
Anionic Polymerization of Para-Diethynylbenzene: Synthesis of a Strictly Linear Polymer

 , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Measurements
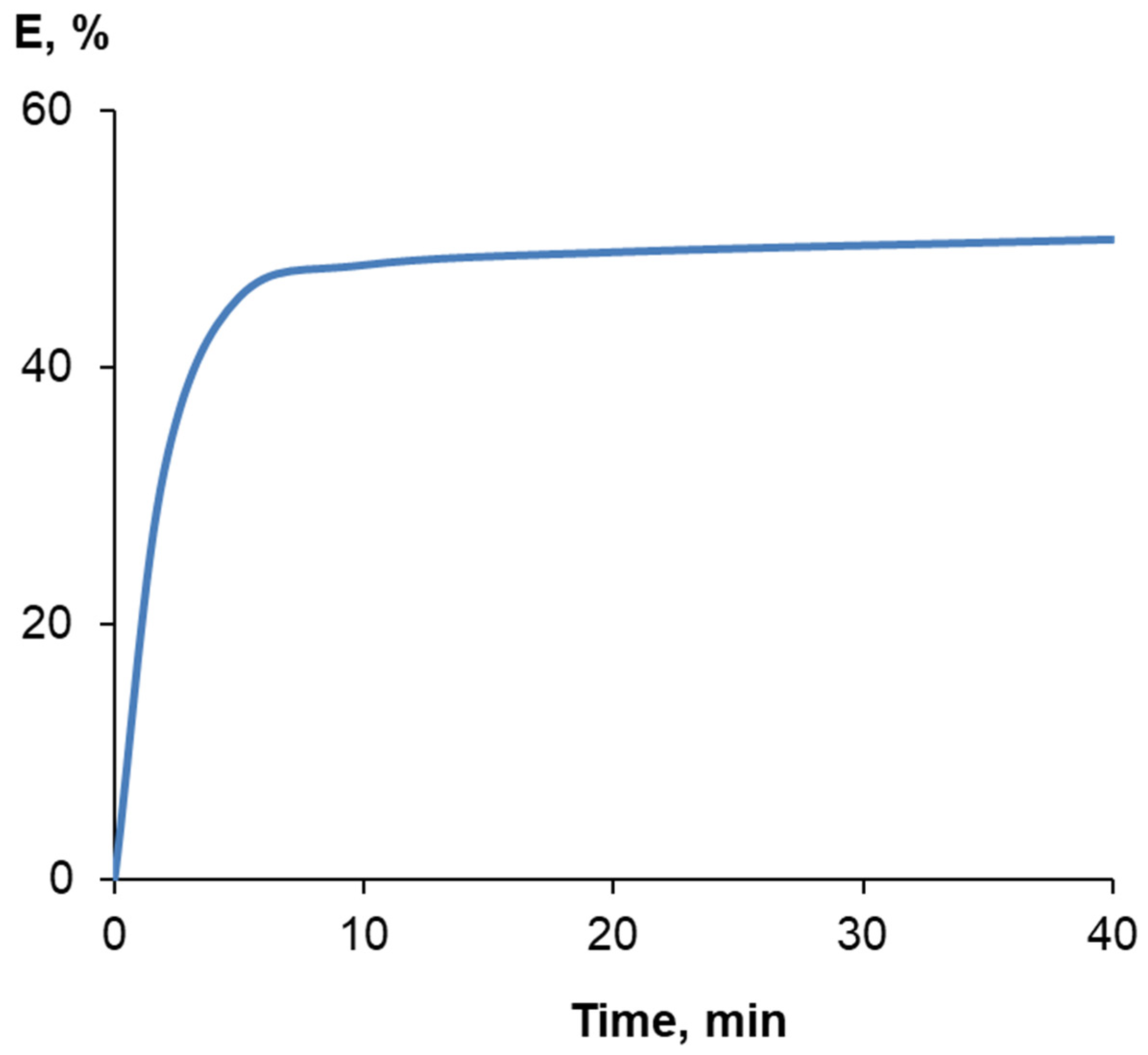
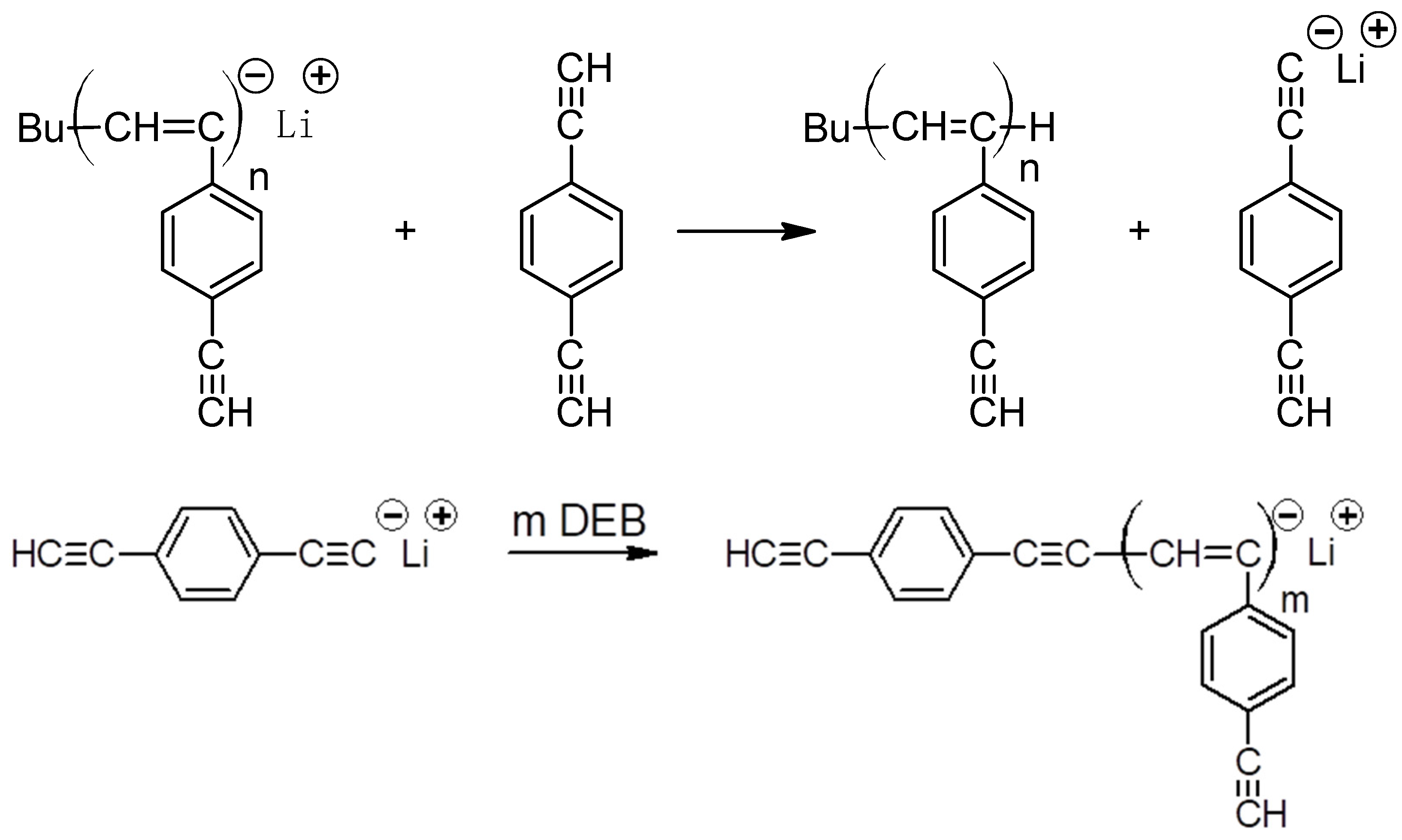
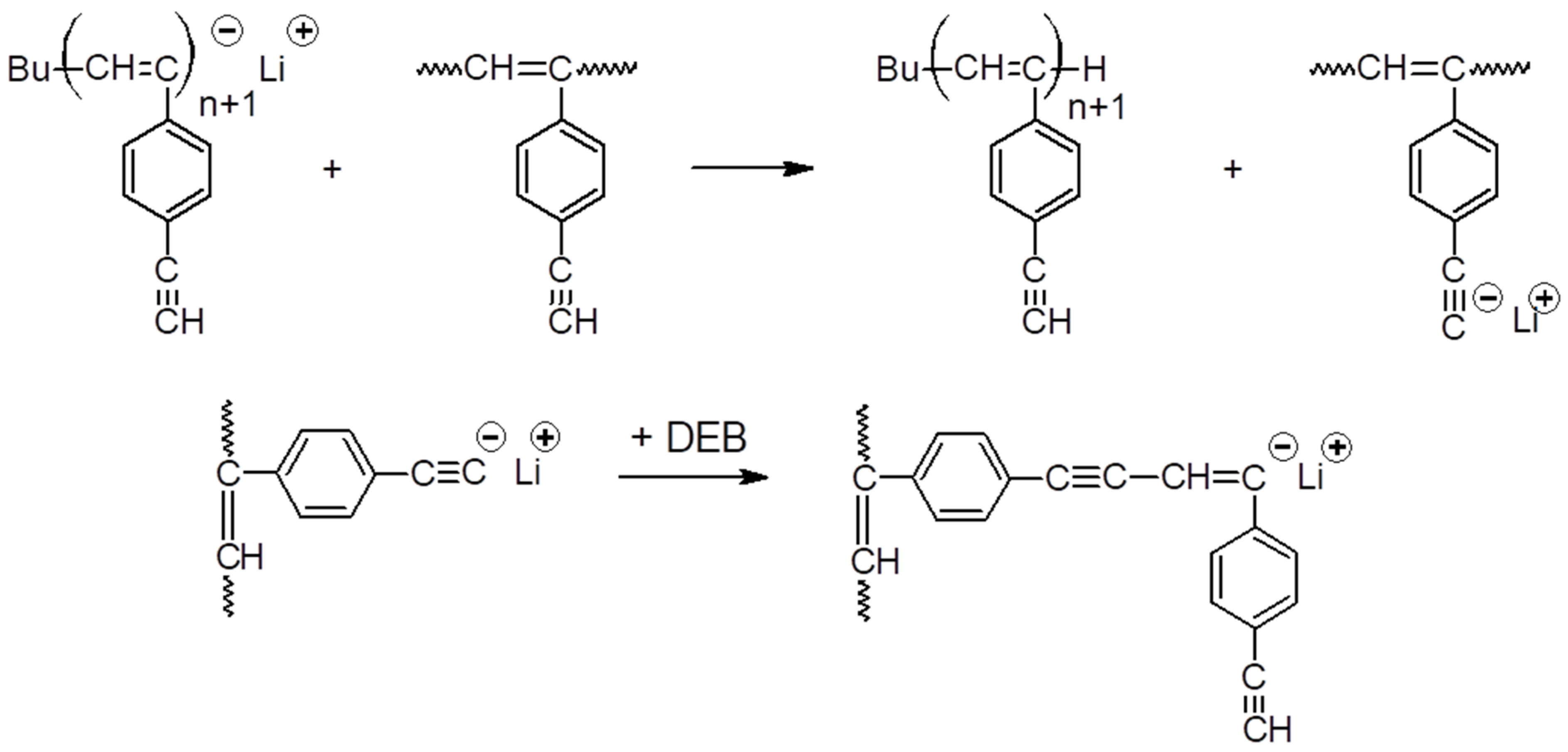
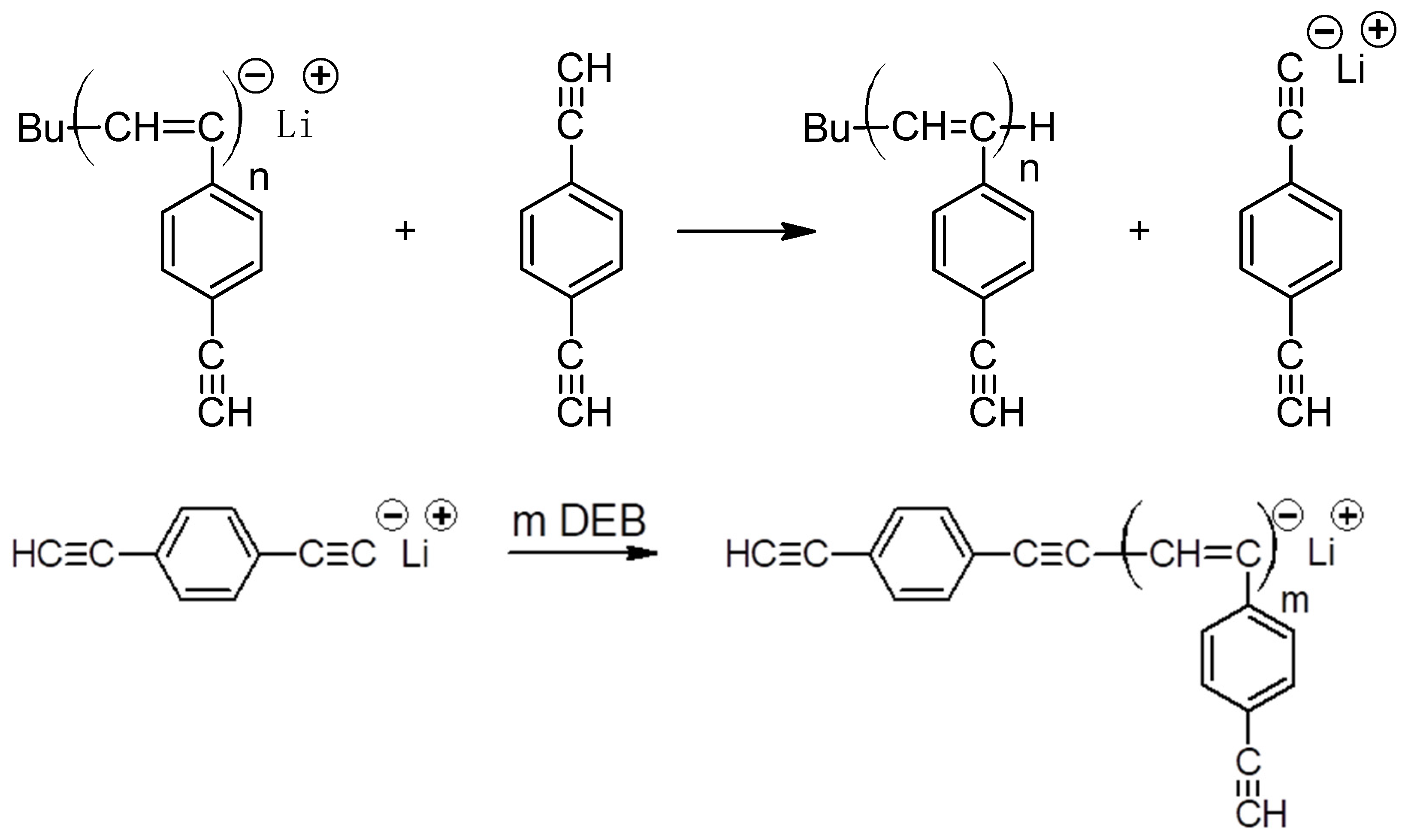
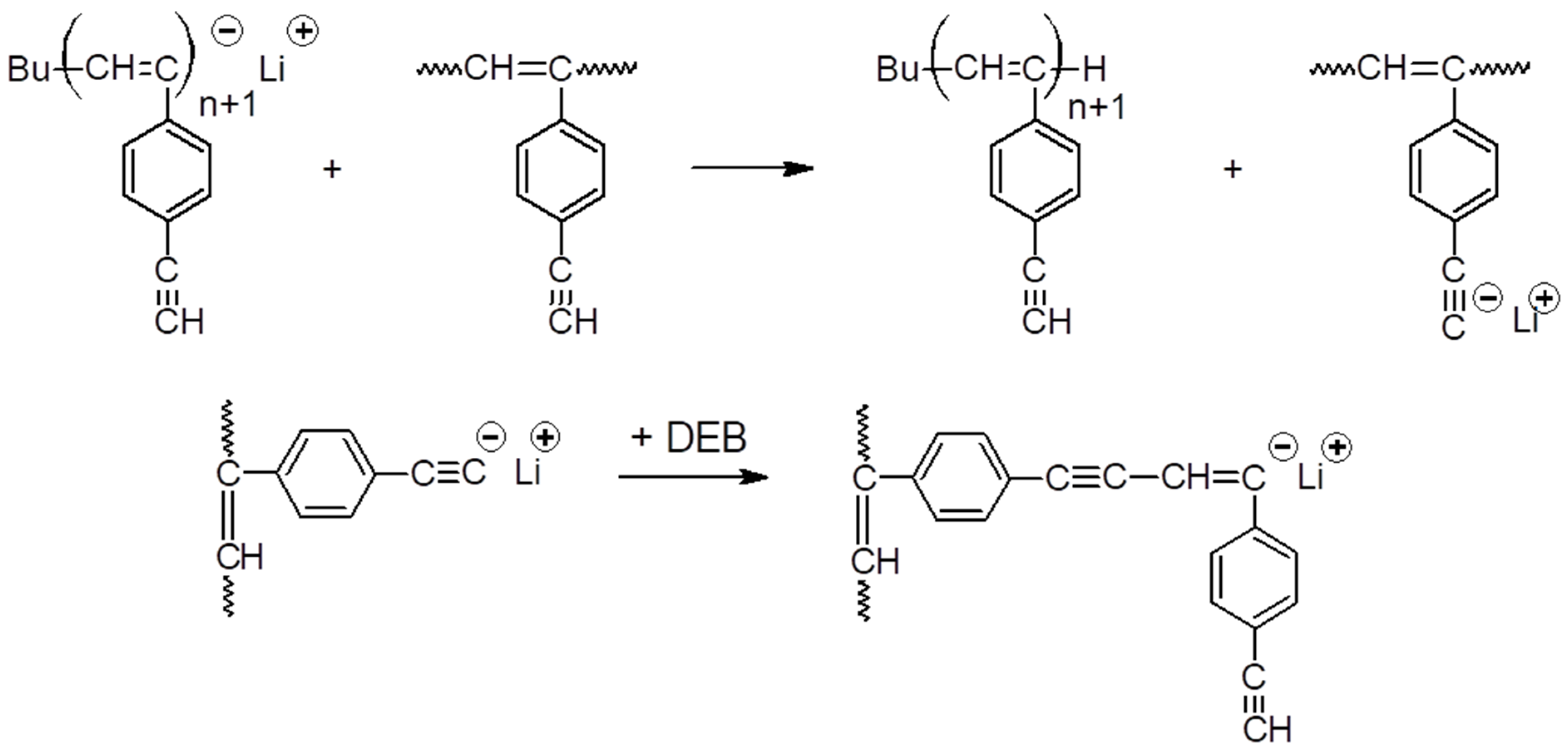
2.3. Polymerization
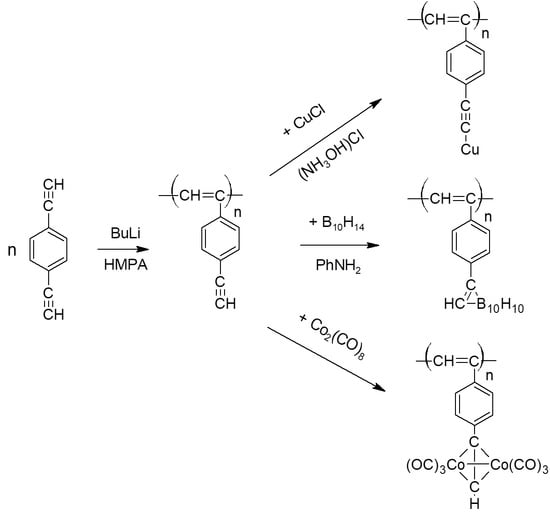
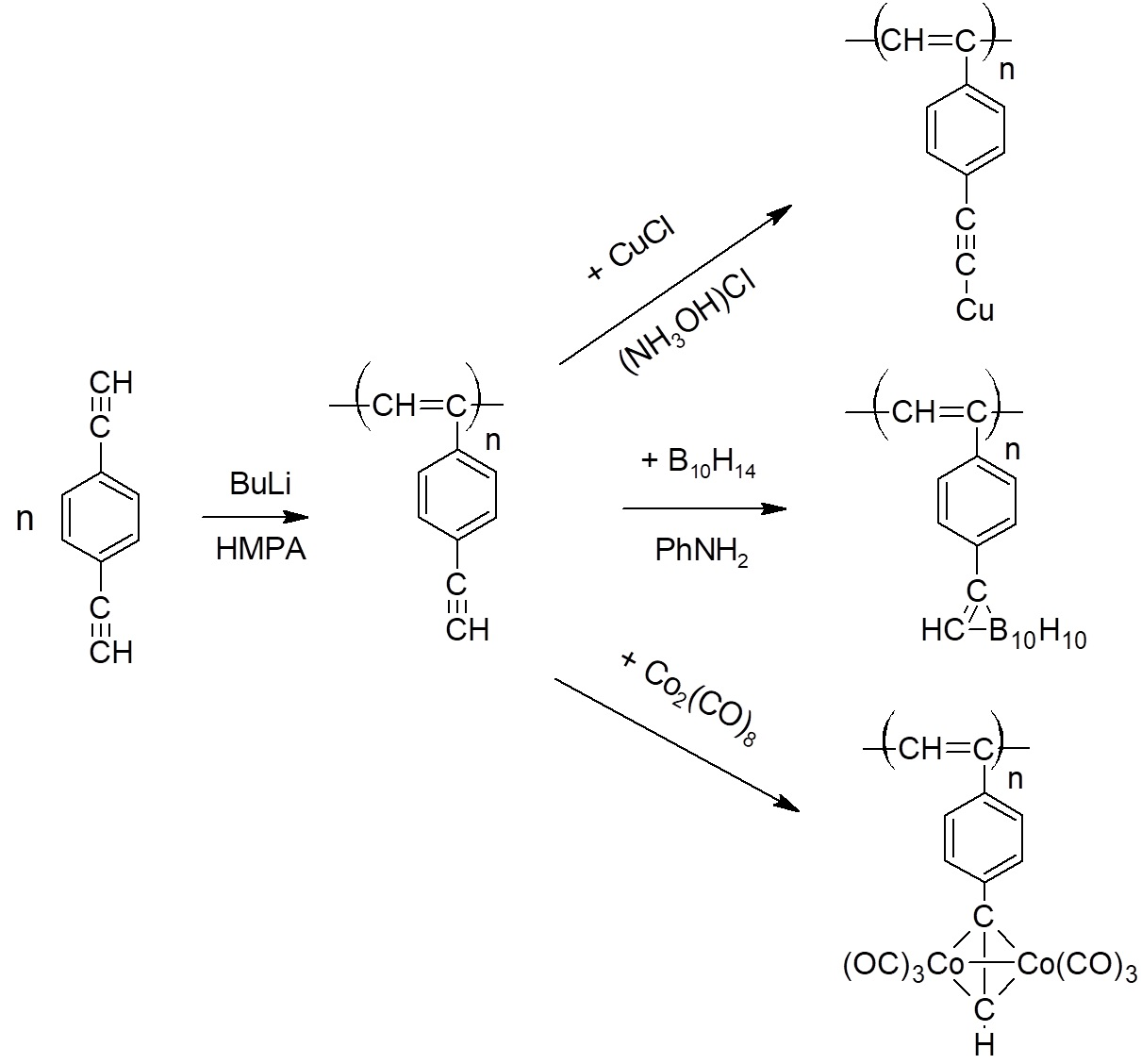
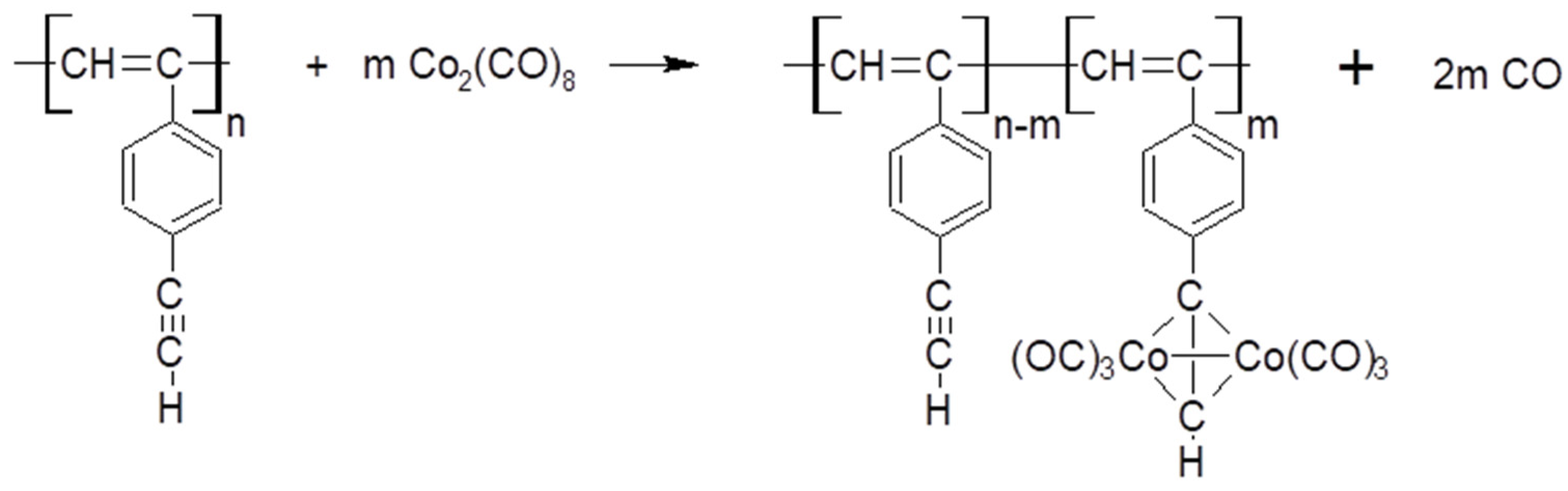
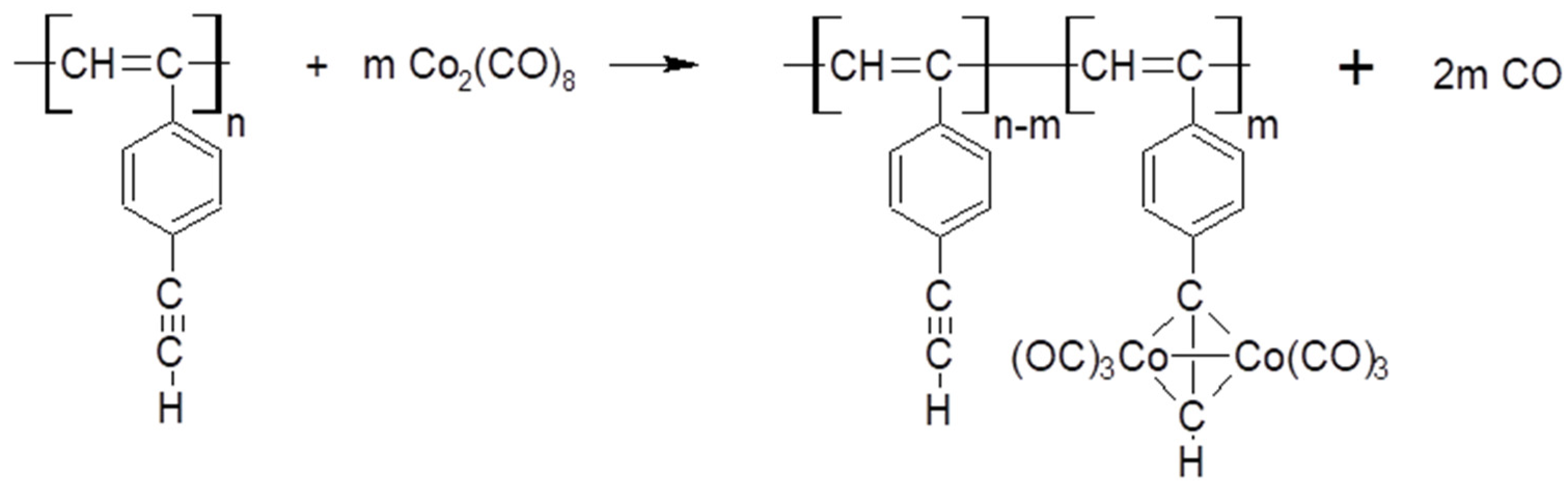
