
Fabrication, Characterization and In Vitro Assessment of Laevistrombus canarium-Derived Hydroxyapatite Particulate-Filled Polymer Composite for Implant Applications

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Materials and Methodology
2.1. Materials
2.2. Cell Line
2.3. Methodology
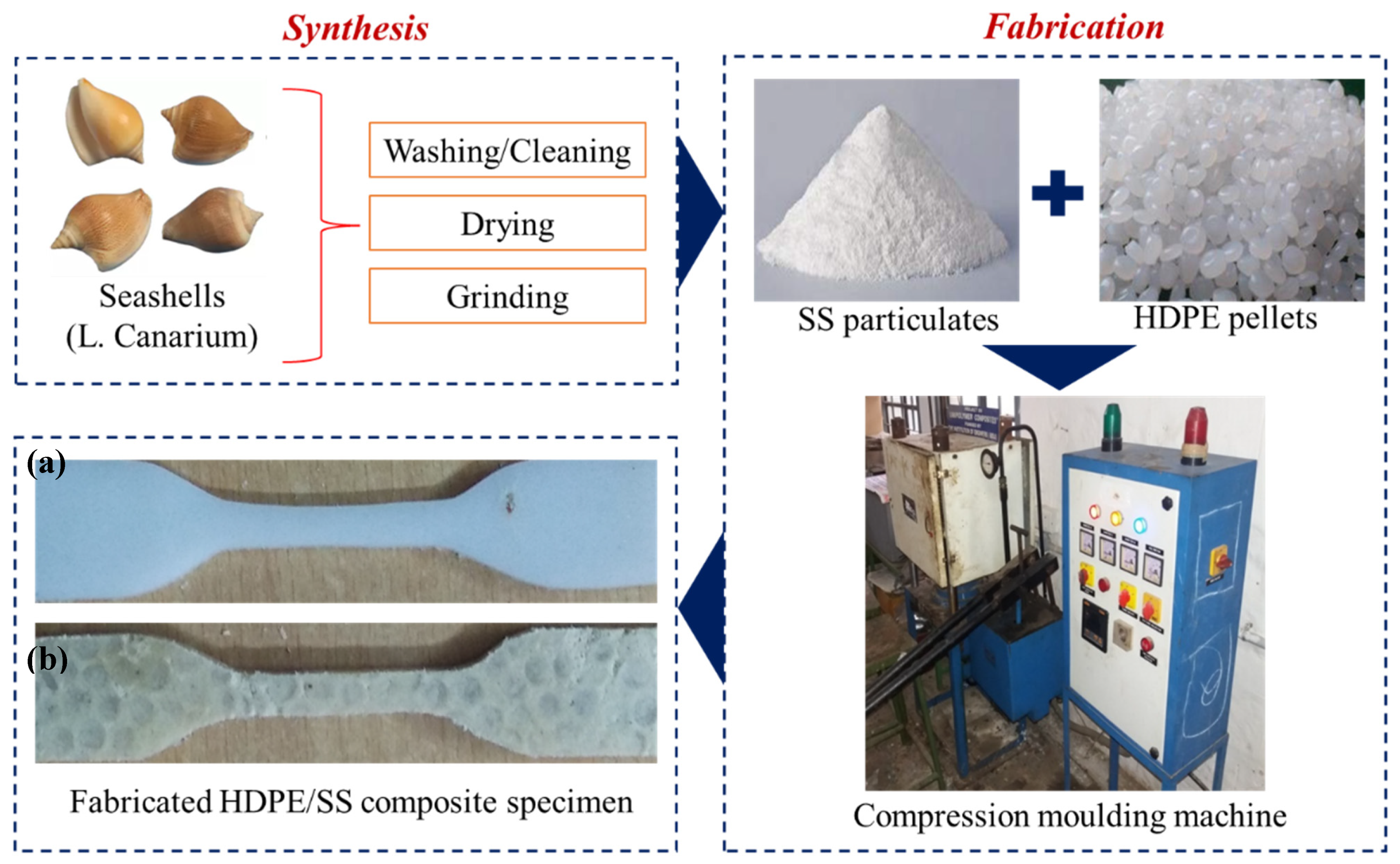
2.3.1. Preparation of Seashell (SS) Particulates
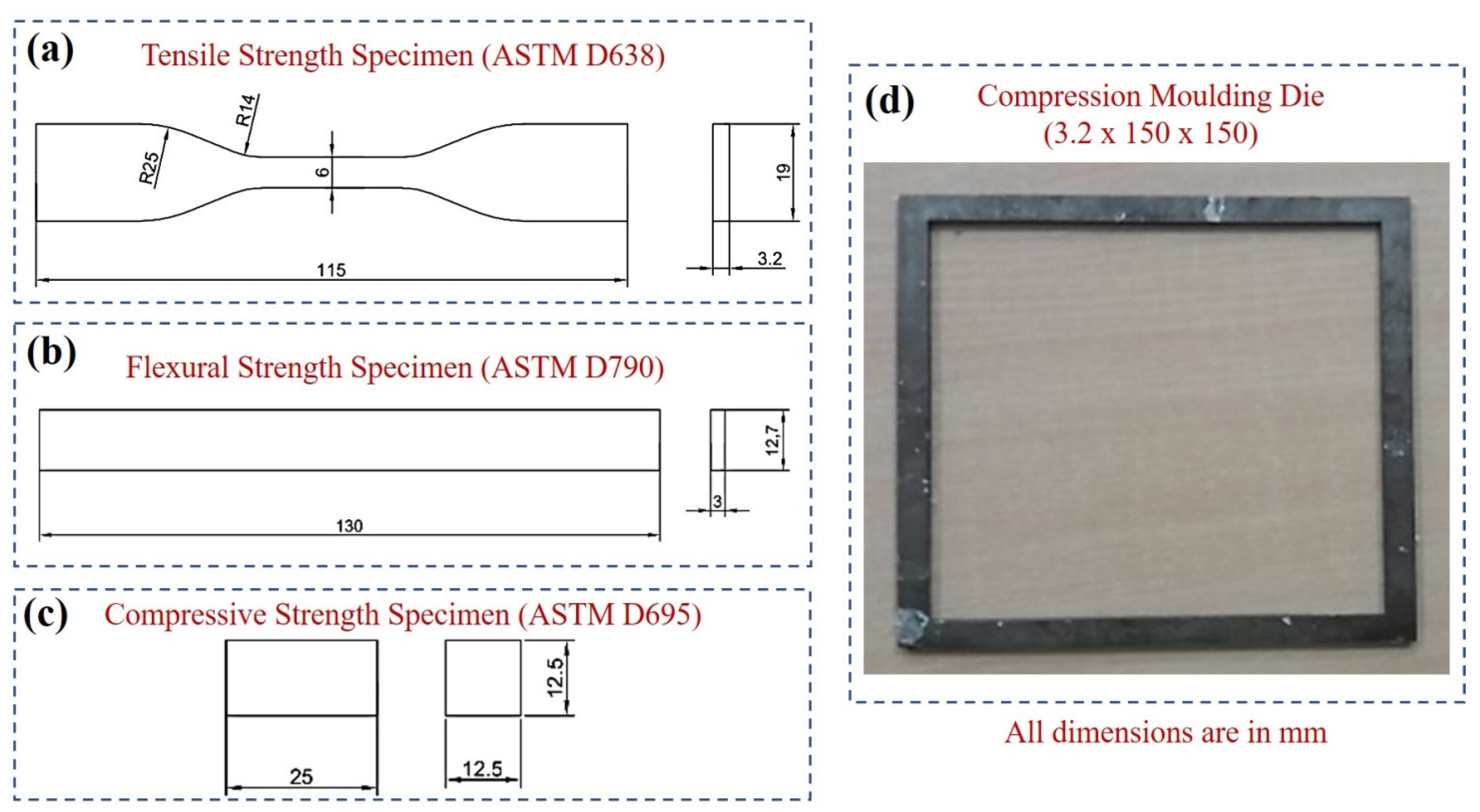
2.3.2. Fabrication of Molded Composite Specimen
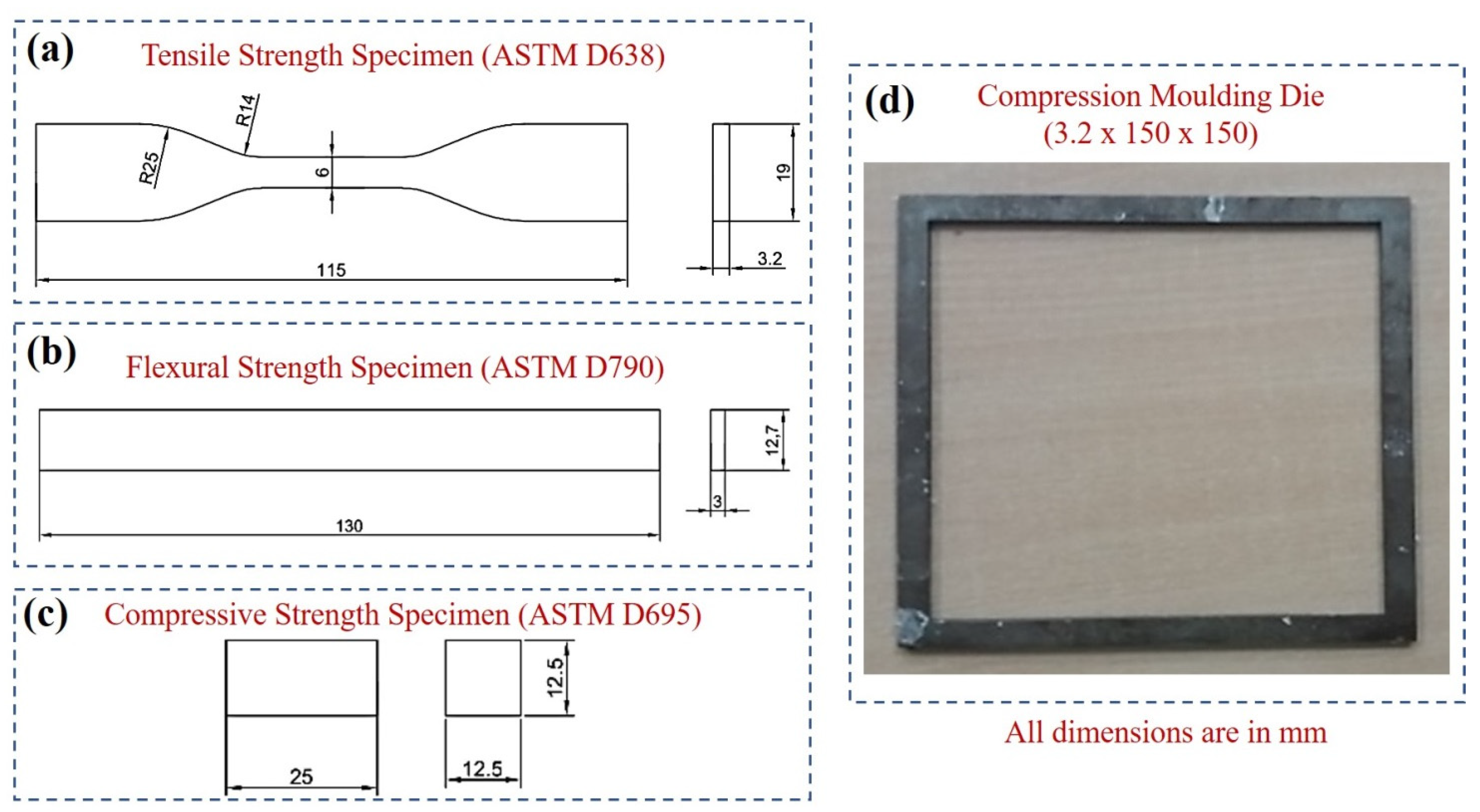
2.4. Characterization Studies
2.5. Biocompatibility Studies (Evaluation of Cytotoxicity and Cell Viability of HDPE/LC Composites)
3. Result and Discussions
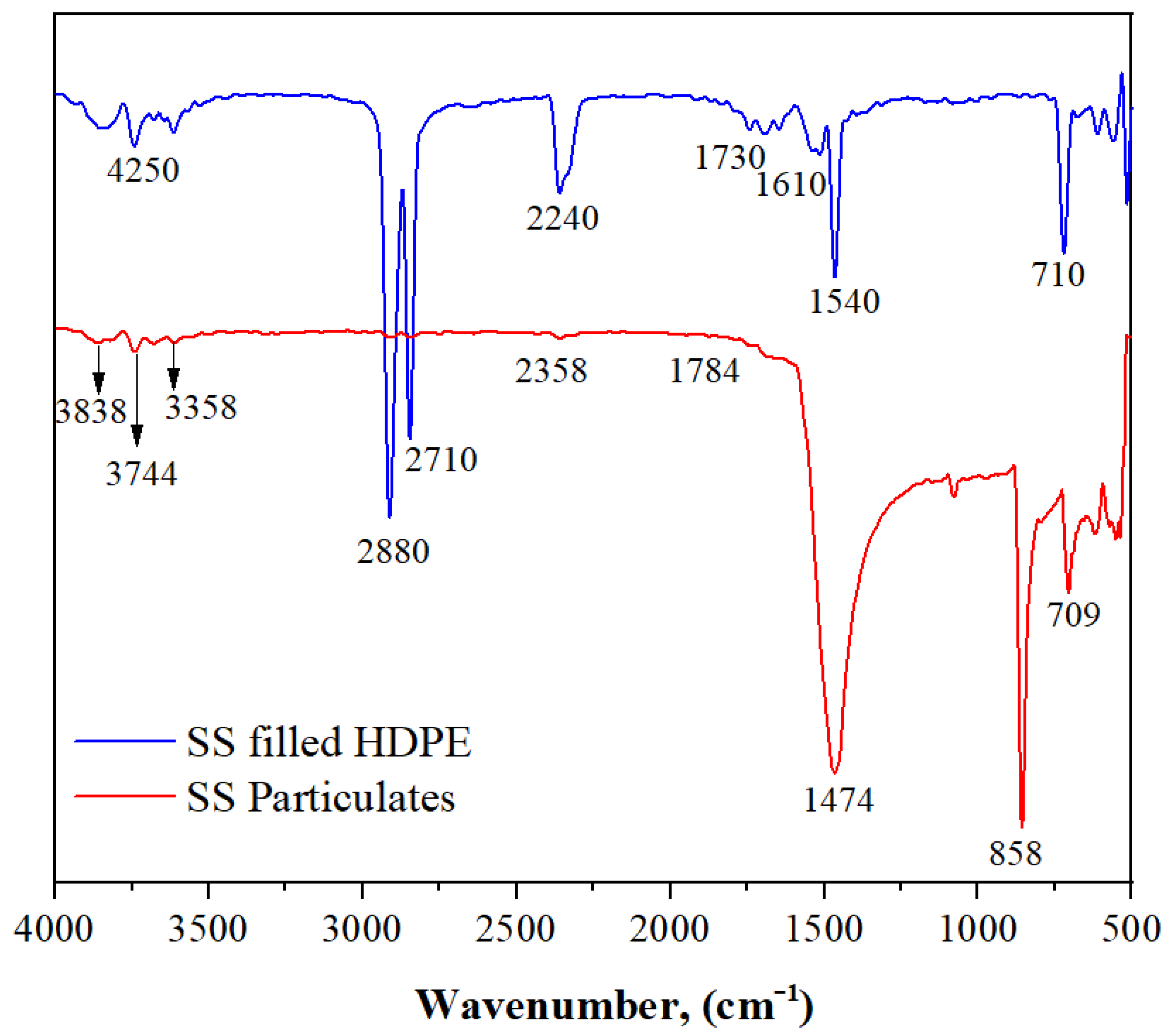
3.1. FTIR Spectra of SS Particulates and SS Particulate-Filled HDPE Composite
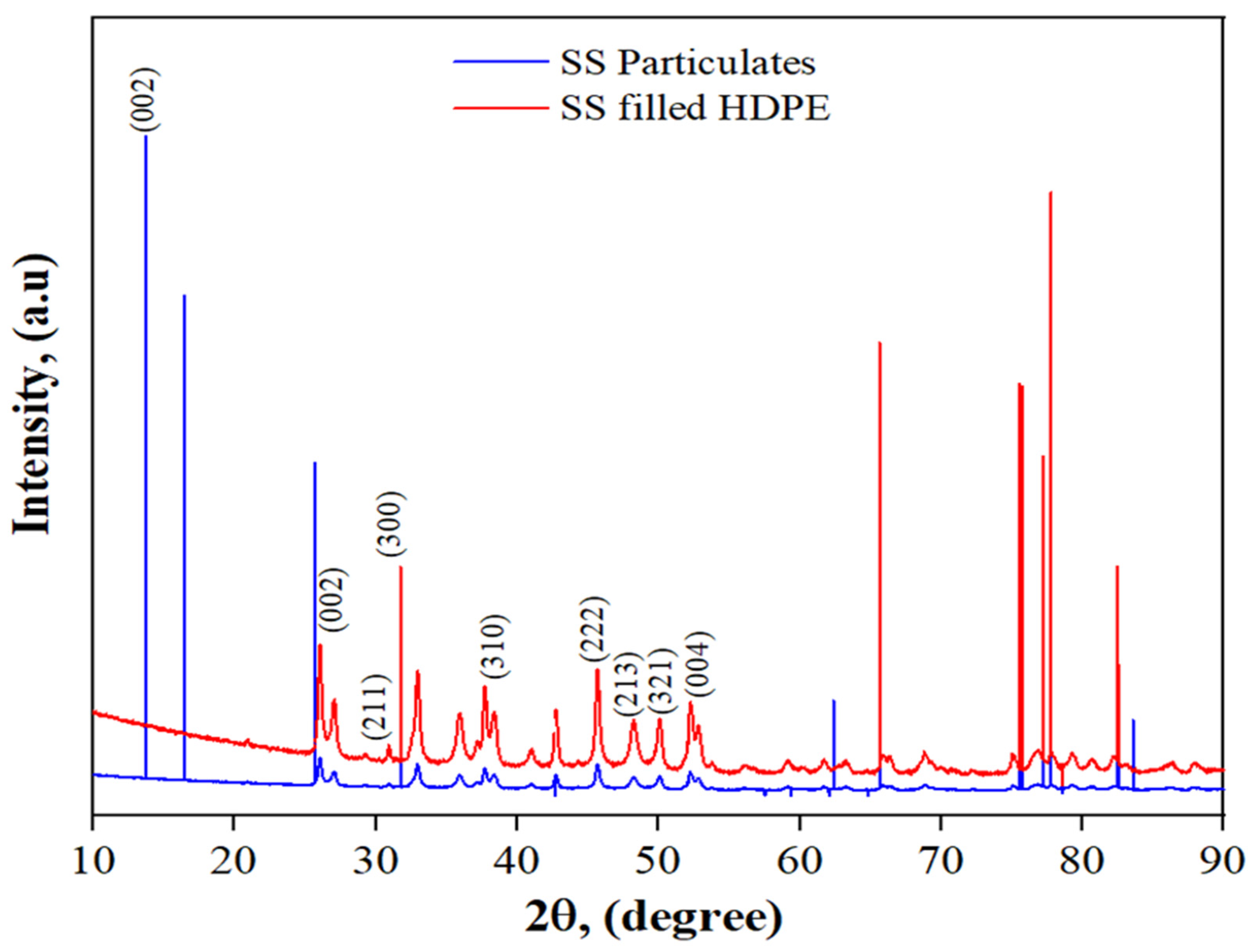
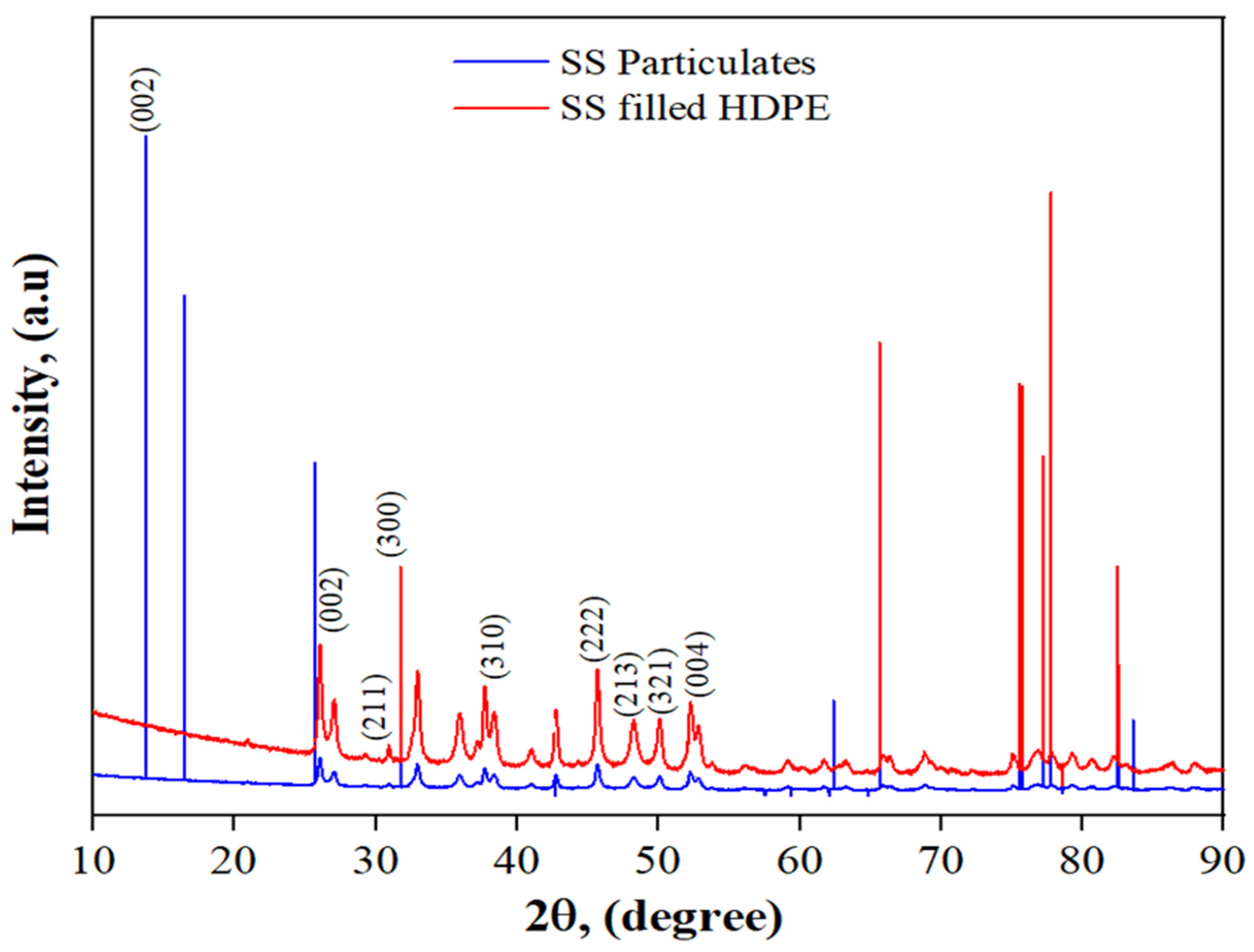
3.2. XRD of SS Particulates and 30 wt% SS Particulate-Filled HDPE Composite
3.3. FESEM Surface Morphology of SS Particulates and SS Particulate-Filled HDPE
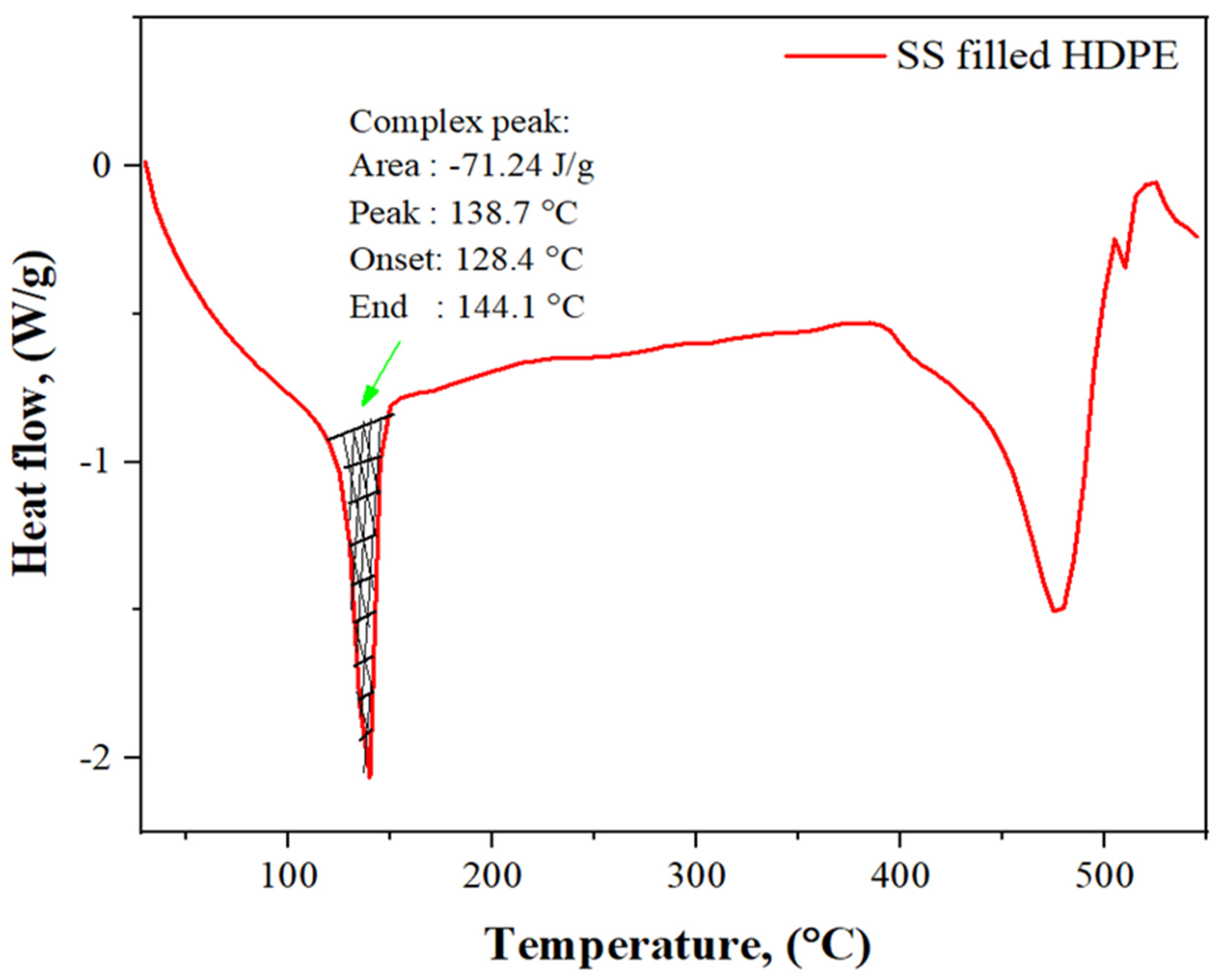
3.4. DSC Analysis of SS Particulate-Filled HDPE Composite
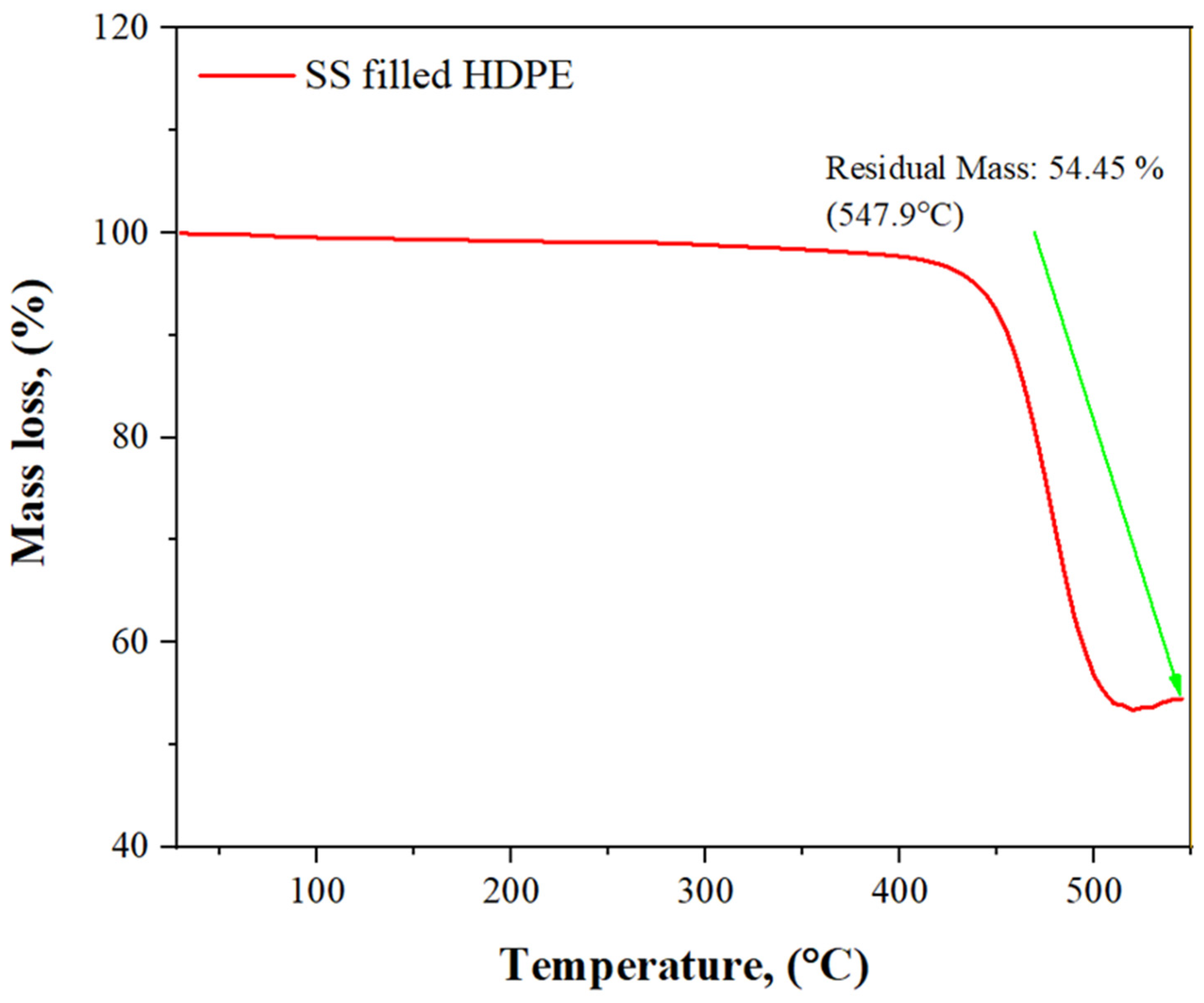
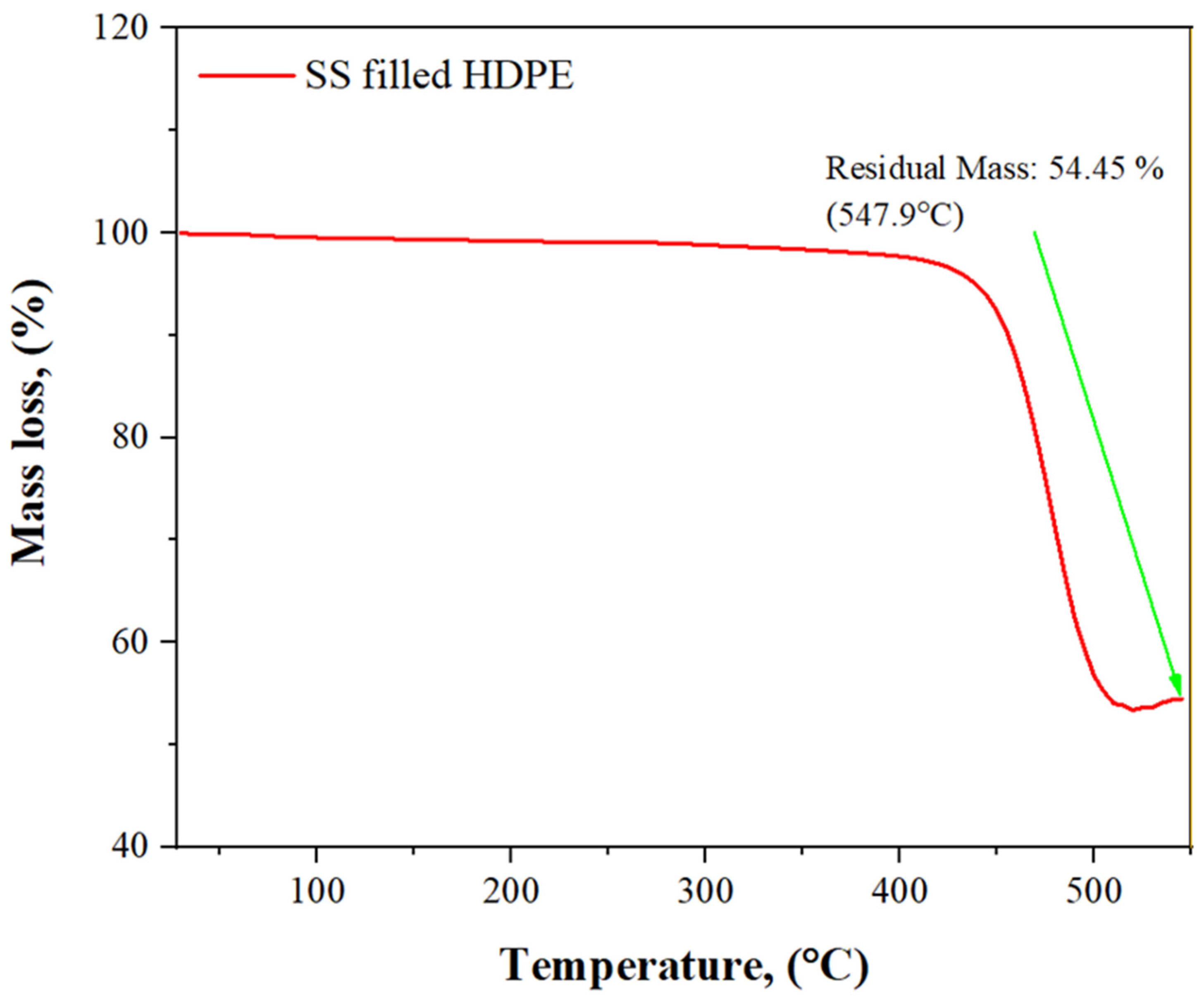
3.5. TGA Analysis of SS Particulate-Filled HDPE Composite
4. Mechanical Characterization
4.1. Tensile Strength
4.2. Compressive Strength
4.3. Shore D Hardness
4.4. Flexural Strength
5. Biocompatibility Study
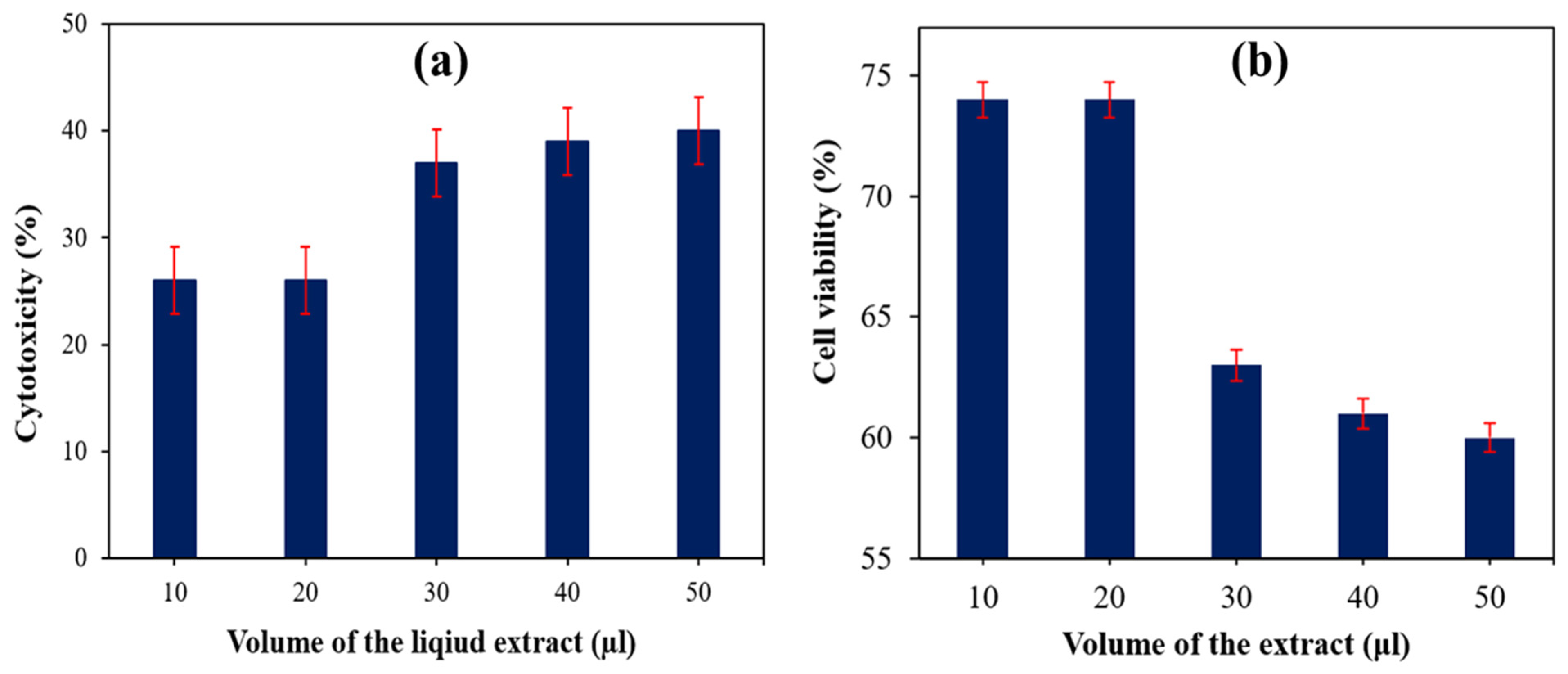
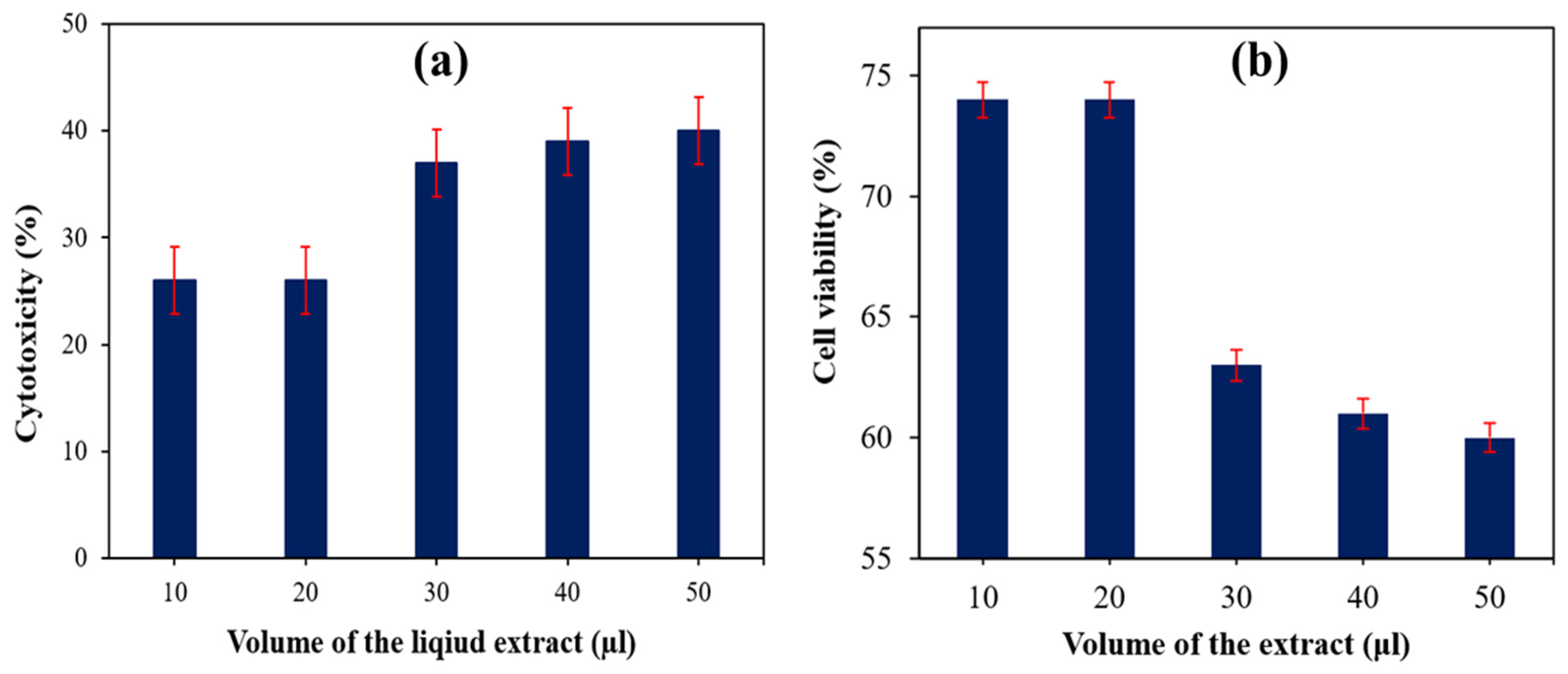
5.1. Evaluation of Cytotoxicity and Cell Viability of SS Particulate-Filled HDPE Composite
