4844–4854. [Google Scholar] [CrossRef]
- Wu, Y.; Feng, C.; Yang, J.; Chen, G. High thermally stable thermosetting polyimides derived from a carborane-containing tetramine. High Perform. Polym. 2018, 31, 548–556. [Google Scholar] [CrossRef]
- Zhou, D.; Yuan, L.; Hong, W.; Zhang, H.; Hu, A.; Yang, S. Molecular design of interpenetrating fluorinated polyimide network with enhanced high performance for heat-resistant matrix. Polymer 2019, 173, 66–79. [Google Scholar] [CrossRef]
- Milhourat-Hammadi, A.; Chayrigues, H.; Merienne, C.; Gaudemerz, A. NMR Study of PMR-15 Prepolymerization Steps. J. Polym. Sci. Part A Polym. Chem. 1994, 32, 203–217. [Google Scholar] [CrossRef]
- Garcia, D.; Serafini, T. FTIR Studies of PMR-15 Polyimides. J. Polym. Sci. Part B Polym. Phys. 1987, 25, 2275–2282. [Google Scholar] [CrossRef]
- Hurwitz, F.I. Influence of excess diamine on properties of PMR polyimide resins and composites. In Proceedings of the 12th National SAMPE Technical Conference, Seattle, WA, USA, 7–9 October 1980. [Google Scholar]
- Morgan, R.; Shin, E.; Lincoln, J.; Zhou, J. Overview of polymer matrix composites performance and materials development for aerospace applications. SAMPE J. 2001, 37, 102–107. [Google Scholar]
- Allred, R.; Wesson, S.; Shin, E.; Inghram, L.; McCorkle, L.; Papadopoulos, D.; Wheeler, D.; Sutter, J. The Influence of Sizings on the Durability of High-Temperature Polymer Composites. High Perform. Polym. 2004, 15, 395–419. [Google Scholar] [CrossRef] [Green Version]
- Ogasawara, T.; Ishida, Y.; Yokota, R.; Watanabe, T.; Aoi, T.; Goto, J. Processing and properties of carbon fiber/Triple-A polyimide composites fabricated from imide oligomer dry prepreg. Compos. Part A Appl. Sci. Manuf. 2007, 38, 1296–1303. [Google Scholar] [CrossRef]
- Liu, H.B.; Simone, C.D.; Scola, D.A. Synthesis and characterization of phenylethynyl-end-capped cooligomides from fluorinated dianhydrides 4,4′-(hexafluoroisopropylidene)dipthalic anhydride and 4,4′-(2,2,2-trifluoro-1-phenylethylidene)dipthalic anhydride and para- and meta-phenylene diamines and cooligomide blends with phenylethynyl-end-capped reactive diluents. J. Polym. Sci. Part A Polym. Chem. 2003, 41, 2630–2649. [Google Scholar]
- Ogasawara, T.; Ishikawa, T.; Yokota, R.; Ozawa, H.; Taguchi, M.; Sato, R.; Shigenari, Y.; Miyagawa, K. Processing and properties of carbon fiber reinforced triple-A polyimide (Tri-A PI) matrix composites. Adv. Compos. Mater. 2003, 11, 277–286. [Google Scholar] [CrossRef]
- Yu, P.; Wang, Y.; Yu, J.; Zhu, J.; Hu, Z. Synthesis and characterization of phenylethynyl-terminated polyimide oligomers derived from 2,3,3′,4′-diphenyl ether tetracarboxylic acid dianhydride and 3,4′-oxydianiline. Chin. J. Polym. Sci. 2016, 34, 122–134. [Google Scholar] [CrossRef]
- Yu, P.; Xue, M.-Z.; Liu, Y.-G.; Yang, X.; Pan, L.-J.; Zhang, Y.; Wang, W. Effect of Blending Modifications for Phenylethynyl-terminated Polyimides. Fibers Polym. 2020, 21, 282–289. [Google Scholar] [CrossRef]