2.4. Synthesis of polyDEB π-Complexes with Co2(CO)8
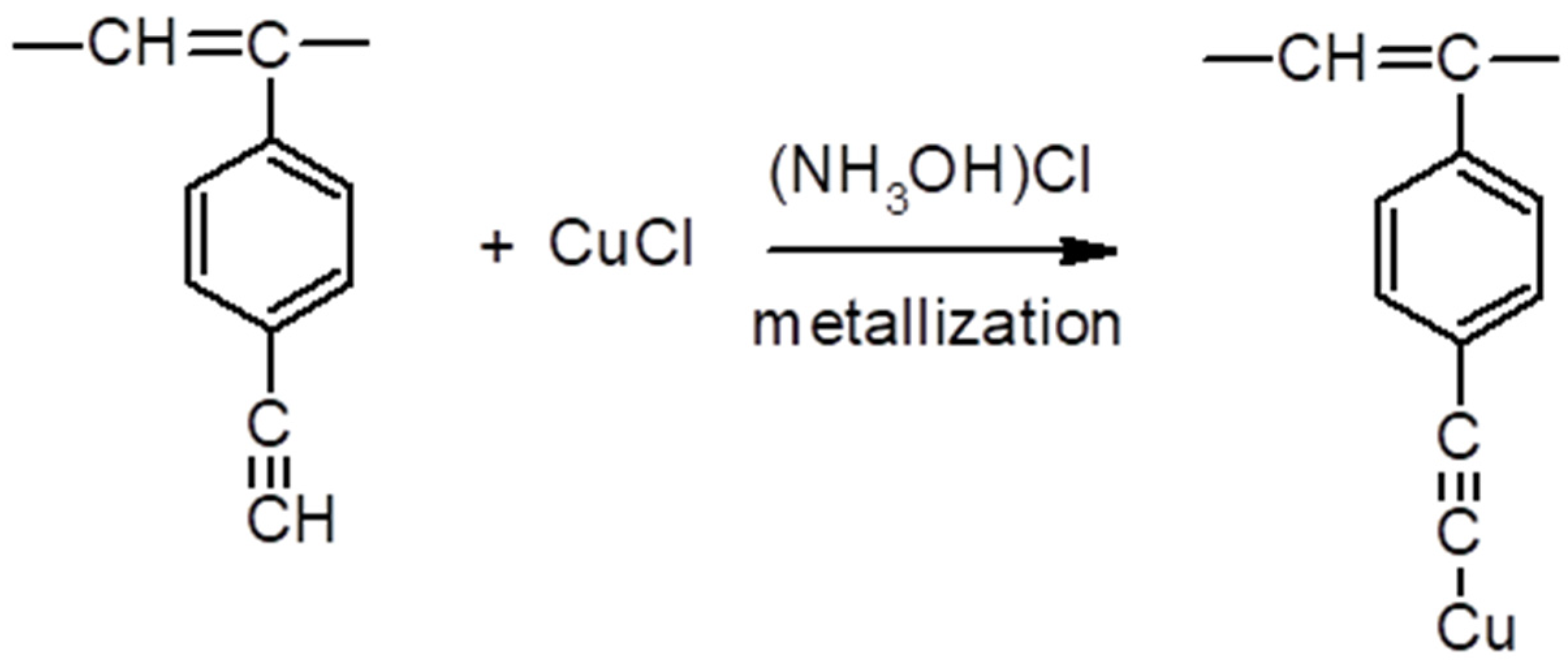
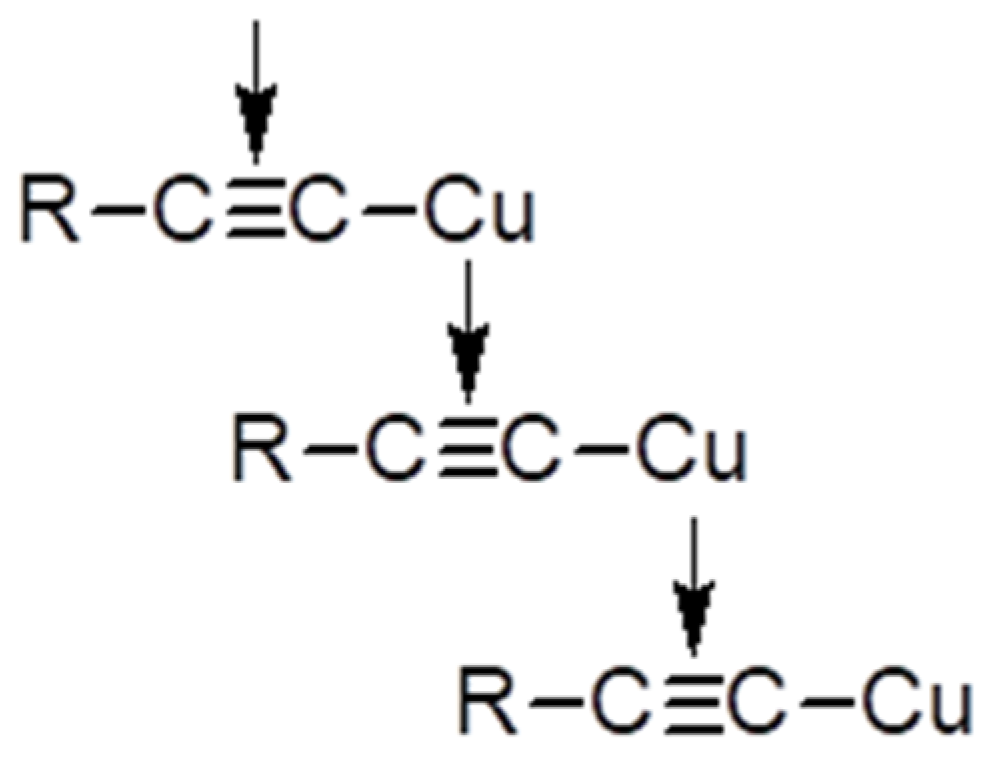
2.5. Synthesis of Polymeric σ-Acetylides of Copper
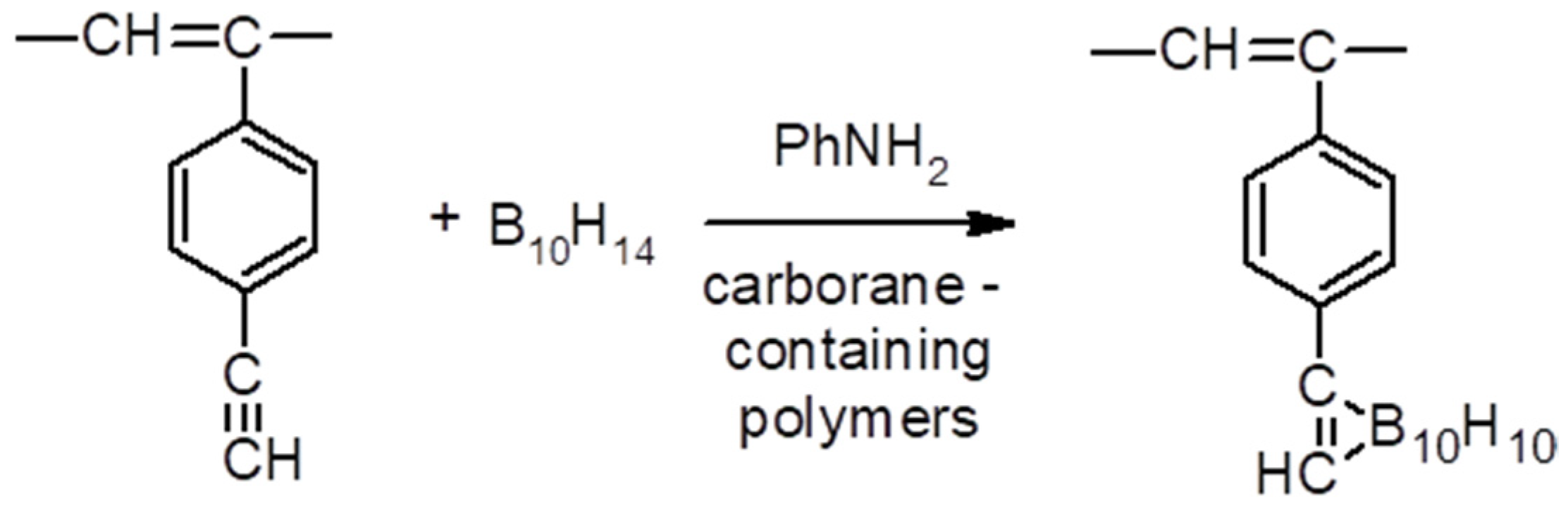
2.6. Synthesis of Carborane-Containing Polymers DEB
3. Results
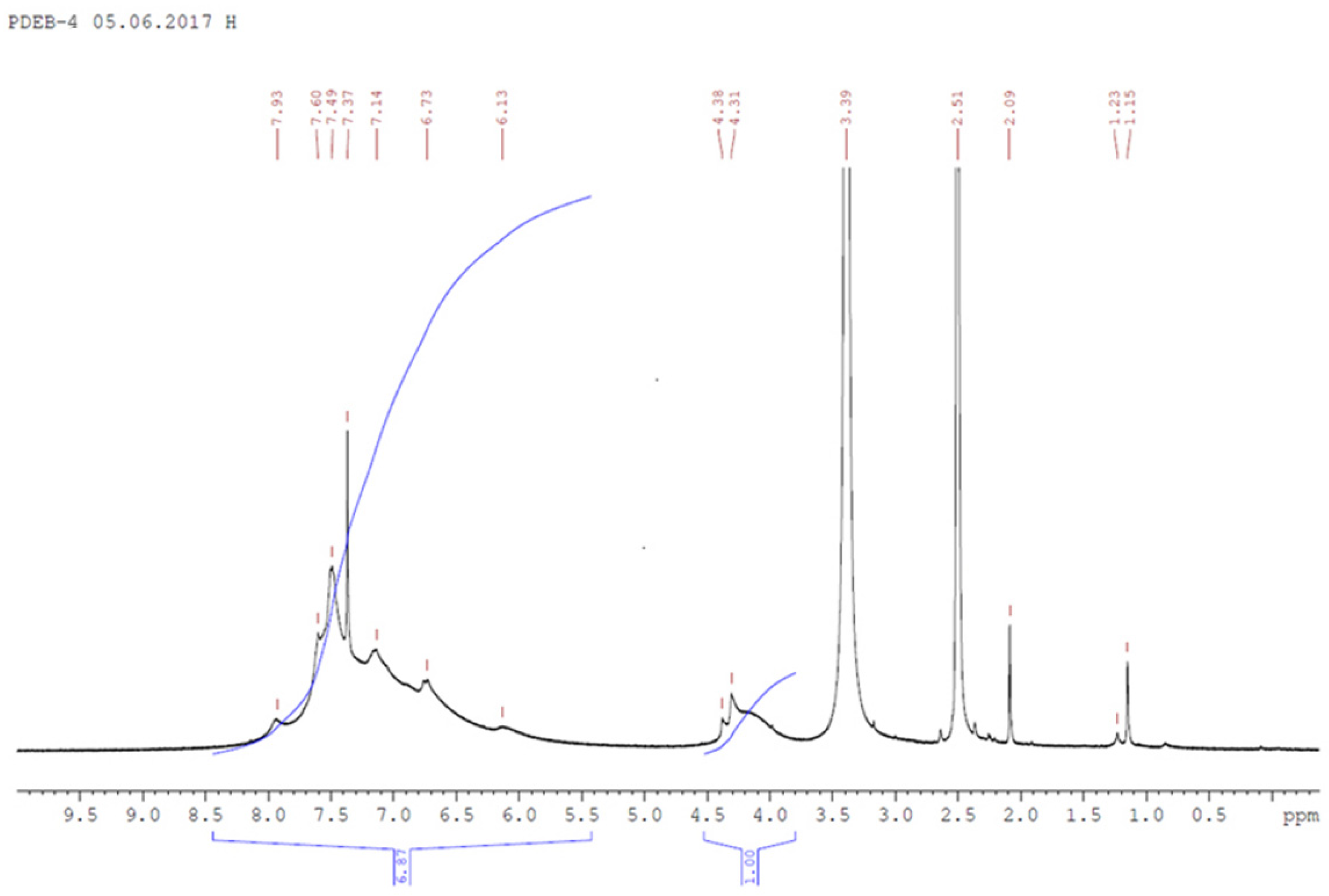
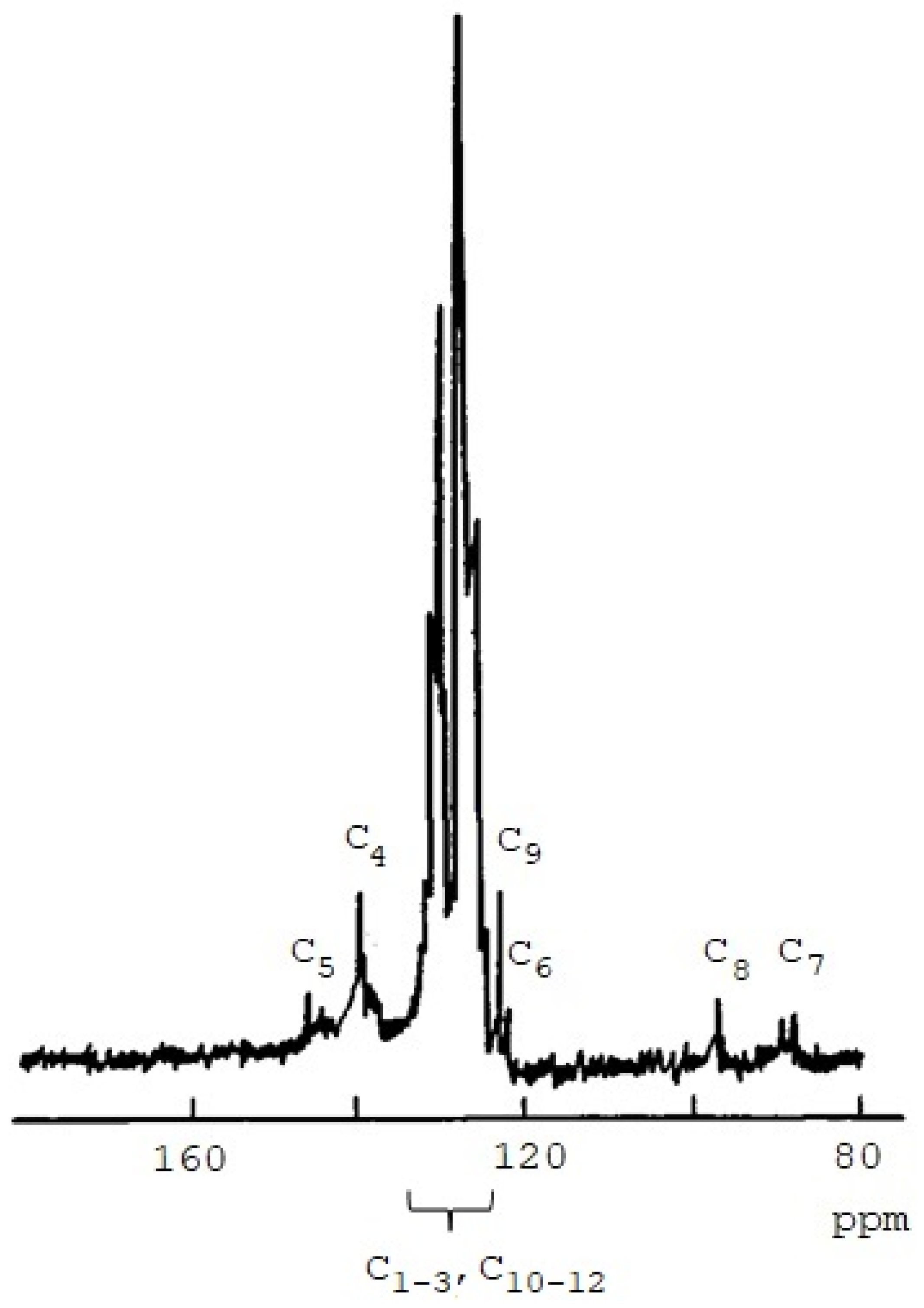
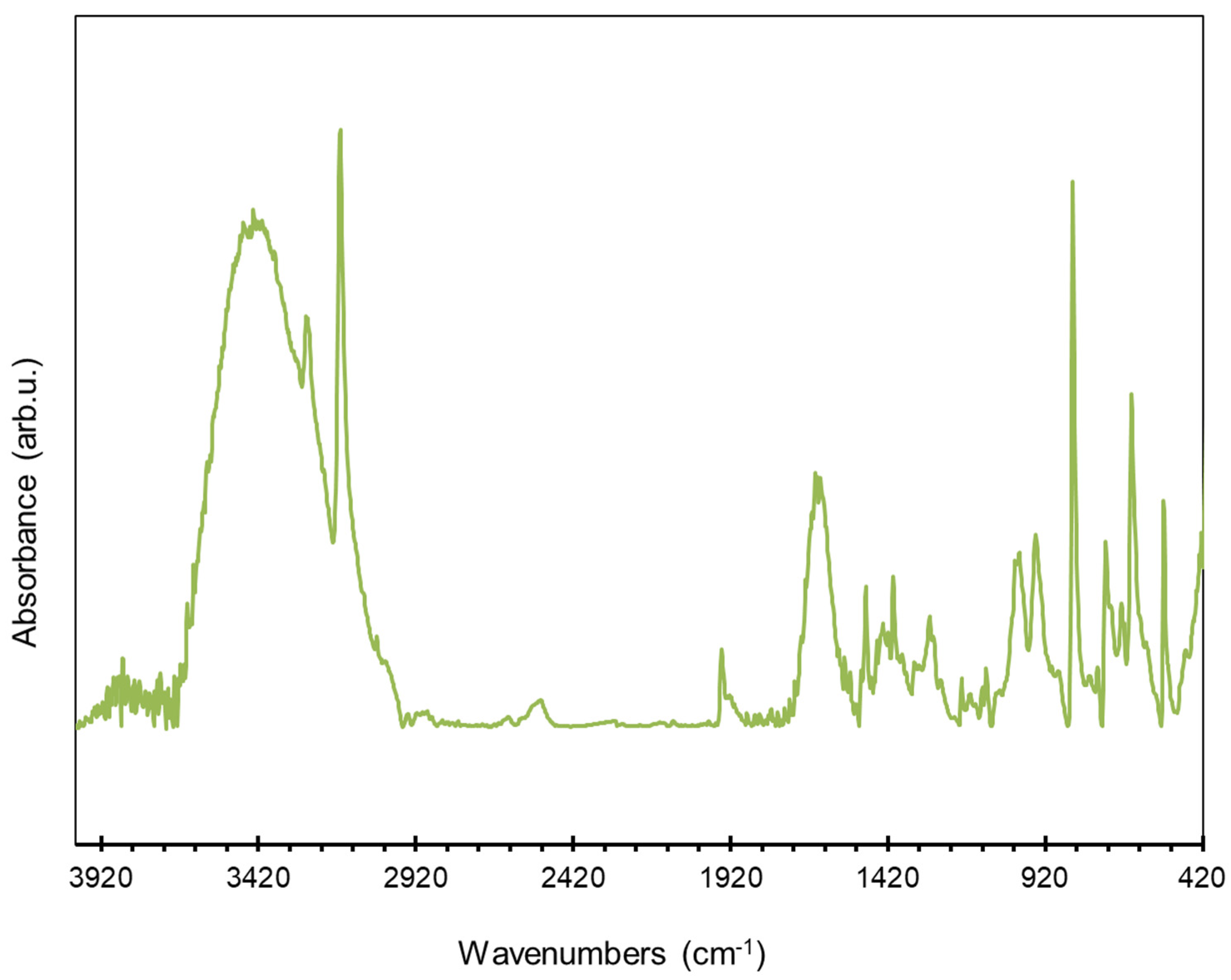
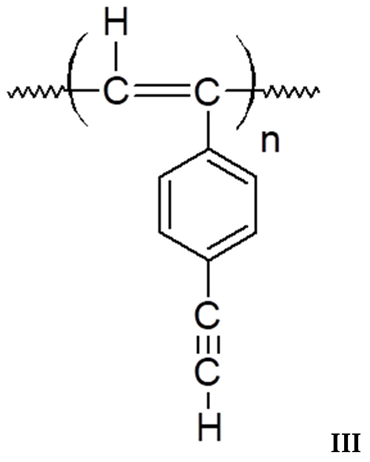
3.1. Synthesis and Characterization of Homo-Polymers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