5.2. Cell Morphology
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Santhosh, S.; Balasivanandha Prabu, S. Thermalstability of nano hydroxyapatite synthesized from seashells through wet chemical synthesis. Mater. Lett. 2013, 97, 121–124. [Google Scholar] [CrossRef]
- Gergely, G.; Weber, F.; Lukacs, I.; Toth, A.L.; Horvath, Z.E.; Mihaly, J.; Balazsi, C. Preparation and characterization of hydroxyapatite from eggshell. Ceram. Int. 2010, 36, 803–806. [Google Scholar] [CrossRef]
- Sobczak-Kupiec, A.; Wzorek, Z. The influence of calcination parameters on free calcium oxide content in natural hydroxyapatite. Ceram. Int. 2012, 38, 641–647. [Google Scholar] [CrossRef]
- Ojeda-Niño, O.H.; Blanco, C.; Daza, C.E. High-temperature CO2 capture of hydroxyapatite extracted from tilapia scales. J. Fac. Sci. 2017, 22, 215–236. [Google Scholar] [CrossRef] [Green Version]
- Anjaneyulu, U.; Pattanayak, D.K.; Vijayalakshmi, U. Snail Shell derived natural Hydroxyapatite: Effects on NIH-3T3 cells for Orthopedic Applications. Mater. Manuf. Processes 2016, 31, 206–216. [Google Scholar] [CrossRef]
- Ozawa, M.; Suzuki, S. Microstructural development of natural hydroxyapatite originated from fish-bone waste through heat treatment. J. Am. Ceram. Soc. 2002, 85, 1315–1317. [Google Scholar] [CrossRef]
- Wu, S.C.; Tsou, H.K.; Hsu, H.C.; Hsu, S.K.; Liou, S.P.; Ho, W.F. A hydrothermal synthesis of eggshell and fruit waste extract to produce nanosized hydroxyapatite. Ceram. Int. 2013, 39, 8183–8188. [Google Scholar] [CrossRef]