- Rasheva, Z.; Sorochynska, L.; Grishchuk, S.; Friedrich, K. Effect of the solvent type and polymerization conditions on the curing kinetics, thermal and viscoelastic performance of poly (amide-imide) resins. Express Polym. Lett. 2015, 9, 196–210. [Google Scholar] [CrossRef]
- Sasaki, T.; Moriuchi, H.; Yano, S.; Yokota, R. High thermal stable thermoplastic–thermosetting polyimide film by use of asymmetric dianhydride (a-BPDA). Polymer 2005, 46, 6968–6975. [Google Scholar] [CrossRef]
- Li, Y.; Obando, N.; Tschen, F.; Morgan, R.J. Thermalanalysis of phenylethynyl end-capped fluorinated imide oligomer afr-pepa-4. J. Therm. Anal. Calorim. 2006, 85, 125–129. [Google Scholar] [CrossRef]
- Miyauchi, M.; Ishida, Y.; Ogasawara, T.; Yokota, R. Novel phenylethynyl-terminated PMDA-type polyimides based on KAPTON backbone structures derived from 2-phenyl-4,4′-diaminodiphenyl ether. Polym. J. 2012, 44, 959–965. [Google Scholar] [CrossRef]
- Connell, J.W.; Smith, J.G., Jr.; Hergenrother, P.M.; Rommel, M.L. Neat resin, adhesive and composite properties of reactive additive/PETI-5 blends. High Perform. Polym. 2000, 12, 323–333. [Google Scholar] [CrossRef]
- Hong, W.; Yuan, L.; Ma, Y.; Cui, C.; Zhang, H.; Yang, S.; Sun, W.-H. Resin Transfer Moldable Fluorinated Phenylethynyl-Terminated Imide Oligomers with High T-g: Structure-Melt Stability Relationship. Polymers 2021, 13, 903. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Blended Matrix Resins | Matrix Resin (50%) | Reactive Dilutes (50%) |
|---|---|---|
| TMR-15/RDm–a–5 | TMR–15 | RDm–a |
| TMR-25/RDm–a–5 | TMR–25 | |
| TMR-35/RDm–a–5 | TMR–35 | |
| TMR–50/RDm–a–5 | TMR–50 | |
| TMR-75/RDm–a–5 | TMR–75 | |
| TMR-100/RDm–a–5 | TMR–100 | |
| TMR–50/RDm–b–5 | TMR–50 | RDm–b |
| TMR–50/RDm–c–5 | TMR–50 | RDm–c |
| TMR–50/RDm–d–5 | TMR–50 | RDm–d |
| Reactive Diluents (RDm–x) | TMR–50/RDm–x–5 | |||
|---|---|---|---|---|
| Calc’d Mw | |η*|min (Pa·s/°C) | ΔT10 (°C) | |η*|min (Pa·s) | ΔT100 (°C) |
| 660 (RDm–a) | 0.1 (311 °C) | 275–375 °C | 14.4 (360 °C) | 320–380 °C |
| 1120 (RDm–b) | 0.4 (347 °C) | 250–375 °C | 3.8 (358 °C) | 293–380 °C |
| 1580 (RDm–c) | 1.5 (355 °C) | 300–375 °C | 39.8 (363 °C) | 335–382 °C |
| 2040 (RDm–d) | 4.7 (370 °C) | 340–380 °C | 86.1 (360 °C) | 363–375 °C |
| Samples | |η*|min (Pa·s/°C) | ΔT100 (°C) |
|---|---|---|
| TMR–15/RDm–a–5 | 1.70 (343 °C) | 317–383 °C |
| TMR–25/RDm–a–5 | 3.45 (358 °C) | 323–388 °C |
| TMR–35/RDm–a–5 | 4.49 (363 °C) | 316–380 °C |
| TMR–50/RDm–a–5 | 14.4 (360 °C) | 320–380 °C |
| TMR–75/RDm–a–5 | 34.3 (368 °C) | 342–379 °C |
| TMR–100/RDm–a–5 | 40.6 (366 °C) | 343–378 °C |
| Sample | β (°C·min−1) | Ti (°C) | Tp (°C) | Tt (°C) | ∆T (°C) |
|---|---|---|---|---|---|
| TMR–50/RDm–a–5 | 5 | 317.0 | 384.9 | 447.2 | 130.2 |
| 10 | 355.0 | 402.0 | 456.2 | 101.2 | |
| 15 | 336.5 | 410.8 | 472.5 | 136.0 | |
| 20 | 329.0 | 418.2 | 470.5 | 141.5 |