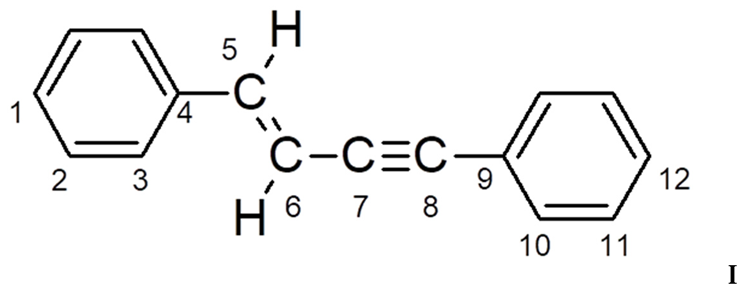
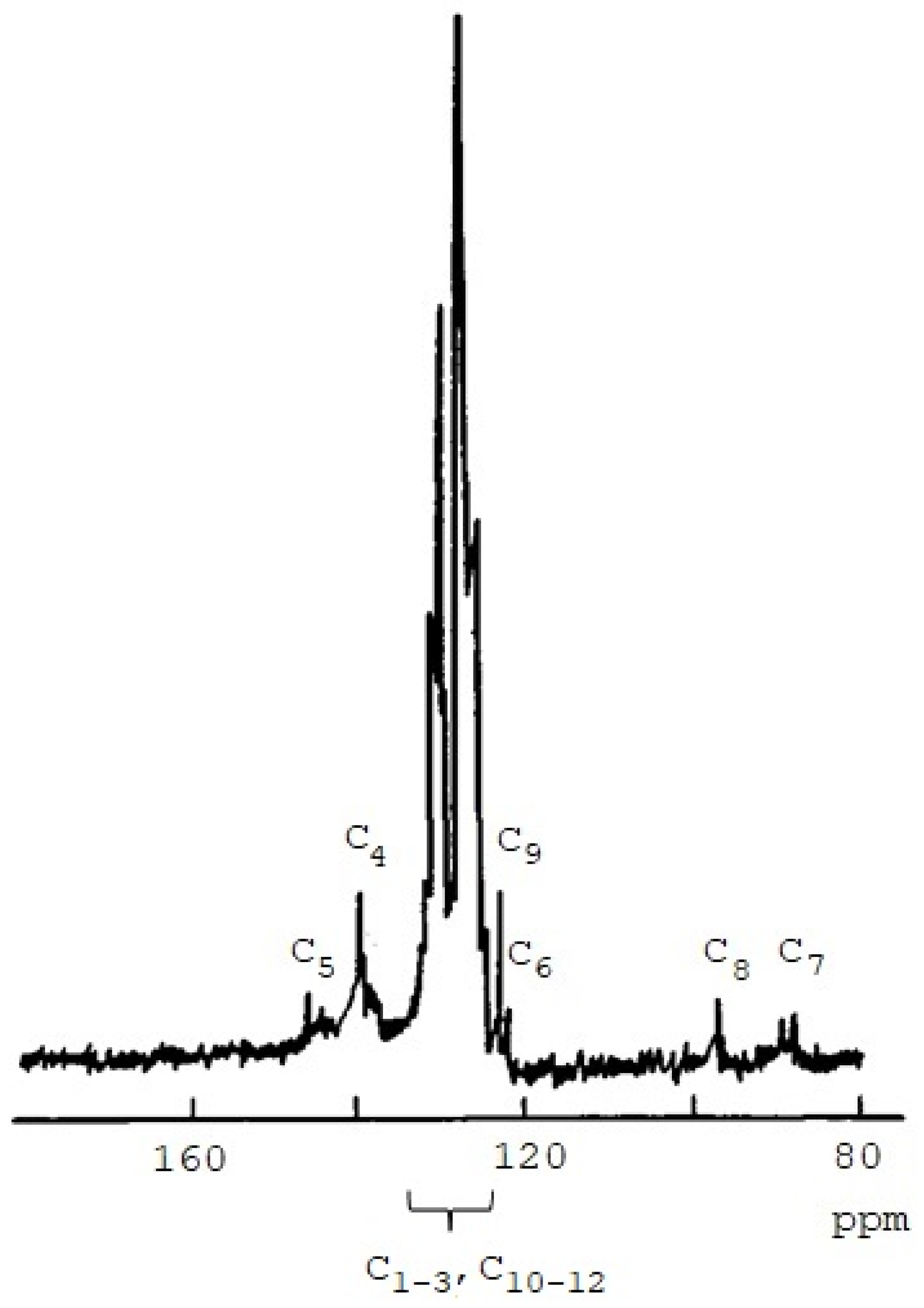
| Atom C | Chemical Shifts δ, ppm | |
|---|---|---|
| Theory [32] | Experiment | |
| C1 | 127.7 | 128.1 |
| C2 | 128.4 | 128.6 |
| C3 | 126.2 | 126.3 |
| C4 | 137.4 | 136.4 |
| C5 | 141.7 | 141.3 |
| C6 | 106.3 | 108.2 |
| C7 | 88.5 | 89.0 |
| C8 | 95.5 | 91.8 |
| C9 | 122.3 | 123.5 |
| C10 | 132.1 | 131.5 |
| C11 | 128.1 | 128.7 |
| C12 | 128.2 | 128.2 |

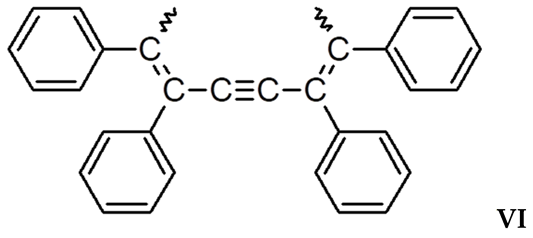
3.2. Synthesis and Characterization of DEB-DPDA Copolymers