- Macha, I.J.; Ozyegin, L.; Chou, J.; Samur, R.; Oktar, F.A.İ.K.; Ben-Nissan, B. An alternative synthesis method for Dicalcium phosphate (monetite) powders from Mediterranean mussel (Mytilus galloprovincialis) shells. J. Aust. Ceram. Soc. 2013, 49, 122–128. [Google Scholar]
- Ozyegin, L.S.; Oktar, F.N.; Goller, G.; Kayali, E.S.; Yazici, T. Plasma-sprayed bovine hydroxyapatite coatings. Mater. Lett. 2004, 58, 2605–2609. [Google Scholar]
- Oladele, I.O.; Agbabiaka, O.G.; Adediran, A.A.; Akinwekomi, A.D.; Balogun, A.O. Structural performance of poultry eggshell derived hydroxyapatite based high-density polyethylene bio-composites. Heliyon 2019, 5, e02552. [Google Scholar] [CrossRef] [Green Version]
- Dhanaraj, K.; Suresh, G. Conversion of waste sea shell (Anadara Granosa) into valuable nanohydroxyapatite (nHAp) for biomedical applications. Vacuum 2018, 152, 222–230. [Google Scholar] [CrossRef]
- Huang, Y.C.; Hsiao, P.C.; Chai, H.J. Hydroxyapatite extracted from fish scale effects on MG63 osteoblast-like cells. Ceram. Int. 2011, 37, 1825–1831. [Google Scholar] [CrossRef]
- Swetha, M.; Sahithi, K.; Moorthi, A.; Srinivasan, N.; Ramasamy, K.; Selvamurugan, N. Review on biocomposites containing natural polymers and hydroxyapatite for bone tissue engineering. Int. J. Biol. Macromol. 2010, 47, 1–4. [Google Scholar] [CrossRef]
- Heinemann, S.; Heinemann, C.; Jager, M.; Neunzehn, J.; Wiesmann, H.P.; Hanke, T. Effect of silica and hydroxyapatite mineralization on the mechanical properties and the biocompatibility of nanocomposite collagen scaffolds. ACS Appl. Mater. Interfaces 2011, 3, 4323–4331. [Google Scholar] [CrossRef] [PubMed]