| Samples | DMA | TGA in N2 | TGA in Air | |||||
|---|---|---|---|---|---|---|---|---|
| To1 (°C) | Tg2 (°C) | Td (°C) | Td5 (°C) | Td10 (°C) | Td (°C) | Td5 (°C) | Td10 (°C) | |
| TMR–15/RDm–a–5 | 434 | 486 | 597.7 | 594.8 | 618.1 | 594.2 | 591.3 | 614.6 |
| TMR–25/RDm–a–5 | 424 | 478 | 595.6 | 591.4 | 614.5 | 592.1 | 587.9 | 611.0 |
| TMR–35/RDm–a–5 | 419 | 467 | 593.2 | 590.6 | 613.3 | 589.7 | 587.1 | 609.8 |
| TMR–50/RDm–a–5 | 423 | 462 | 591.5 | 591.2 | 612.9 | 588.0 | 587.7 | 609.4 |
| TMR–75/RDm–a–5 | 414 | 458 | 589.9 | 586.9 | 610.4 | 586.4 | 583.4 | 606.9 |
| TMR–100/RDm–a–5 | 413 | 454 | 588.5 | 585.8 | 608.8 | 585.0 | 582.3 | 605.3 |
| Samples | Tensile Properties | Flexural Properties | |||
|---|---|---|---|---|---|
| Strength (MPa) | Modulus (GPa) | Elongation (%) | Strength (MPa) | Modulus (GPa) | |
| TMR–15/RDm–a–5 | 53.5 (±3.10) | 2.7 (±0.08) | 2.6 (±0.48) | 111.3 (±5.00) | 3.1 (±0.09) |
| TMR–25/RDm–a–5 | 63.0 (±2.55) | 2.2 (±0.09) | 3.5 (±0.40) | 121.3 (±5.60) | 3.5 (±0.08) |
| TMR–35/RDm–a–5 | 76.2 (±4.31) | 2.5 (±0.08) | 3.2 (±0.32) | 148.2 (±5.33) | 3.4 (±0.08) |
| TMR–50/RDm–a–5 | 84.0 (±4.36) | 2.4 (±0.10) | 4.1 (±0.50) | 169.2 (±4.58) | 3.5 (±0.10) |
| TMR–75/RDm–a–5 | 77.2 (±3.18) | 2.4 (±0.08) | 3.9 (±0.44) | 163.9 (±4.90) | 3.5 (±0.10) |
| TMR–100/RDm–a–5 | 77.9 (±2.99) | 2.4 (±0.10) | 3.8 (±0.41) | 164.8 (±2.70) | 3.5 (±0.09) |
| Property | Test Temperature (°C) | Composite As-Prepared (380 °C/2 h) | Composite after Post-Cured (420 °C/1 h) |
|---|---|---|---|
| Volume fraction of carbon fiber | -- | 62 (±1) | 62(±1) |
| Flexural strength (MPa) | 25 | 1552 (±48.4) | 1663 (±34.9) |
| 370 | 690 (±15.1) | 955 (±23.5) | |
| Flexural modulus (GPa) | 25 | 119 (±4.2) | 136 (±4.0) |
| 370 | 117 (±1.1) | 135 (±5.9) | |
| Interlaminar shear strength (MPa) | 25 | 95.2 (±2.4) | 85.3 (±3.8) |
| 370 | 37.4 (±2.4) | 41.3 (±1.0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.-Y.; Yuan, L.-L.; Hong, W.-J.; Yang, S.-Y. Improved Melt Processabilities of Thermosetting Polyimide Matrix Resins for High Temperature Carbon Fiber Composite Applications. Polymers 2022, 14, 965. https://doi.org/10.3390/polym14050965
Zhang H-Y, Yuan L-L, Hong W-J, Yang S-Y. Improved Melt Processabilities of Thermosetting Polyimide Matrix Resins for High Temperature Carbon Fiber Composite Applications. Polymers. 2022; 14(5):965. https://doi.org/10.3390/polym14050965
Chicago/Turabian StyleZhang, Hao-Yang, Li-Li Yuan, Wei-Jie Hong, and Shi-Yong Yang. 2022. "Improved Melt Processabilities of Thermosetting Polyimide Matrix Resins for High Temperature Carbon Fiber Composite Applications" Polymers 14, no. 5: 965. https://doi.org/10.3390/polym14050965
APA StyleZhang, H.-Y., Yuan, L.-L., Hong, W.-J., & Yang, S.-Y. (2022). Improved Melt Processabilities of Thermosetting Polyimide Matrix Resins for High Temperature Carbon Fiber Composite Applications. Polymers, 14(5), 965. https://doi.org/10.3390/polym14050965