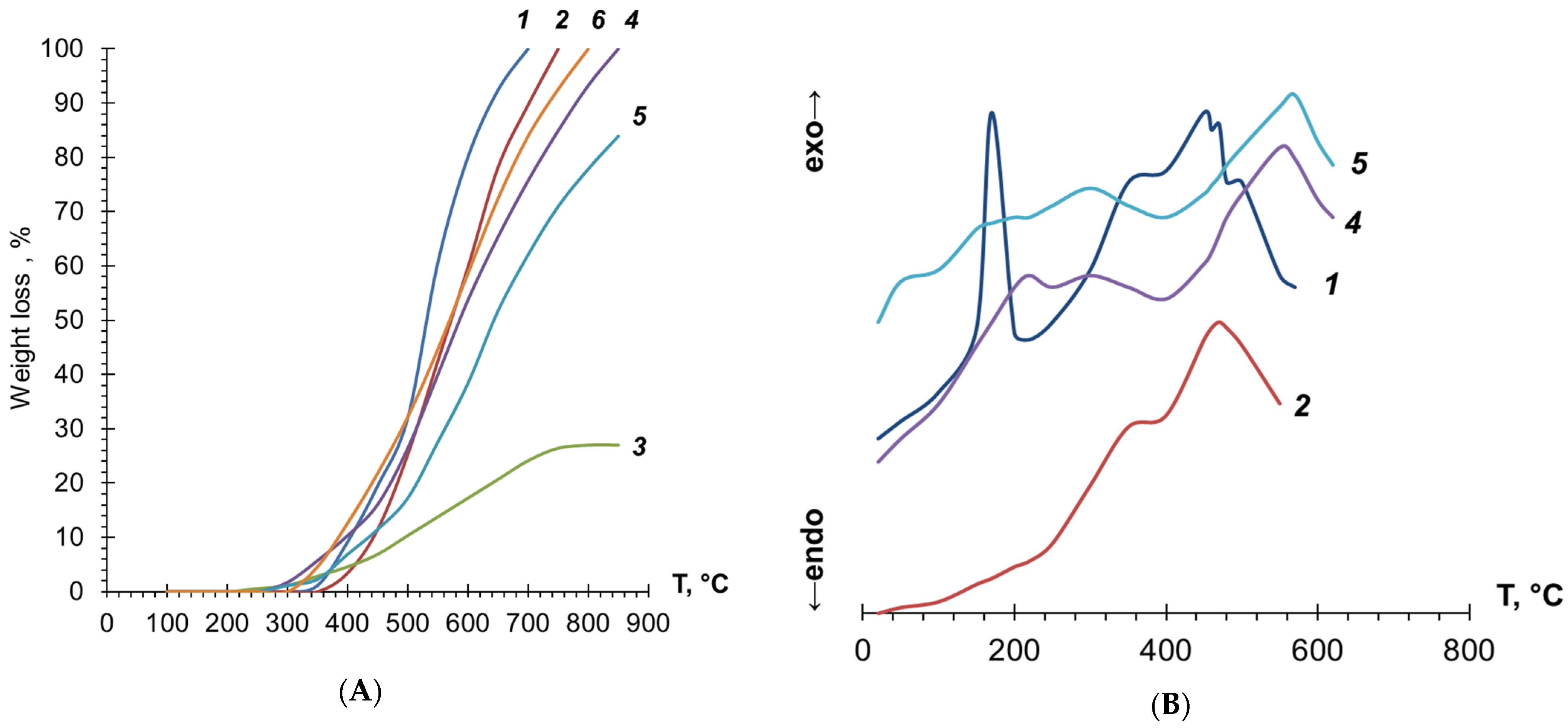
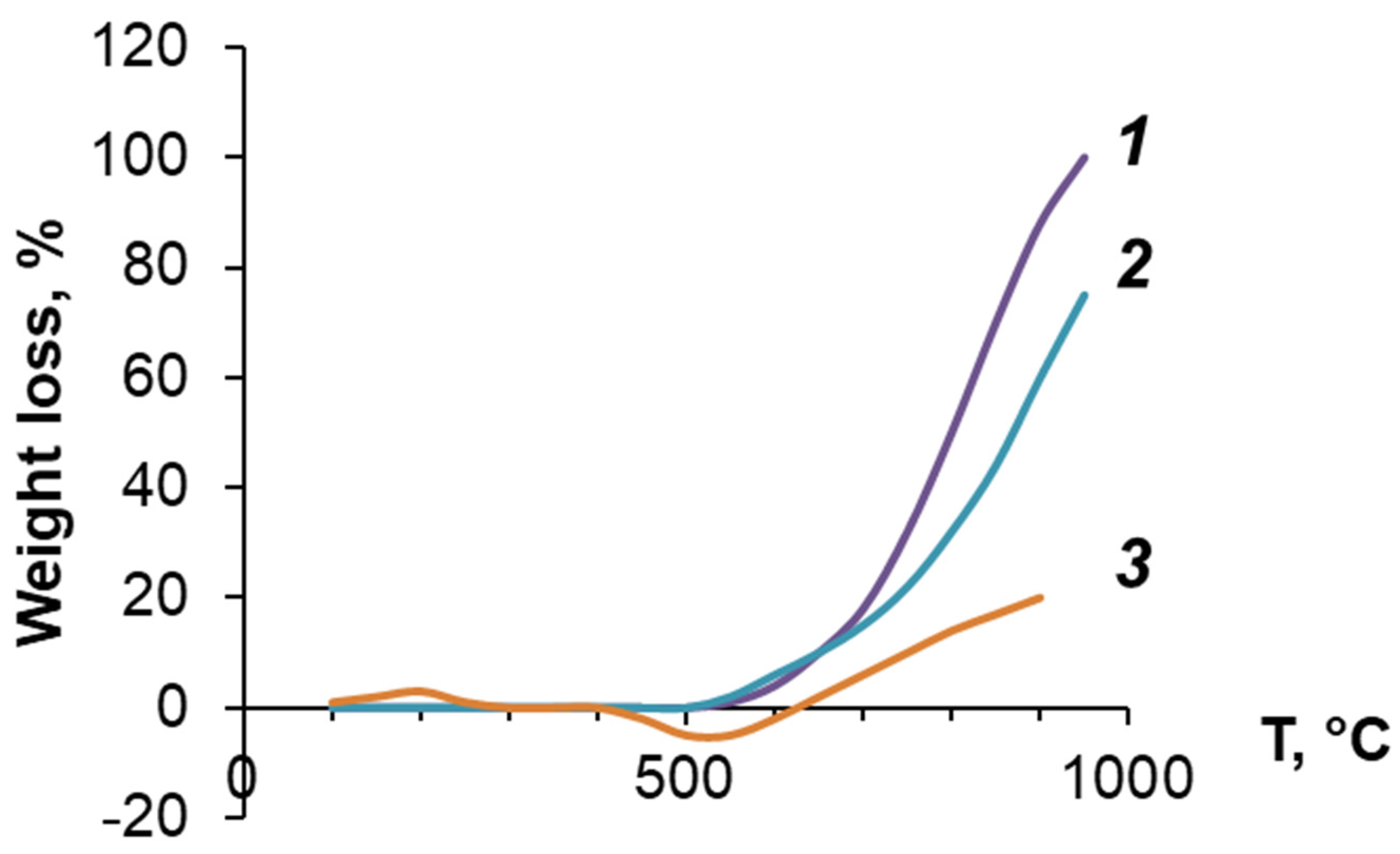
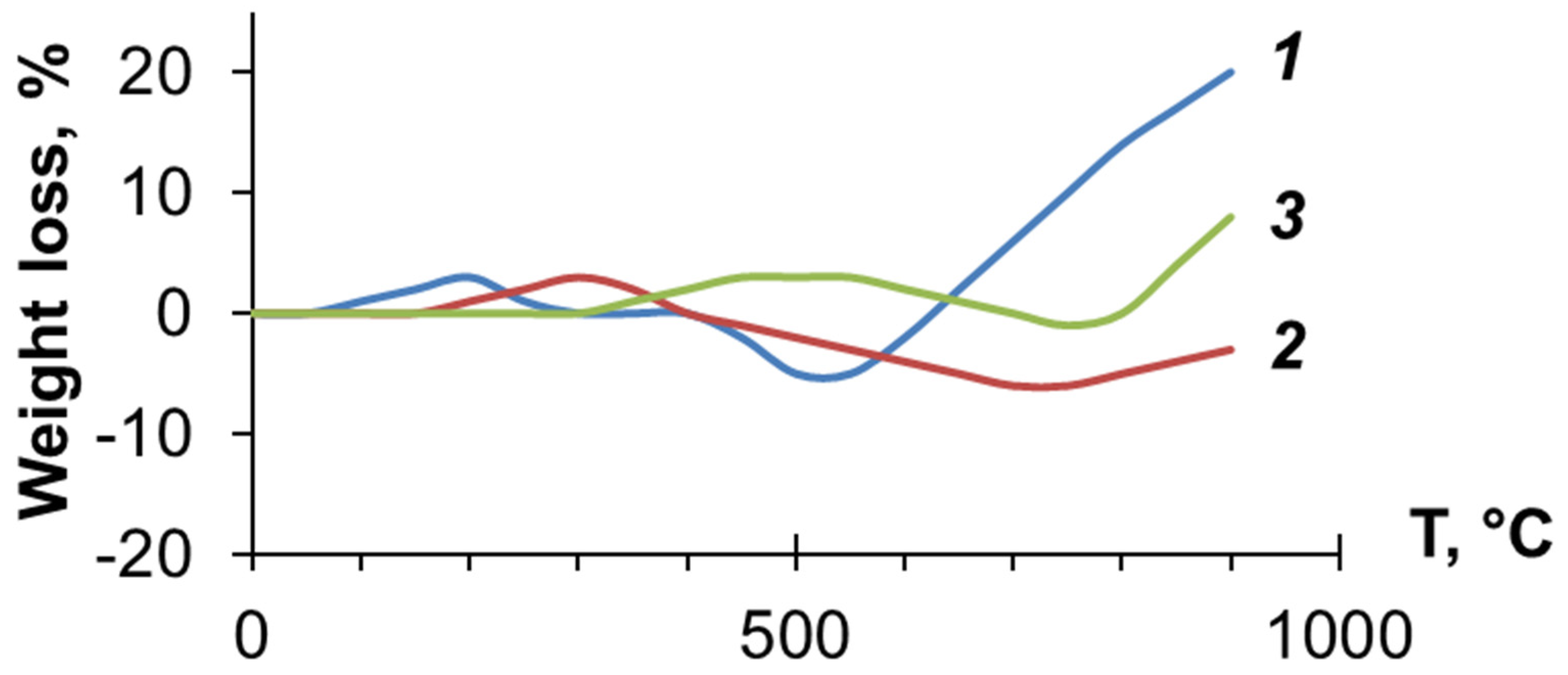
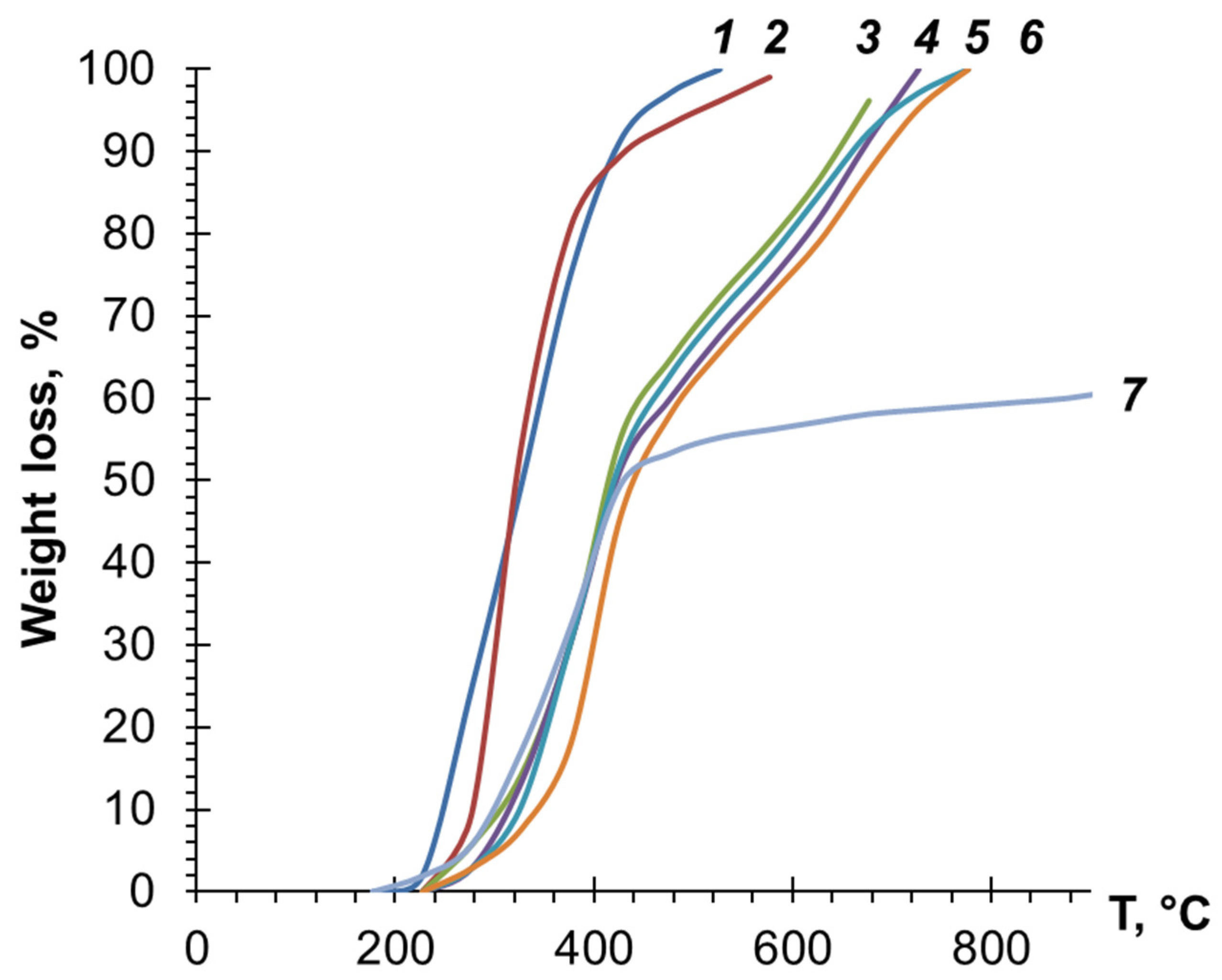
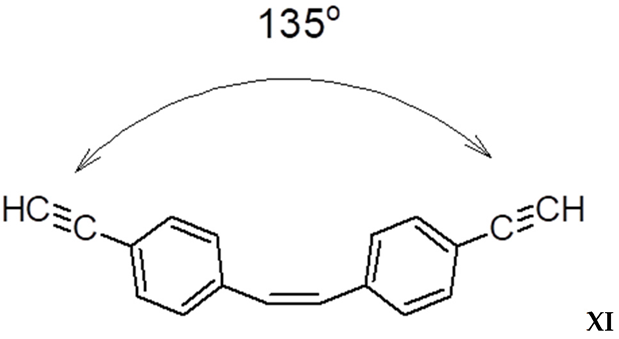
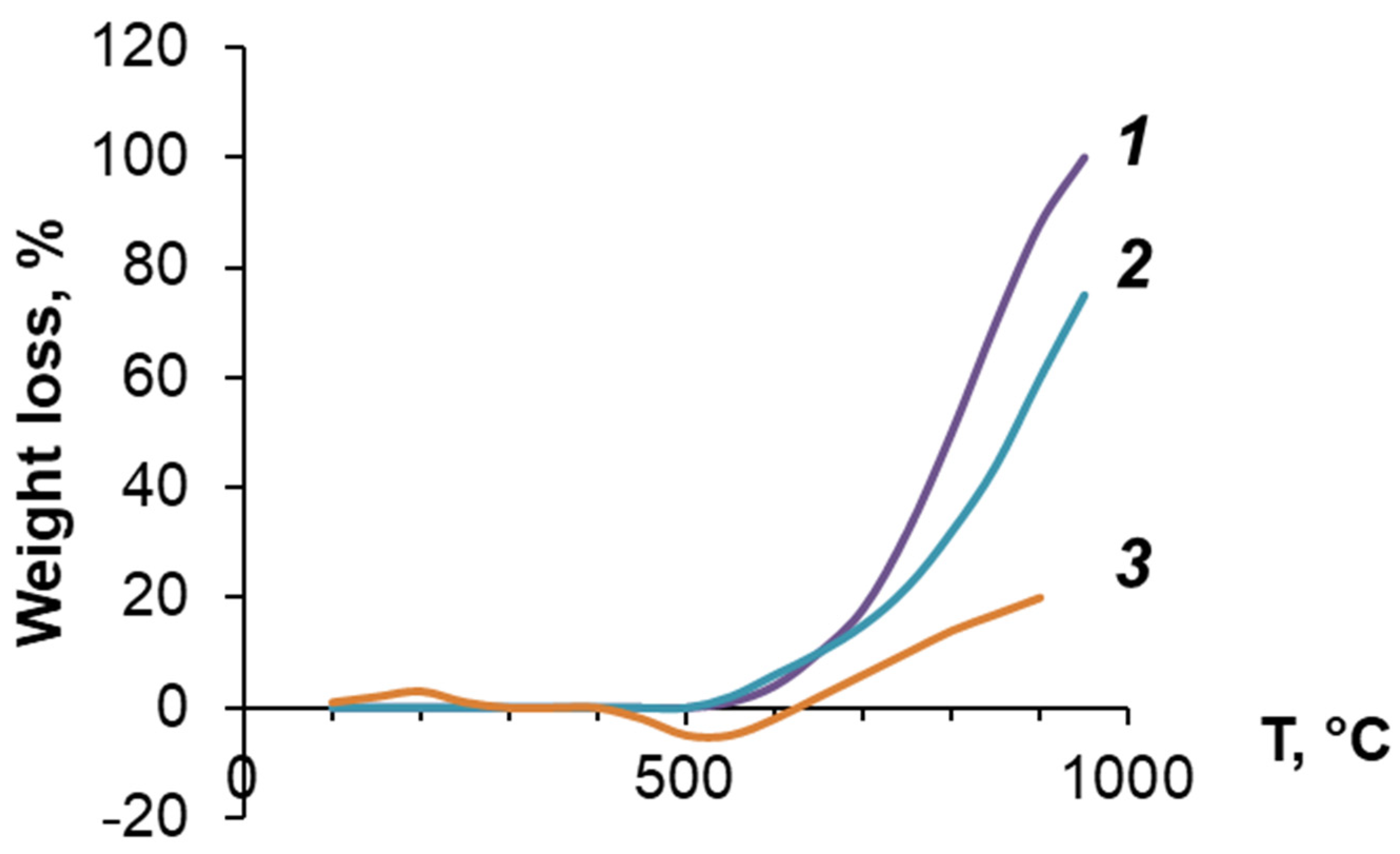
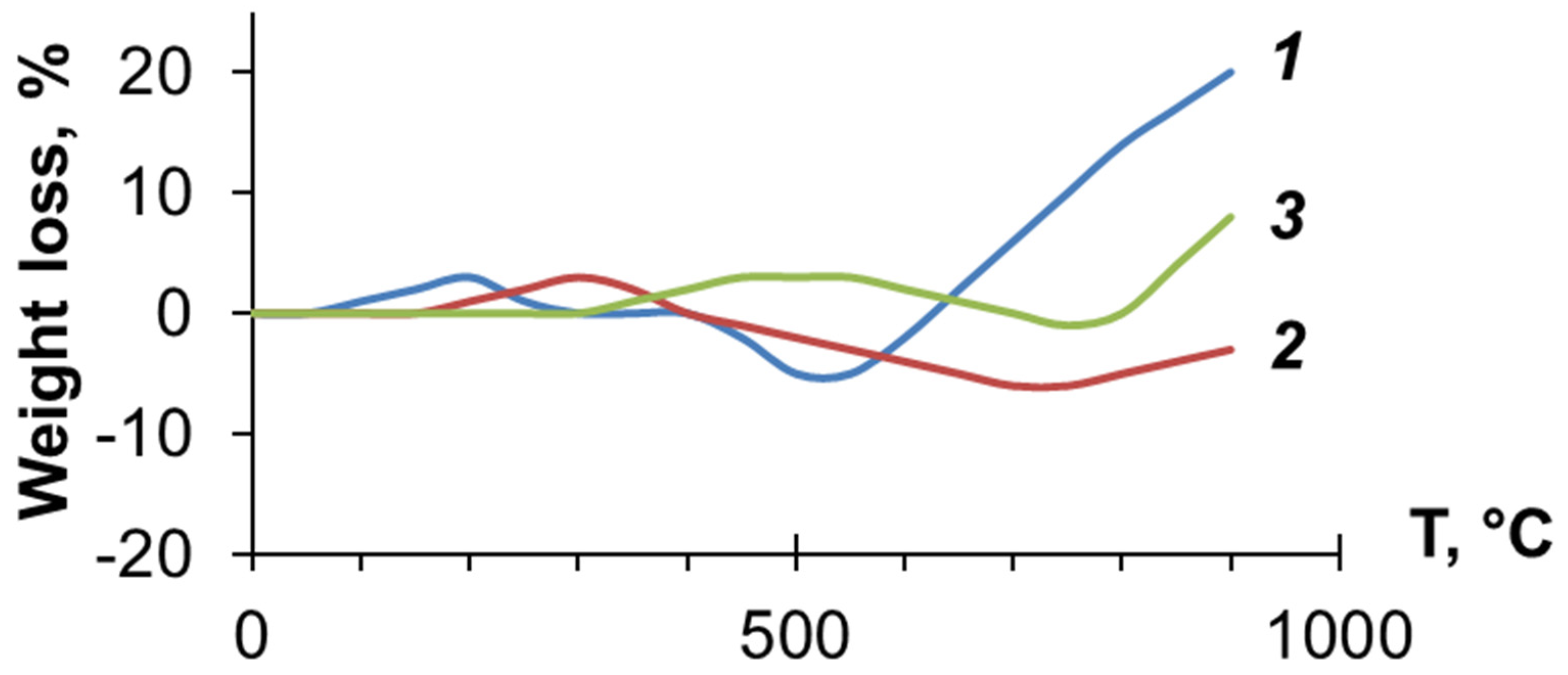
3.3. Investigation of PDEBA and CPA Thermal Degradation
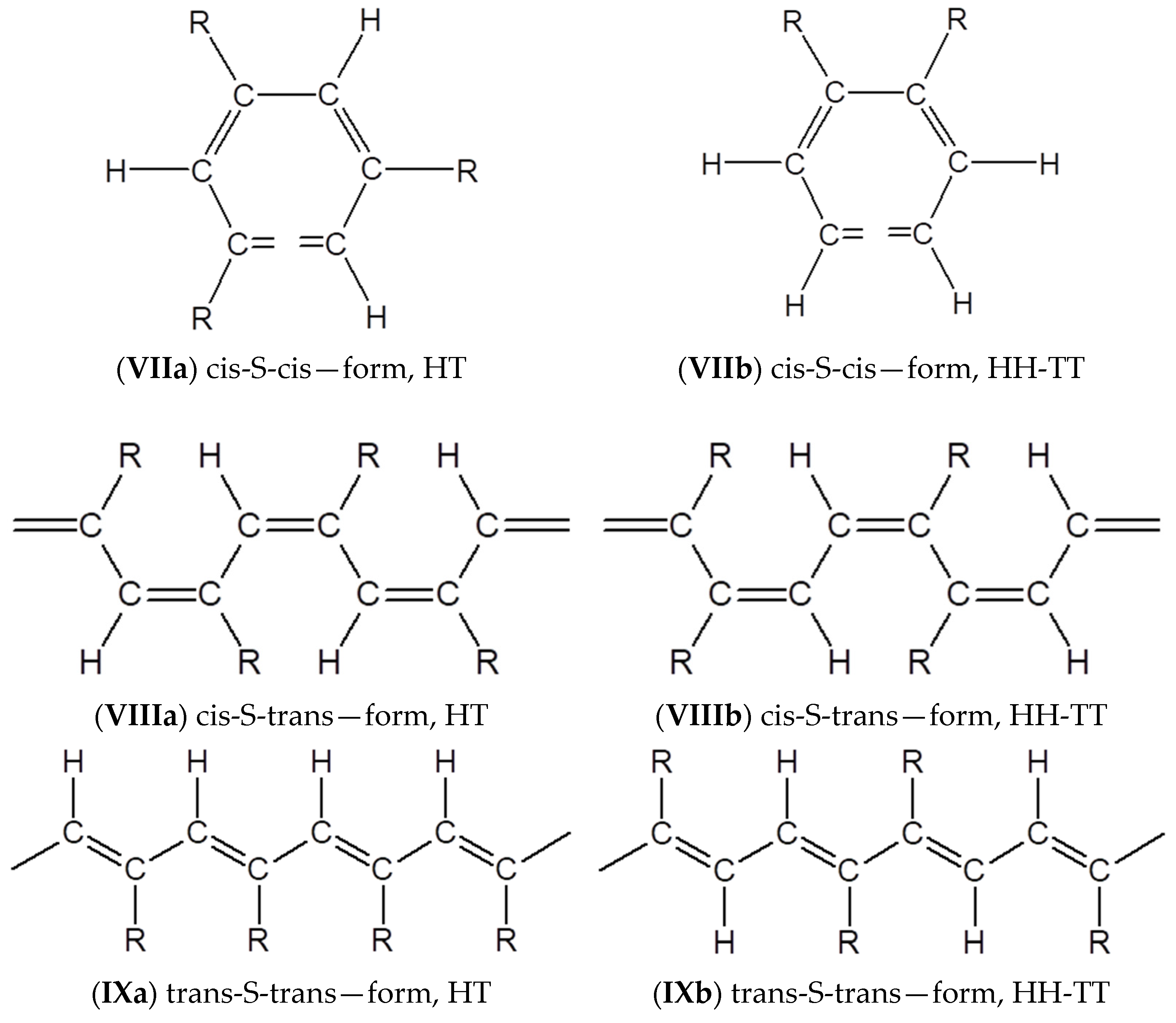
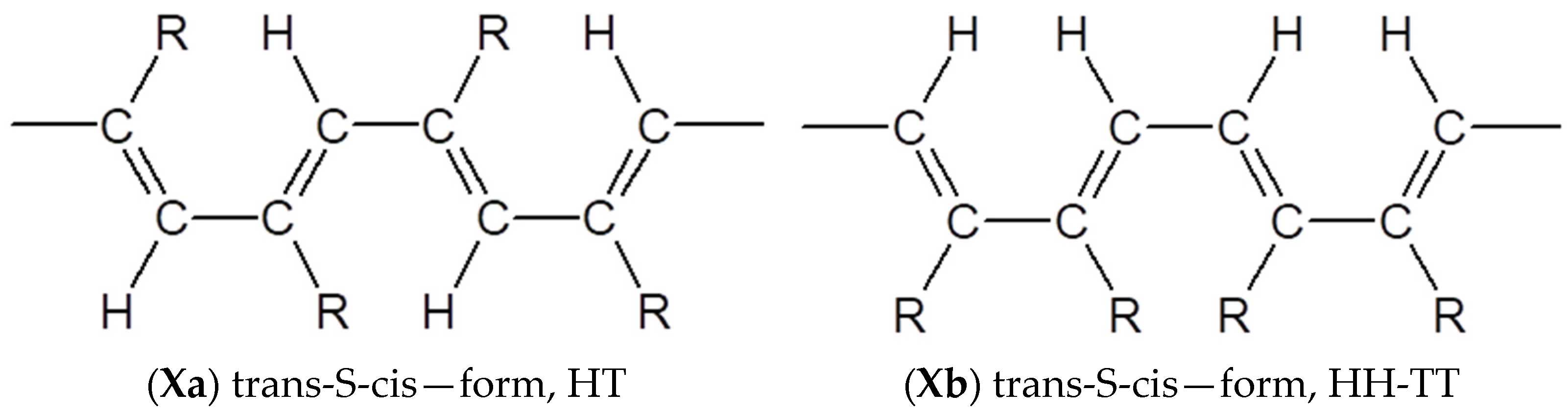
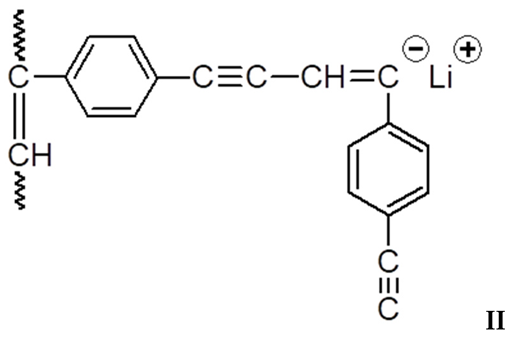
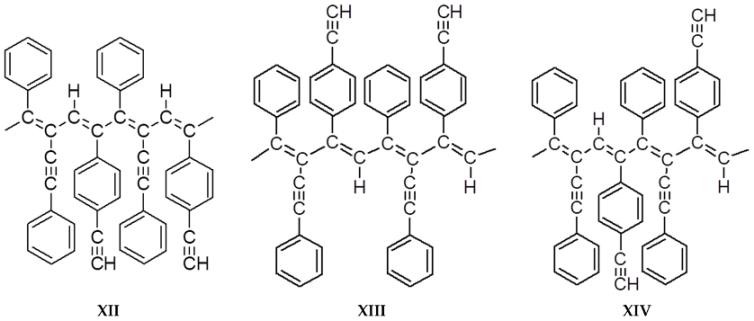
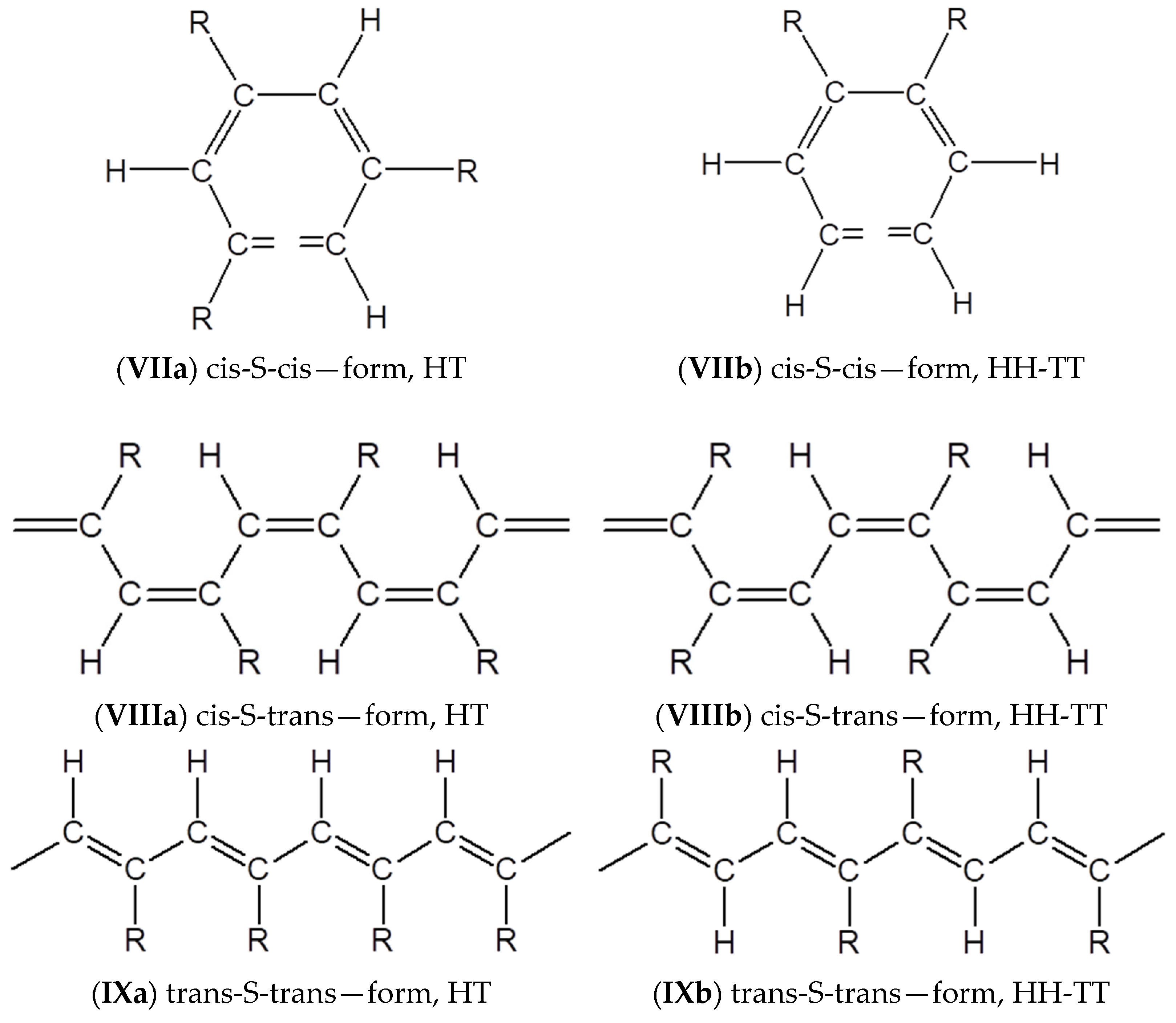