- Ozkan, S.; Kalyon, D.M.; Yu, X. Functionally graded beta-TCP/PCL nanocomposite scaffolds: In vitro evaluation with human fetal osteoblast cells for bone tissue engineering. J. Biomed. Mater. Res. Part A 2010, 92, 1007–1018. [Google Scholar]
- Salerno, A.; Zeppetelli, S.; Di Maio EIannace, S.; Netti, P.A. Design of bimodal PCL and PCL-HA nanocomposite scaffolds by two-step depressurization during solid-state supercritical CO2 foaming. Macromol. Rapid Commun. 2011, 32, 1150–1156. [Google Scholar] [CrossRef]
- Pon-On, W.; Suntornsaratoon, P.; Charoenphandhu, N.; Thongbunchoo, J.; Krishnamra, N.; Tang, I.M. Hydroxyapatite from fish scale for potential use as a bone scaffold for regenerative material. Mater. Sci. Eng. C 2016, 62, 183. [Google Scholar] [CrossRef]
- Okada, M.; Furuzono, T. Hydroxylapatite nanoparticles: Fabrication methods and medical applications. Sci. Technol. Adv. Mater. 2012, 13, 1468–6996. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.C.; Hsu, H.C.; Wu, Y.N.; Ho, W.F. Hydroxyapatite synthesized from oyster shell powders by ball milling and heat treatment. Mater. Charact. 2011, 62, 1180–1187. [Google Scholar] [CrossRef]
- Ramakrishna, S.; Mayer, J.; Wintermantel, E.; Leong, K.W. Biomedical applications of polymer composite materials: A Review. Compos. Sci. Technol. 2001, 61, 1124–1189. [Google Scholar] [CrossRef]
- Aggarwal, P.K.; Chauhan, S.; Raghu, N.; Karmarkar, S.; Shashidhar, G.M. Mechanical properties of bio-fibers-reinforced high-density polyethylene composites: Effect of coupling agents and bio-fillers. J. Reinf. Plast. Compos. 2013, 32, 1722–1732. [Google Scholar] [CrossRef]