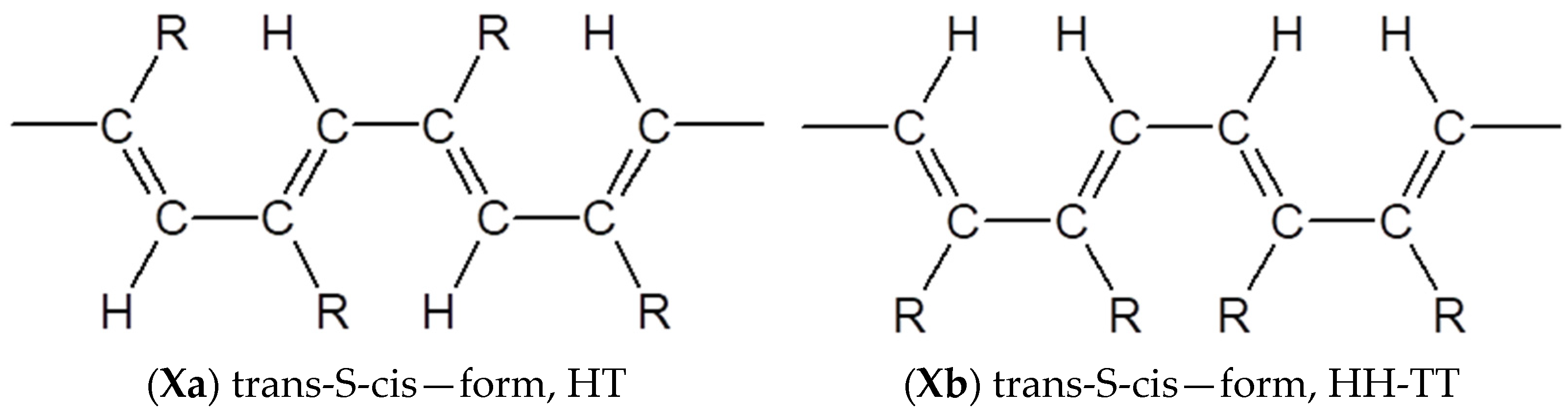
3.4. Steric Features of Linear PDEBA Macromolecules
- formation of cis-S-transoid structure VIII is not possible;
- the trans-S-cisoid structure cannot be realized when connecting the head-head-tail-tail links Xb;
- other types of structures VII, IX, Xa may be formed.
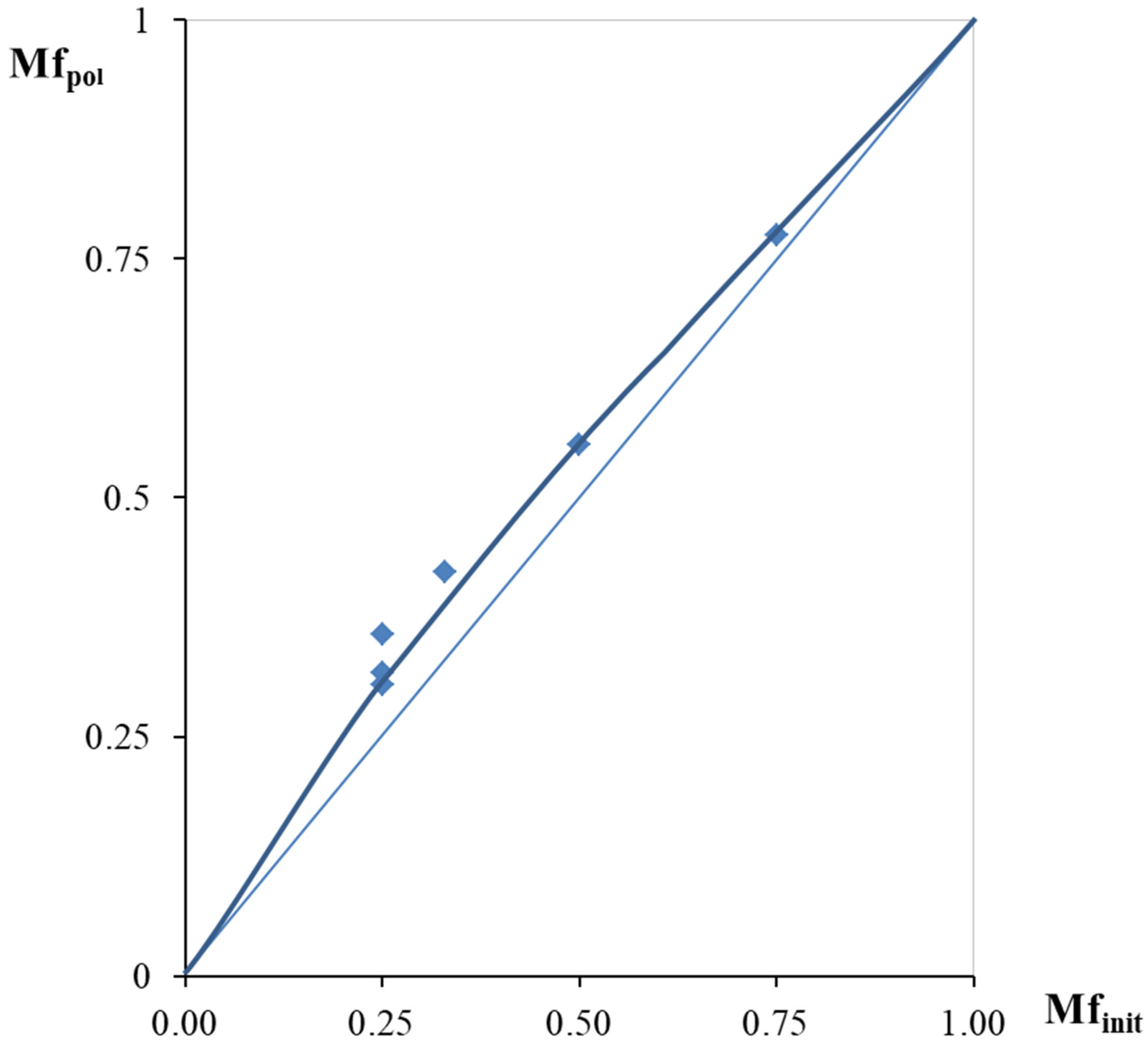
3.5. Steric Features of DEB-DPDA Copolymers Molecules
- the ratio of monomers in the copolymers;
- a different combination of two types of attachment (head-to-tail, head-to-head) for each of the substituents.

- C≡C for –C≡CPh and –PhC≡CH substituents;
- –Ph for –C≡CPh substituents
3.6. Modified Polymers PDEBA
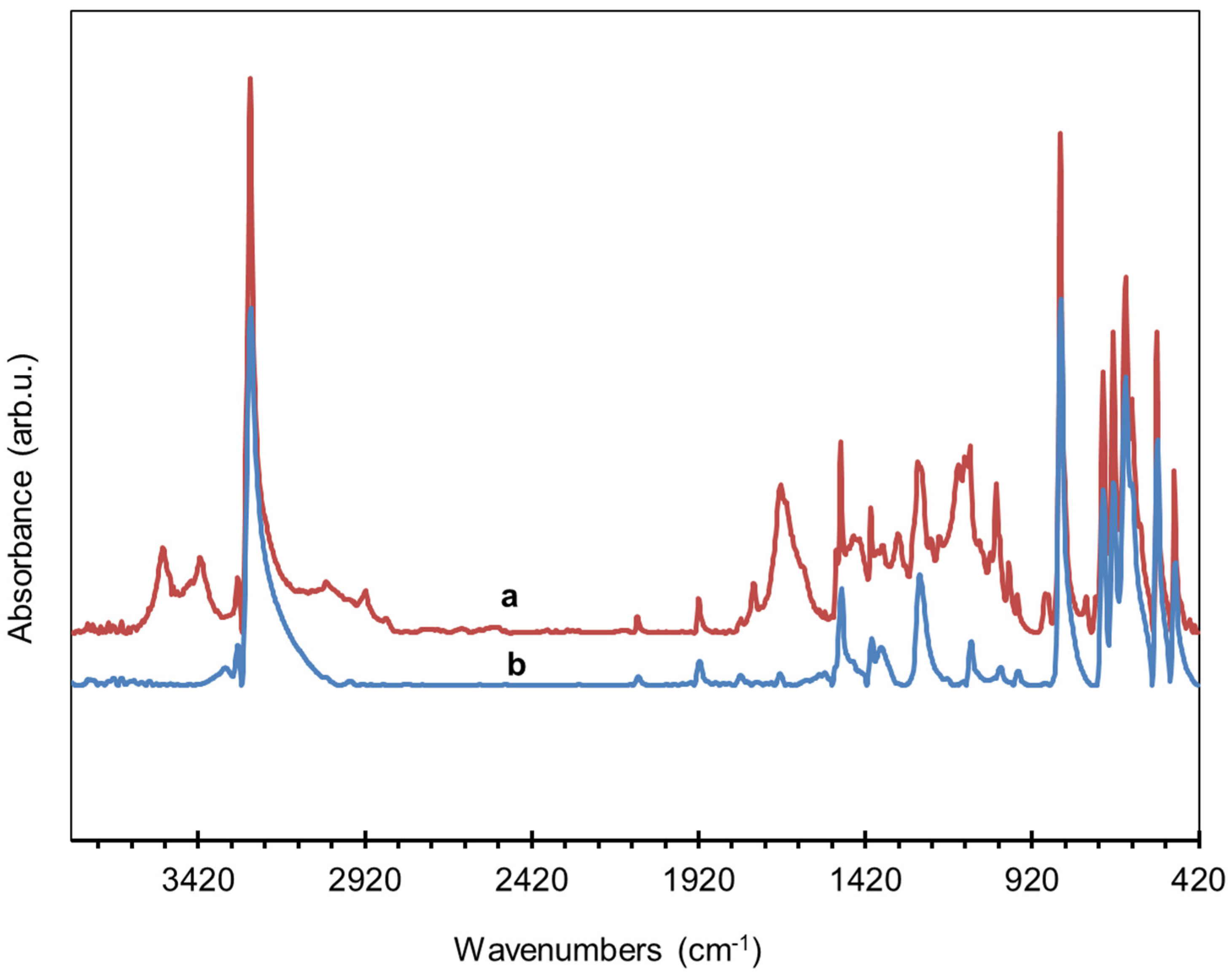
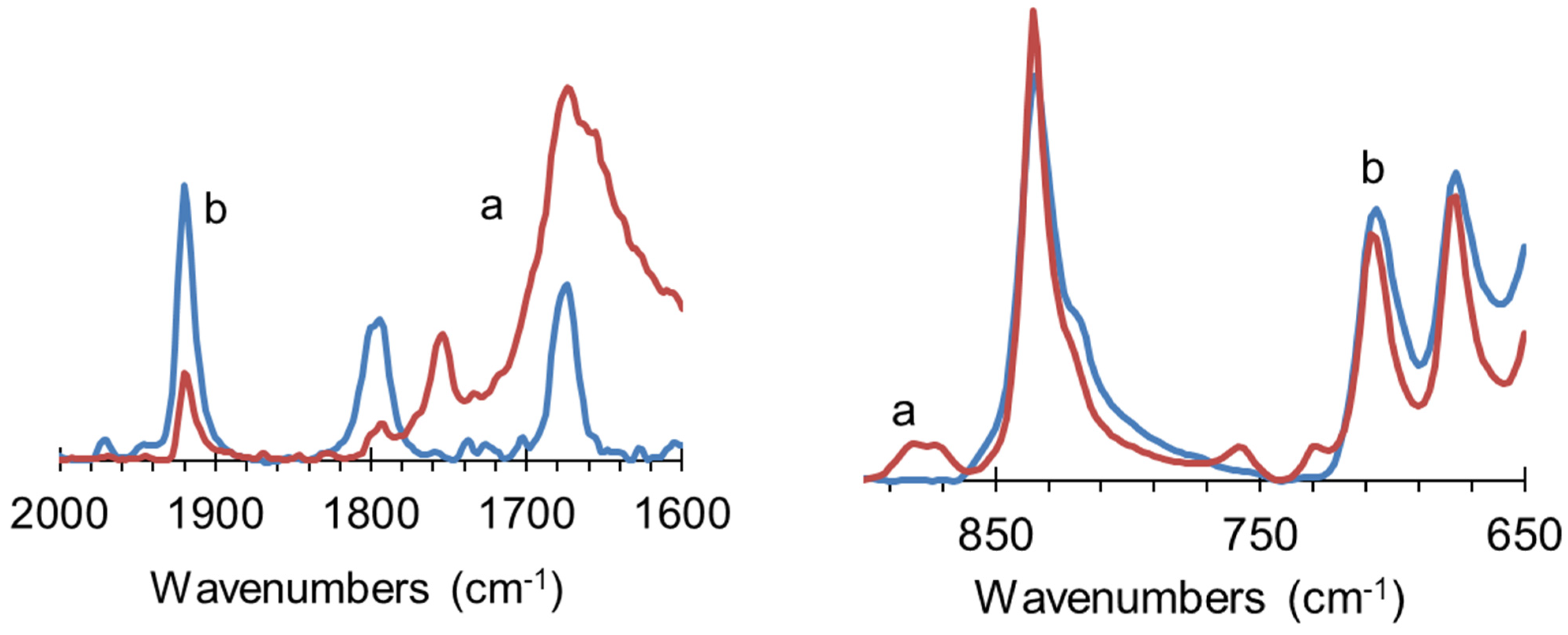
3.6.1. Synthesis of Dicobalt Hexacarbonyl π-Complexes
3.6.2. Copper σ-Acetylides of Poly-p-Diethynylbenzene
- to obtain a soluble copper-containing polymer material capable of forming films;
- create an extended cluster of copper atoms.
3.6.3. Carborane-Containing PDEBA and CPA
3.7. Polymers of DEB as an Industrial Material Modifier
3.7.1. Modification of Industrial Oligoester Acrylates
- TGM-3 CH2=C(CH3)–C(O)–(OCH2CH2)3–O–C(O)–C(CH3)=CH2
- OCM-2 CH2=C(CH3)–C(O)–OCH2CH2OC(O)–(CH2CH2O)2C(O)OCH2CH2O–C(O)C(CH3)=CH2
- the heat resistance on the air of both cured oligoesteracrylates increases significantly with the addition of PDEBA-6;
- the viscosity of compositions with PDEBA-6 increased significantly with the addition of ≥32% PDEBA-6;
- the heat resistance of the cured oligoesteracrylates increased with an increase in the amount of PDEBA-6 added;
- the decrease in mass loss of compositions with PDEBA-6 compared to cross-linked OEA was most significant at the highest temperatures;
- in the case of using PDEBA-6B1, the mass loss of TGM-3 even >1200 °C was less than 60%.
3.7.2. Modification of Industrial Epoxy Novolac Resin
3.7.3. Modification of Oriented Carbon Fibers
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chauser, M.G.; Rodionov, Y.M.; Misin, V.M.; Cherkashin, M.I. Polymerisation of Acetylenes. The Structure and Electrophysical Properties of Polyvinylenes. Russ. Chem. Rev. 1976, 45, 348–374. [Google Scholar] [CrossRef]
- Misin, V.M.; Cherkashin, M.I. The Solid-phase Polymerisation of Monomers with Conjugated Acetylenic Groups. Russ. Chem. Rev. 1985, 54, 562–593. [Google Scholar] [CrossRef]
- Shiotsuki, M.; Sanda, F.; Masuda, T. Polymerization of substituted acetylenes and features of the formed polymers. Polym. Chem. 2011, 2, 1044–1058. [Google Scholar] [CrossRef]
- Liu, Y.; Qin, A.; Tang, B.Z. Polymerizations based on triple-bond building blocks. Prog. Polym. Sci. 2018, 78, 92–138. [Google Scholar] [CrossRef]
- Masuda, T.; Higashimura, T. Polyacetylenes with substituents: Their synthesis and properties. In Catalytical and Radical Polymerization; Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 1986; Volume 81, pp. 121–165. ISBN 978-3-540-16754-9. [Google Scholar]
- Zhang, D.; Zhang, Y. An Overview of Polyarylacetylene as the Resin Matrix for High Temperature Composites. J. Solid Rocket Technol. 2001, 1, 53–59. [Google Scholar]
- Wang, M.; Yu, R.; Li, W.; Li, J.; Feng, Z. Study on Performance of Modified Polyarylacetylene Resins. Aerospace Mater. Technol. 2003, 4, 43–48. [Google Scholar]
- Wang, S.; Li, M.; Gu, Y.; Zhang, Z. Experimental Study on Crack Defects Formation in Polyarylacetylene Composites and Modification Improvement of Resin. J. Compos. Mater. 2010, 44, 3017–3032. [Google Scholar] [CrossRef]
- Chernick, E.T.; Tykwinski, R.R. Carbon-rich nanostructures: The conversion of acetylenes into materials. J. Phys. Org. Chem. 2013, 26, 742–749. [Google Scholar] [CrossRef]
- Rondeau-Gagné, S.; Morin, J.-F. Preparation of carbon nanomaterials from molecular precursors. Chem. Soc. Rev. 2014, 43, 85–98. [Google Scholar] [CrossRef]
- Li, Q.; Yu, R.; Zhu, C.; Jiao, Z. 60Co γ-rays irradiation modified p-diethynylbenzene as prepolymers to prepare polyarylacetylene with excellent heat resistance. Polym. Degrad. Stab. 2015, 114, 81–88. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Y.; Li, A.; Chen, Q.; Shi, X.; Huang, J.; Hu, Z. Carbon Composites. In Composite Materials Engineering, Volume 2; Yi, X.-S., Du, S., Zhang, L., Eds.; Springer: Singapore, 2018; pp. 531–617. ISBN 978-981-10-5689-5. [Google Scholar]
- Cai, W.; Li, M.; Wang, S.; Gu, Y.; Li, Q.; Zhang, Z. Strong, flexible and thermal-resistant CNT/polyarylacetylene nanocomposite films. RSC Adv. 2016, 6, 4077–4084. [Google Scholar] [CrossRef]
- Jiang, Z.X.; Huang, Y.D.; Liu, L.; Long, J. Effects of roughness on interfacial performances of silica glass and non-polar polyarylacetylene resin composites. Appl. Surf. Sci. 2007, 253, 9357–9364. [Google Scholar] [CrossRef]
- Domínguez, C.; Metz, K.M.; Hoque, M.K.; Browne, M.P.; Esteban-Tejeda, L.; Livingston, C.K.; Lian, S.; Perova, T.S.; Colavita, P.E. Continuous Flow Synthesis of Platinum Nanoparticles in Porous Carbon as Durable and Methanol-Tolerant Electrocatalysts for the Oxygen Reduction Reaction. ChemElectroChem 2018, 5, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Yang, M.; Ling, M.; Zhu, Y. Surface acoustic wave humidity sensors based on poly(p-diethynylbenzene) and sodium polysulfonesulfonate. Sens. Actuators, B 2007, 122, 560–563. [Google Scholar] [CrossRef]
- Slováková, E.; Zukal, A.; Brus, J.; Balcar, H.; Brabec, L.; Bondarev, D.; Sedláček, J. Transition-Metal-Catalyzed Chain-Growth Polymerization of 1,4-Diethynylbenzene into Microporous Crosslinked Poly(phenylacetylene)s: The Effect of Reaction Conditions. Macromol. Chem. Phys. 2014, 215, 1855–1869. [Google Scholar] [CrossRef]
- Misin, V.M.; Cherkashin, M.I. Synthesis of linear soluble polymer of para-diethynylbenzene. Vysokomol. Soedin. Ser. B 1981, 23, 130–131. [Google Scholar]
- Hay, A. Oxidative Coupling of Acetylenes. J. Org. Chem. 1960, 25, 1275–1276. [Google Scholar] [CrossRef]
- Kern, R.J. Preparation and properties of isomeric polyphenylacetylenes. J. Polym. Sci. A-1 Polym. Chem. 1969, 7, 621–631. [Google Scholar] [CrossRef]
- Sazanov, Y.N.; Dobrovol’skaya, I.P.; Lysenko, V.A.; Sal’nikova, P.Y.; Kosyakov, D.S.; Pokryshkin, S.A.; Fedorova, G.N.; Kulikova, E.M. Thermochemical structural transformations of polyoxadiazoles. Russ. J. Appl. Chem. 2015, 88, 1304–1310. [Google Scholar] [CrossRef]
- Guimarães, C.J.B.; de Aguiar, A.P.; Castro, A.T. de Accurate measurement of pitch-based carbon fiber electrical resistivity. Polímeros 2021, 31, e2021011. [Google Scholar] [CrossRef]
- Misin, V.M.; Kisilitsa, P.P.; Bolondayeva, N.I.; Cherkashin, M.I. Anionic polymerization of tolane and of diphenylbutadi-yne. Polym. Sci. USSR 1976, 18, 1973–1981. [Google Scholar] [CrossRef]
- Glagolev, N.N.; Misin, V.M.; Cherkashin, M.I. Method for Obtaining a Soluble Linear Polymer of P-diethylbenzene. USSR Patent SU-910662-A1, 15 July 1980. issued 1981. [Google Scholar]
- Berlin, A.A.; Kadantseva, A.I.; Mukhin, M.A.; Ivanov, A.A. Investigation of the anionic polymerization of phenylacetylene in the presence of lithium initiators. Polym. Sci. U.S.S.R. 1975, 17, 942–947. [Google Scholar] [CrossRef]
- Zaliznaya, N.F.; Geiderikh, M.A.; Davydov, B.E.; Kubasova, N.A. The effect of polymers with a system of conjugated bonds on the anionic polymerization of acetylene hydrocarbons. Vysokomol. Soedin. Ser. B 1981, 23, 33–35. [Google Scholar]
- Dyer, J.R. Applications of Absorption Spectroscopy of Organic Compounds; Prentice-Hall: Englewood Cliffs, NJ, USA; London, UK, 1965. [Google Scholar]
- Sedláček, J.; Balcar, H. Substituted Polyacetylenes Prepared with Rh Catalysts: From Linear to Network-Type Conjugated Polymers. Polym. Rev. 2017, 57, 31–51. [Google Scholar] [CrossRef]
- Donda, A.F.; Cervone, E.; Biancifiori, M.A. Cyclic trimerization of phenylacetylene with the catalytic system TiCl4/Al(C2H5)3. Recl. Trav. Chim. Pays-Bas 2010, 81, 585–590. [Google Scholar] [CrossRef]
- Szwarc, M.; Van Beylen, M. Ionic Polymerization and Living Polymers; Springer: Dordrecht, The Netherlands, 1993; ISBN 978-94-010-4649-7. [Google Scholar]
- Geiderikh, M.A.; Davydov, B.E.; Zaliznaya, N.F.; Minayeva, V.S. Kinetics of the anionic polymerization of phenylacetylene. Polym. Sci. USSR 1976, 18, 1451–1457. [Google Scholar] [CrossRef]
- Pretsch, E.; Bühlmann, P.; Affolter, C. Structure Determination of Organic Compounds Tables of Spectral Data; Springer: Berlin/Heidelberg, Germany, 2000; ISBN 978-3-662-04201-4. [Google Scholar]