- Ranjan, N.; Singh, R.; Ahuja, I.P.S. Development of PLA-HAp-CS-based biocompatible functional prototype: A case study. J. Thermoplast. Compos. Mater. 2020, 33, 305–323. [Google Scholar] [CrossRef]
- Liu, X.; Miao, Y.; Liang, H.; Diao, J.; Hao, L.; Shi, Z.; Zhao, N.; Wang, Y. 3D-printed bioactive ceramic scaffolds with biomimetic micro/nano-HAp surfaces mediated cell fate and promoted bone augmentation of the bone–implant interface in vivo. Bioact. Mater. 2021, 12, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Nakonieczny, D.S.; Kern, F.; Dufner, L.; Antonowicz, M.; Matus, K. Alumina and Zirconia-Reinforced Polyamide PA-12 Composites for Biomedical Additive Manufacturing. Materials 2021, 14, 6201. [Google Scholar] [CrossRef] [PubMed]
- Oladapo, B.I.; Zahedi, S.A.; Ismail, S.O.; Omigbodun, F.T. 3D printing of PEEK and its composite to increase biointerfaces as a biomedical material—A review. Colloids Surf. B Biointerfaces 2021, 203, 111726. [Google Scholar] [CrossRef]
- Nakonieczny, D.S.; Antonowicz, M.; Paszenda, Z.K.; Radko, T.; Drewniak, S.; Bogacz, W.; Krawczyk, C. Experimental investigation of particle size distribution and morphology of alumina-yttria-ceria-zirconia powders obtained via sol–gel route. Biocybern. Biomed. Eng. 2018, 38, 535–543. [Google Scholar] [CrossRef]
- Oladapo, B.I.; Zahedi, S.A. Improving bioactivity and strength of PEEK composite polymer for bone application. Mater. Chem. Phys. 2021, 266, 124485. [Google Scholar] [CrossRef]