- Percéc, V.; Rinaldi, P.L. A 13C-NMR study of the microstructure of polyphenylacetylenes prepared with MoCl5 and WCl6. Polym. Bull. 1983, 9, 548–555. [Google Scholar] [CrossRef]
- Glagolev, N.N.; Misin, V.M.; Zaichenko, N.L.; Khandozhko, V.N.; Kolobova, N.Y.; Cherkashin, M.I. Polymerization of diphenyldiacetylene and of tolan in the presence of carbonyl cobalt complexes. Structure and properties of the polymer. Polym. Sci. USSR 1986, 28, 2359–2366. [Google Scholar] [CrossRef]
- Berlin, A.A.; Vakulskaya, T.I.; Zadontsev, B.G.; Chauser, M.G.; Cherkashin, M.I.; Chibrikin, V.M.; Chigir, A.N. Hyperfine structure of EPR spectra of nitropolyarylvinylenes. Dokl. Akad. Nauk SSSR 1968, 182, 581–584. [Google Scholar]
- Fineman, M.; Ross, S.D. Linear method for determining monomer reactivity ratios in copolymerization. J. Polym. Sci. 1950, 5, 259–262. [Google Scholar] [CrossRef]
- Müller, H.; Eckhardt, C.J.; Chance, R.R.; Baughman, R.H. Optical and electrical properties of a polydiacetylene crystal: Poly(5,7-dodecadiyne-1,12-diol-bis phenylurethane). Chem. Phys. Lett. 1977, 50, 22–25. [Google Scholar] [CrossRef]
- Babbitt, G.E.; Patel, G.N. Carbon-13 nuclear magnetic resonance studies on soluble poly(diacetylenes). Macromolecules 1981, 14, 554–557. [Google Scholar] [CrossRef]
- Glagolev, N.N.; Misin, V.M.; Cherkashin, M.I. Synthesis of Organometallic Acetylene Compounds Based on an Oligomer with a Free Ethynyl Group; USSR: Yerevan, Armenia, 1984; p. 211. [Google Scholar]
- Misin, V.M.; Glagolev, N.N.; Misin, M.V. Material Based on Poly-p-diethynylbenzene for Protection from Electromagnetic Fields; Ivanovo State Power University named after V. I. Lenin: Ples, Russia, 2004; pp. 341–344. [Google Scholar]
- Vohlídal, J.; Sedláček, J.; Patev, N.; Lavastre, O.; Dixneuf, P.H.; Cabioch, S.; Balcar, H.; Pfleger, J.; Blechta, V. New Substituted Polyacetylenes with Phenyleneethynylene Side Groups [−(C6H4−C⋮C)n−SiiPr3; n = 1, 2]: Synthesis, Characterization, Spectroscopic, and Photoelectric Properties. Macromolecules 1999, 32, 6439–6449. [Google Scholar] [CrossRef]
- Ding, X.; Wang, W.; Qi, H.; Huang, Y.; Zhuang, Y.; Wang, J.; Jiao, Y. Rheological Behavior of Polyarylacetylene Prepolymer. J. Funct. Polym. 2001, 14, 105–108. [Google Scholar]
- Yang, M.; Zhan, X. Polymerization of p-diethynylbenzene initiated by Ni(C≡CC6H4C≡CH)2(PPH3)2. Chin. J. Polym. Sci. 2001, 19, 303–309. [Google Scholar]
- Zhan, X.; Yang, M.; Lei, Z. Transition metal acetylide catalysts for polymerization of p-diethynylbenzene 4: Effect of transition metals on catalytic activity of complexes. J. Mol. Catal. A Chem. 2002, 184, 139–145. [Google Scholar] [CrossRef]
- Slater, J.C. Atomic Radii in Crystals. J. Chem. Phys. 1964, 41, 3199–3204. [Google Scholar] [CrossRef]
- Abd-El-Aziz, A.S.; Strohm, E.A. Transition metal-containing macromolecules: En route to new functional materials. Polymer 2012, 53, 4879–4921. [Google Scholar] [CrossRef]
- Vidal, F.; Jäkle, F. Functional Polymeric Materials Based on Main-Group Elements. Angew. Chem. Int. Ed. 2019, 58, 5846–5870. [Google Scholar] [CrossRef]
- Liu, J.; Lam, J.W.Y.; Häußler, M.; Qin, A.; Tang, B.Z. Cobalt-Containing Hyperbranched Poly(silylenearylene)s. J. Inorg. Organomet. Polym. 2009, 19, 133–138. [Google Scholar] [CrossRef]
- Häußler, M.; Qin, A.; Tang, B.Z. Acetylenes with multiple triple bonds: A group of versatile An-type building blocks for the construction of functional hyperbranched polymers. Polymer 2007, 48, 6181–6204. [Google Scholar] [CrossRef] [Green Version]
- Scholz, S.; Leech, P.J.; Englert, B.C.; Sommer, W.; Weck, M.; Bunz, U.H.F. Cobalt-Carbon Spheres: Pyrolysis of Dicobalthexacarbonyl-Functionalized Poly(p-phenyleneethynylene)s. Adv. Mater. 2005, 17, 1052–1055. [Google Scholar] [CrossRef]
- Draper, S.M.; Delamesiere, M.; Champeil, E.; Twamley, B.; Byrne, J.J.; Long, C. Novel acetylene-linked di-cobalt and tetra-cobalt carbonyl clusters. J. Organomet. Chem. 1999, 589, 157–167. [Google Scholar] [CrossRef]
- Iwashita, Y.; Ishikawa, A.; Kainosho, M. Spectroscopic study on bond hybridization of co-ordinated acetylenes. Spectrochim. Acta Part A 1971, 27, 271–277. [Google Scholar] [CrossRef]
- Bauer, H.; Faust, J.; Froböse, R.; Füssel, J.; Faust, J.; Füssel, J. Organocopper Compounds. In Cu Organocopper Compounds; Faust, J., Füssel, J., Eds.; Springer: Berlin/Heidelberg, Germany, 1986; pp. 1–247. ISBN 978-3-662-06441-2. [Google Scholar]
- Lang, H.; Jakob, A.; Milde, B. Copper(I) Alkyne and Alkynide Complexes. Organometallics 2012, 31, 7661–7693. [Google Scholar] [CrossRef]
- Mylnikov, V.S.; Terenin, A.N. Photovoltaic properties of metal acetylenides. Dokl. Akad. Nauk SSSR 1963, 153, 1381–1384. [Google Scholar]
- Korshak, V.V.; Sladkov, A.M.; Kudryavtsev, Y.P. The synthesis of polymeric acetylides. Polym. Sci. USSR 1962, 3, 503–507. [Google Scholar] [CrossRef]
- Jin, X.; Wu, Y.; Lin, Z.; Liang, D.; Wang, F.; Zheng, X.; Liu, H.; Lv, W.; Liu, G. Plasmonic Ag nanoparticles decorated copper-phenylacetylide polymer for visible-light-driven photocatalytic reduction of Cr(VI) and degradation of PPCPs: Performance, kinetics, and mechanism. J. Hazard. Mater. 2022, 425, 127599. [Google Scholar] [CrossRef]
- Jiang, H.-Y.; Zhou, P.; Wang, Y.; Duan, R.; Chen, C.; Song, W.; Zhao, J. Copper-Based Coordination Polymer Nanostructure for Visible Light Photocatalysis. Adv. Mater. 2016, 28, 9776–9781. [Google Scholar] [CrossRef]
- Berg, R.; Straub, B.F. Advancements in the mechanistic understanding of the copper-catalyzed azide–alkyne cycloaddition. Beilstein J. Org. Chem. 2013, 9, 2715–2750. [Google Scholar] [CrossRef] [Green Version]
- Mykhalichko, B.M.; Temkin, O.N.; Mys’kiv, M.G. Polynuclear complexes of copper(I) halides: Coordination chemistry and catalytic transformations of alkynes. Russ. Chem. Rev. 2000, 69, 957–984. [Google Scholar] [CrossRef]
- Temkin, O.N. “Golden Age” of Homogeneous Catalysis Chemistry of Alkynes: Dimerization and Oligomerization of Alkynes. Kinet. Catal. 2019, 60, 689–732. [Google Scholar] [CrossRef]
- Grimes, R.N. Carboranes; Elsevier: London, UK, 2016; ISBN 978-0-12-801905-4. [Google Scholar]
- Núñez, R.; Romero, I.; Teixidor, F.; Viñas, C. Icosahedral boron clusters: A perfect tool for the enhancement of polymer features. Chem. Soc. Rev. 2016, 45, 5147–5173. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Jäkle, F. Incorporation of Group 13 Elements into Polymers. In Main Group Strategies towards Functional Hybrid Materials; Baumgartner, T., Jäkle, F., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2018; pp. 79–110. ISBN 978-1-119-23594-1. [Google Scholar]
- Helten, H. Doping the Backbone of π-Conjugated Polymers with Tricoordinate Boron: Synthetic Strategies and Emerging Applications. Chem. Asian J. 2019, 14, 919–935. [Google Scholar] [CrossRef]
- Sergeyev, V.A.; Kirilenko, Y.K.; Plyashkevich, L.A.; Kalinin, V.N.; Perepechkina, Y.P.; Shitikov, V.K.; Kudryavtsev, G.I.; Parfenov, B.P.; Zakharkin, L.I. Carborane-containing poly(arylacetylenes). Polym. Sci. USSR 1986, 28, 2638–2641. [Google Scholar] [CrossRef]
- Cherkashin, M.I.; Chauser, M.G.; Dyumaev, K.M.; Kirilenko, Y.K.; Vlasenko, T.Y.; Plyashkevich, L.A.; Kudryavtsev, G.I.; Belitsin, M.N. Synthesis of a fibre-forming carbon-chain polymer based on para-diethynylbenzene. Vysokomol. Soedin. Ser. B 1985, 27, 9–11. [Google Scholar]
- Cessna, L.C.; Jabloner, H. A New Class of Easily Moldable Highly Stable Thermosetting Resins. J. Elastomers Plast. 1974, 6, 103–113. [Google Scholar] [CrossRef]
- Jabloner, H.; Cessna, L.C. Thermo setting polymers of p-diethenylbenzene. Am. Chem. Soc. Polym. Prepr. 1976, 17, 169. [Google Scholar]
- Martín, C.; Hunt, B.J.; Ebdon, J.R.; Ronda, J.C.; Cádiz, V. Synthesis, polymerization, and effects on the flame retardancy of boron-containing styrenic monomers. J. Polym. Sci. A Polym. Chem. 2005, 43, 6419–6430. [Google Scholar] [CrossRef]
- Khodeir, M.; Antoun, S.; Ruymbeke, E.; Gohy, J. Temperature and Redox-Responsive Hydrogels Based on Nitroxide Radicals and Oligoethyleneglycol Methacrylate. Macromol. Chem. Phys. 2020, 221, 1900550. [Google Scholar] [CrossRef]
- Yudin, V.V.; Kovylin, R.S.; Baten’kin, M.A.; Kulikova, T.I.; Aleynik, D.Y.; Egorikhina, M.N.; Rubtsova, Y.P.; Charykova, I.N.; Mlyavykh, S.G.; Chesnokov, S.A.; et al. Visible-light induced synthesis of biocompatible porous polymers from oligocarbonatedimethacrylate (OCM-2) in the presence of dialkyl phthalates. Polymer 2020, 192, 122302. [Google Scholar] [CrossRef]
- Ebnesajjad, S.; Landrock, A.H. Characteristics of Adhesive Materials. In Adhesives Technology Handbook; Elsevier: London, UK, 2015; pp. 84–159. ISBN 978-0-323-35595-7. [Google Scholar]
- Ewen J.C., K. Key issues in selecting the right adhesive. In Advances in Structural Adhesive Bonding; Elsevier: Padstow, Cornwall, UK, 2010; pp. 3–19. ISBN 978-1-84569-435-7. [Google Scholar]
- Binegar, G.A.; Noblet, J.A.; Zaldivar, R.D.; Sheaffer, P.M.; Rellick, G.S. Effects of Heat Treatment on Microstructure Sand Flexural Properties of Unidirectional Carbon-Carbon Composites; Space Systems Division Air Force Systems Command: Los Angeles, CA, USA, 1989. [Google Scholar]
- Rajesh, S.; Suresh, G.; Mohan, R.C. A Review on Material Selection and Fabrication of Composite Solid Rocket Motor (SRM) Casing. J. Mech. Sol. 2017, 12, 125–138. [Google Scholar]
- Hwang, J.U.; Ahn, W.J.; Im, J.S.; Lee, J.D. Properties of synthetic graphite from boric acid-added pitch: Performance as anode in lithium-ion batteries. SN Appl. Sci. 2021, 3, 600. [Google Scholar] [CrossRef]
- Ariharan, A.; Viswanathan, B.; Nandhakumar, V. Hydrogen storage on boron substituted carbon materials. Int. J. Hydrogen Energy 2016, 41, 3527–3536. [Google Scholar] [CrossRef]
- Zaldivar, R.J.; Kobayashi, R.W.; Rellick, G.S.; Yang, J.-M. Carborane-catalyzed graphitization in polyarylacetylene-derived carbon-carbon composites. Carbon 1991, 29, 1145–1153. [Google Scholar] [CrossRef]
- Kozykina, M.A.; Fainberg, E.Z.; Papkov, S.P.; Varshavskii, V.Y.; Kumok, I.L.; Konkin, A.A. Thermochemical study of carbon fibres based on polyacrylonitrile modified by boron. Polym. Sci. USSR 1980, 22, 2849–2856. [Google Scholar] [CrossRef]
- Cherkashin, M.I.; Misin, V.M.; Kazakov, M.E.; Kirilenko, Y.K.; Stanko, V.I.; Valetsky, P.M. Carbon Materials with Increased Electrical Conductivity; USSR: Aghveran, Armenia, 1984; pp. 87–88. [Google Scholar]
- Naumkin, A.A.; Kraut-Vass, A.; Gaarenstroom, S.W.; Powell, C.J. X-ray Photoelectron Spectroscopy Database XPS; Version 4.1; NIST Standard Reference Database: Gaithersburg, MD, USA, 1989; Volume 20. [Google Scholar] [CrossRef]
- Moulder, J.F.; Stickle, W.F.; Sobol, W.M.; Bomben, K.D. Handbook of X-ray Photoelectron Spectroscopy; Perkin-Elmer Corporation: Eden Prairie, MN, USA, 1992. [Google Scholar]
- Zaldivar, R.J.; Kobayashi, R.W.; Rellick, G.S.; Yang, J.-M. Boron migration in boron-doped carbon/carbon composites. Carbon 1992, 30, 711–714. [Google Scholar] [CrossRef]
- Forintos, N.; Czigany, T. Multifunctional application of carbon fiber reinforced polymer composites: Electrical properties of the reinforcing carbon fibers—A short review. Compos. Part B 2019, 162, 331–343. [Google Scholar] [CrossRef]
- Babu, A.; Kumar, N. Electrical Conductivity for Carbon Fiber: A Review. J. Crit. Rev. 2020, 7, 958–963. [Google Scholar] [CrossRef]
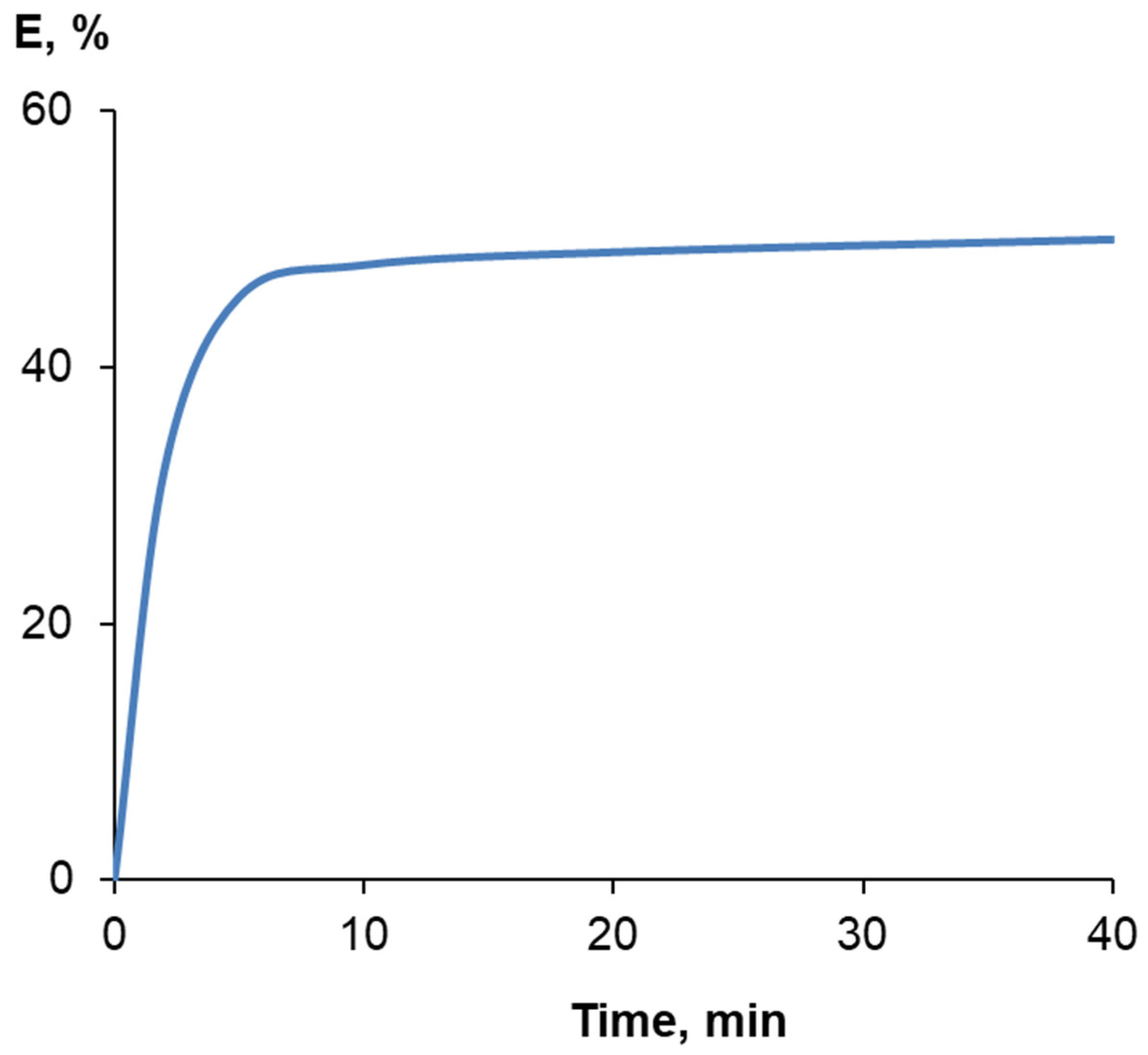

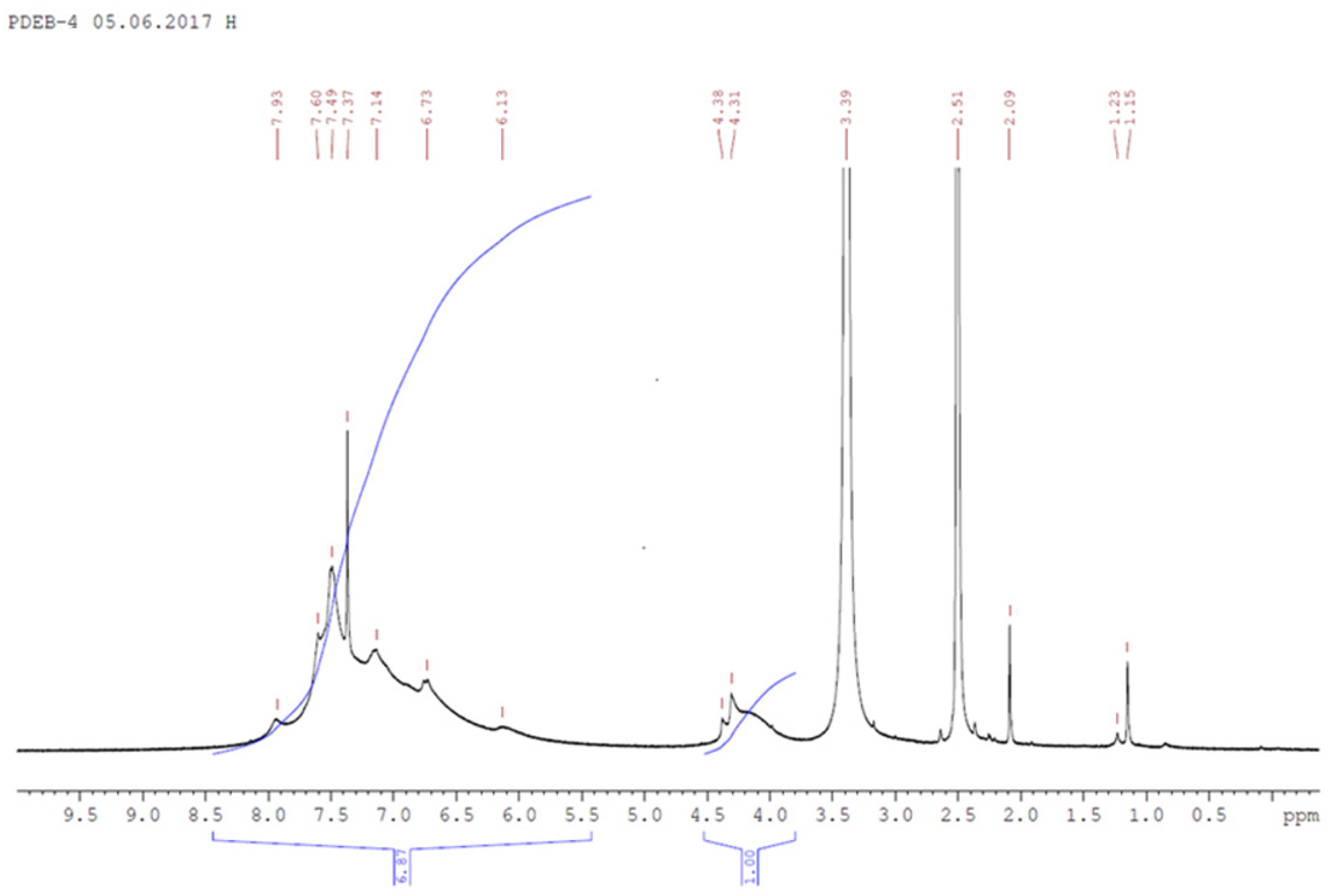



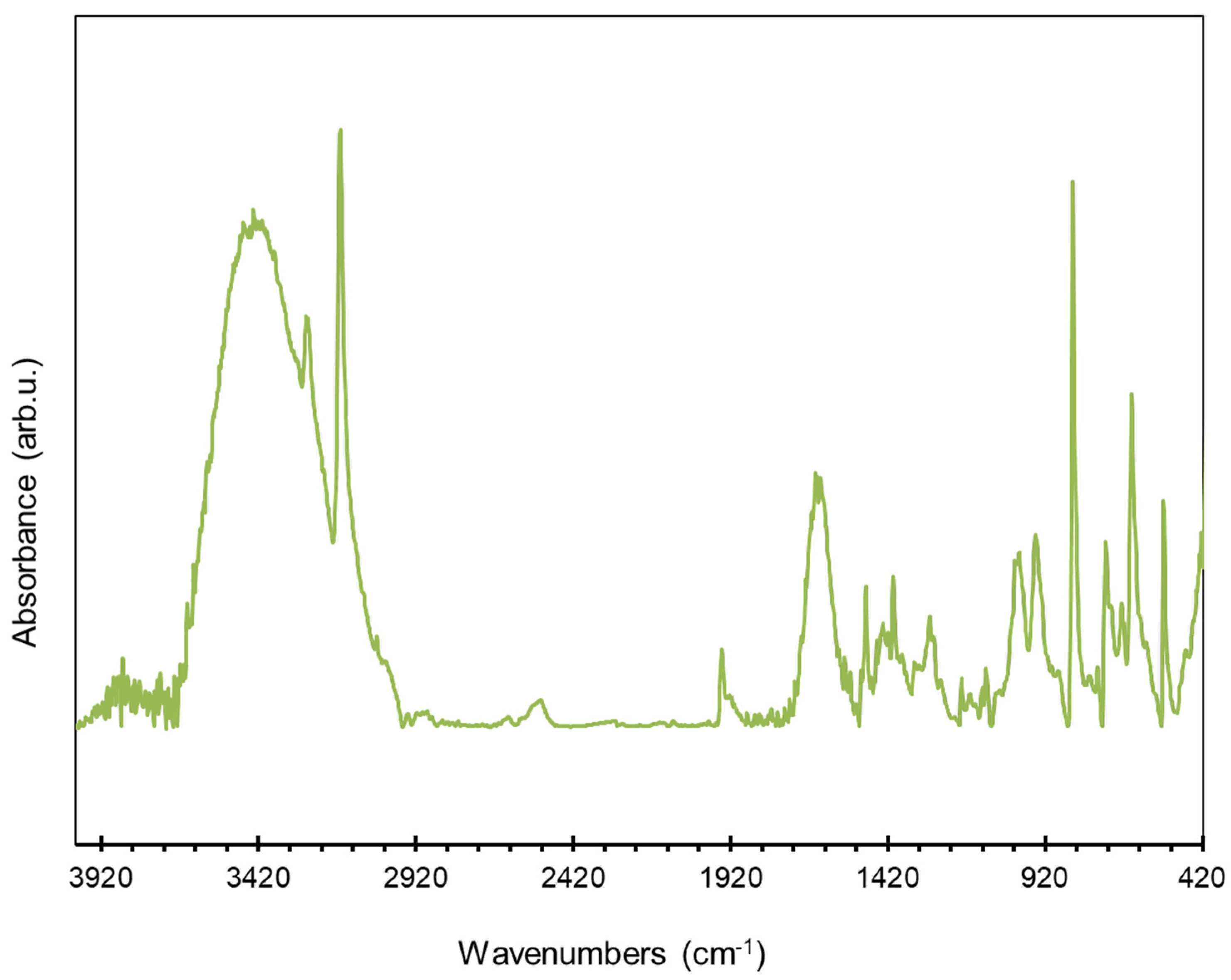
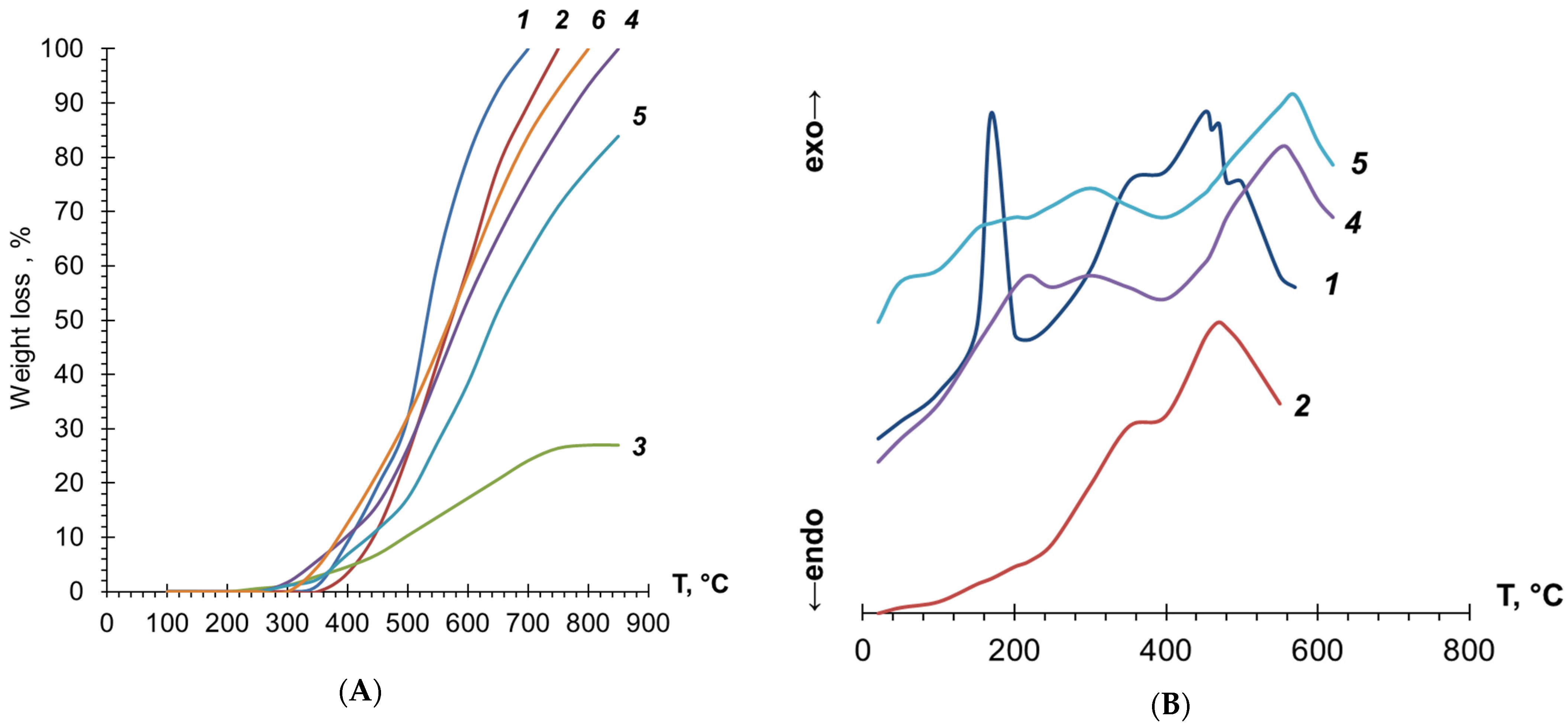





| Samples | Solvent | τ, Min | Y, % | Har+ol/Heth d | ||
|---|---|---|---|---|---|---|
| Sol a | Ins b | |||||
| PDEBA-1 | HMPA | 5 | 47 | - | 1300/10.3 | 5/1 |
| PDEBA-2 | HMPA | 20 | 48 | - | 1340/10.6 | 5/1 |
| PDEBA-3 | HMPA | 40 | 49 | - | 1370/10.9 | 5.3/1 |
| PDEBA-4 | DMSO | 1 | 5 | - | 1800/14 | 7/1 |
| PDEBA-5 | DMSO | 20 | 58 | 23 | 3160/25 | 7/1 |
| PDEBA-6 | DMSO | 60 | 63 | 26 | 3730/29.6 | 8/1 |
| Degree of Polymerization | Polymer | ||
|---|---|---|---|
| PDEBA-1 | PDEBA-2 | PDEBA-3 | |
| 7.05 | 7.20 | 7.26 | |
| 10.32 | 10.63 | 10.87 | |
| Samples | |||||
|---|---|---|---|---|---|
| PDEBA-1 | 1300 | 2380 | 7200 | 1.82 | 3.02 |
| PDEBA-2 | 1340 | 2430 | 7300 | 1.82 | 3.01 |
| PDEBA-3 | 1370 | 2700 | 8700 | 1.97 | 3.20 |
| PDEBA-4 a | 1800 | 2400 | 4400 | 1.33 | 1.83 |
| PDEBA-5 a | 3160 | 5400 | 14,000 | 1.70 | 2.66 |
| PDEBA-6 a | 3730 | 10,800 | 24,000 | 2.89 | 2.22 |
| Samples | Initial Ratio | τ, Min | Y, % | –C≡CH, % Mass b | Quantity –C≡CH in a Macromolecule | ||
|---|---|---|---|---|---|---|---|
| CPA-1 | 1:3 | 1 | 3 | 860 | 1:31 | 3.3 | 1.1 |
| CPA-2 | 1:3 | 60 | 41 | 1070 | 1:40 | 3.0 | 1.3 |
| CPA-3 | 1:1 | 1 | 4 | 1650 | 1:18 | 6.3 | 4.2 |
| CPA-4 | 1:1 | 60 | 30 | 1940 | 1:21 | 5.7 | 4.5 |
| CPA-5 | 3:1 | 1 | 5 | 1500 | 1:11 | 10.2 | 6.1 |
| CPA-6 | 3:1 | 60 | 65 | 1890 | 1:13 | 9.4 | 7.1 |
| Samples a | b | Y, % | c | Ratio in CPA | ||||
|---|---|---|---|---|---|---|---|---|
| CPA-1 | 1:3 | 3.5 | 1:31.0 | 1:1.80 | 0.33 | 0.556 | 0.200 | −0.267 |
| CPA-7 | 1:3 | 3.5 | 1:34.0 | 1:2.16 | 0.33 | 0.463 | 0.240 | −0.387 |
| CPA-8 | 1:3 | 3.0 | 1:35.8 | 1:2.28 | 0.33 | 0.439 | 0.253 | −0.427 |
| CPA-9 | 1:2 | 4.2 | 1:25.3 | 1:1.37 | 0.50 | 0.730 | 0.343 | −0.185 |
| CPA-3 | 1:1 | 3.8 | 1:17.6 | 1:0.80 | 1.00 | 1.250 | 0.800 | 0.200 |
| CPA-5 | 3:1 | 4.6 | 1:11.0 | 1:0.29 | 3.00 | 3.448 | 2.610 | 2.130 |
| Samples | Initial Ratio mol per mol | Reaction Time, Hour | Content Co, % Mass a | Polymer Complex Ratio mol per mol | |
|---|---|---|---|---|---|
| Theor. | Exp. | ||||
| PDEBA-Co-1 | 3:1 | 3 | 14.50 | 12.55 | 2.45:1 |
| PDEBA-Co-2 | 2:1 | 8 | 17.32 | 15.90 | 1.26:1 |
| PDEBA-Co-3 | 1.5:1 | 15 | 21.50 | 19.55 | 0.65:1 |
| PDEBA-Co-4 | 1:1 | 20 | 25.61 | 24.59 | 0.04:1 |
| Initial Ratio: | PDEBA-Cu Polymer Complex Ratio: |
|---|---|
| 1:10 | 1:10.2 |
| 3:10 | 1:11.7 |
| № | Composition Formulation, % mass. | Coke Yield, % | ||
|---|---|---|---|---|
| CPA-5 | Poly-DPDA | EN-6 | ||
| 1 | − | − | 100 | 29.95 |
| 2 | 10 | − | 90 | 62.80 |
| 3 | 20 | − | 80 | 55.84 |
| 4 | 30 | − | 70 | 59.61 |
| 5 | − | 10 | 90 | 44.91 |
| 6 | − | 20 | 80 | 49.66 |
| 7 | − | 30 | 70 | 50.81 |
| Sample | Treatment Condition | B a, % mass. | XPS Surface Analysis b | ||||
|---|---|---|---|---|---|---|---|
| T, °C | Medium | B | N | ||||
| Binding Energy, eV | Relative Content, % | Binding Energy, eV | Relative Content, at. % | ||||
| 1 | 500 | N2 | 0.63 | 190.1; 189.1 | 1 | - | - |
| 2 | 1000 | N2 | 0.53 | 187.7; 186.2 | 2 | 399.0 | 5 |
| 3 | 1250 | N2 | 0.48 | 190.6 | 10 | 398.4 | 30 |
| 4 | 1500 | N2 | 0.62 | 190.9 | 20 | 398.7 | 80 |
| 5 | 2000 | N2 | 0.76 | 190.7 | 2 | 398.7 | 5 |
| 6 | 2400 | N2 | 0.30 | 190.3 | Traces | 398.5 | Traces |
| 7 | 2400 | Ar | 0.10 | 188.5 | Traces | - | - |
| Sample Processing Temperature, °C | Specific Electrical Resistance, Ω·mm2/m |
|---|---|
| Without modification | 35 |
| 500 | 120 |
| 1000 | 88 |
| 1250 | 41 |
| 1500 | 32 |
| 2000 | 31 |
| 2400 | 7 |
| 2400 (argon) | 18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Misin, V.M.; Maltseva, I.E.; Maltsev, A.A.; Naumkin, A.V.; Kazakov, M.E. Anionic Polymerization of Para-Diethynylbenzene: Synthesis of a Strictly Linear Polymer. Polymers 2022, 14, 900. https://doi.org/10.3390/polym14050900
Misin VM, Maltseva IE, Maltsev AA, Naumkin AV, Kazakov ME. Anionic Polymerization of Para-Diethynylbenzene: Synthesis of a Strictly Linear Polymer. Polymers. 2022; 14(5):900. https://doi.org/10.3390/polym14050900
Chicago/Turabian StyleMisin, Vyacheslav M., Irina E. Maltseva, Alexander A. Maltsev, Alexander V. Naumkin, and Mark E. Kazakov. 2022. "Anionic Polymerization of Para-Diethynylbenzene: Synthesis of a Strictly Linear Polymer" Polymers 14, no. 5: 900. https://doi.org/10.3390/polym14050900
APA StyleMisin, V. M., Maltseva, I. E., Maltsev, A. A., Naumkin, A. V., & Kazakov, M. E. (2022). Anionic Polymerization of Para-Diethynylbenzene: Synthesis of a Strictly Linear Polymer. Polymers, 14(5), 900. https://doi.org/10.3390/polym14050900