- Mondal, S.; Pal, U.; Dey, A. Natural origin hydroxyapatite scaffold as potential bone tissue engineering substitute. Ceram. Int. 2016, 42, 18338–18346. [Google Scholar] [CrossRef]
- Kantharia, N.; Naik, S.; Apte, S.; Kheur, M.; Kheur, S.; Kale, B. Nano-hydroxyapatite and its contemporary applications. Int. J. Dent. Res. Dev. 2014, 1, 15–19. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Daly, M.; Geever, L.M.; Major, I.; Higginbotham, C.L.; Devine, D.M. Synthesis and characterization of high-density polyethylene/peat ash composites. Compos. Part B Eng. 2016, 94, 312–321. [Google Scholar] [CrossRef]
- Cesur, S.; Oktar, F.N.; Ekren, N.; Kilic, O.; Alkaya, D.B.; Seyhan, S.A.; Ege, Z.E.; Lin, C.-C.; Kuruca, S.E.; Erdemir, G.; et al. Preparation and characterization of electrospun polylactic acid/sodium alginate/orange oyster shell composite nanofiber for biomedical application. J. Aust. Ceram. Soc. 2020, 56, 533–543. [Google Scholar] [CrossRef]
- Sapuan, S.M.; Jawaid, M.; Hoque, M.E. Biopolymers and Biocomposites: Chemistry and Technology. Curr. Anal. Chem. 2018, 14, 184. [Google Scholar] [CrossRef]
- Hoque, M.E.; Rayhan, A.M.; Shaily, S.I. Natural Fiber-Based Green Composites: Processing, Properties and Biomedical Applications. Appl. Sci. Eng. Prog. 2021, 14, 689–718. [Google Scholar] [CrossRef]
- Lett, J.A.; Sagadevan, S.; Fatimah, I.; Hoque, M.E.; Lokanathan, Y.; Léonard, E.; Alshahateet, S.F.; Schirhagl, R.; Oh, W.C. Recent advances in natural polymer-based hydroxyapatite scaffolds: Properties and applications. Eur. Polym. J. 2021, 148, 110360. [Google Scholar] [CrossRef]
- Biswas, M.C.; Jony, B.; Nandy, P.K.; Chowdhury, R.A.; Halder, S.; Kumar, D.; Ramakrishna, S.; Hassan, M.; Ahsan, M.A.; Hoque, M.E.; et al. Recent Advancement of Biopolymers and Their Potential Biomedical Applications. J. Polym. Environ. 2021, 30, 51–74. [Google Scholar] [CrossRef]
- Ayyanar, C.B.; Marimuthu, K.; Gayathri, B.; Sankarraja. Characterization and in vitro cytotoxicity evaluation of fish scale and seashell derived nano-hydroxyapatite high-density polyethylene composite. Polym. Polym. Compos. 2021, 29, 1534–1542. [Google Scholar] [CrossRef]
- Wu, D.Y.; Wang, S.S.; Wu, C.S. Antibacterial properties and cytocompatibility of biobased nanofibers of fish scale gelatine, modified polylactide, and freshwater clam shell. Int. J. Biol. Macromol. 2020, 165, 1219–1228. [Google Scholar] [CrossRef]
- Thanigachalam, M.; Muthusamy Subramanian, A.V. Evaluation of PEEK-TiO2-SiO2 nanocomposite as biomedical implants with regard to in-vitro biocompatibility and material characterization. J. Biomater. Sci. Polym. Ed. 2021, 1–20. [Google Scholar] [CrossRef]
- Ayyanar, C.B.; Mohan, S.P.; Bharathiraj, C.; Mavinkere Rangappa, S.; Siengchin, S. Characterization of Syzygium cumini particulates filled E-glass fiber-reinforced epoxy composites. Polym. Compos. 2021, 42, 6298–6309. [Google Scholar] [CrossRef]
- Mitran, V.; Ion, R.; Miculescu, F.; Necula, M.G.; Mocanu, A.C.; Stan, G.E.; Antoniac, I.V.; Cimpean, A. Osteoblast cell response to naturally derived calcium phosphate-based materials. Materials 2018, 11, 1097. [Google Scholar] [CrossRef] [Green Version]
- Pokhrel, S. Hydroxyapatite: Preparation, properties and its biomedical applications. Adv. Chem. Eng. Sci. 2018, 8, 225. [Google Scholar] [CrossRef] [Green Version]
- Balaji Ayyanar, C.; Marimuthu, K. Investigation on the morphology, thermal properties, and in-vitro cytotoxicity of the fish scale particulates filled high-density polyethylene composite. Polym. Polym. Compos. 2020, 28, 285–296. [Google Scholar] [CrossRef]
- Thanigachalam, M.; Muthusamy Subramanian, A.V. In-Vitro Cytotoxicity Assessment and Cell Adhesion Study of Functionalized nTiO2 reinforced PEEK Biocompatible Polymer Composite. Polym. Technol. Mater. 2021, 61, 566–576. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl. No | Different wt% of Fillers | Dry Amount of Matrix and Filler (gm) | |
|---|---|---|---|
| HDPE | SS | ||
| 1 | 10 | 67.5 | 7.5 |
| 2 | 20 | 60 | 15 |
| 3 | 30 | 52.5 | 22.5 |
| 4 | 40 | 45 | 30 |
| 5 | 50 | 37.5 | 37.5 |
| Sample Particulars | Cytotoxicity (%) | Cell Viability (%) | Cytotoxicity Reactivity | |
|---|---|---|---|---|
| Description | The Volume of the Extract (μL) | |||
| 30 wt% SS particulate-filled HDPE | 10 | 26 | 74 | Mild |
| 20 | 26 | 74 | Mild | |
| 30 | 37 | 63 | Mild | |
| 40 | 39 | 61 | Mild | |
| 50 | 40 | 60 | Mild | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chinnappan, B.A.; Krishnaswamy, M.; Thanigachalam, M.; Xu, H.; Khan, S.I.; Hoque, M.E. Fabrication, Characterization and In Vitro Assessment of Laevistrombus canarium-Derived Hydroxyapatite Particulate-Filled Polymer Composite for Implant Applications. Polymers 2022, 14, 872. https://doi.org/10.3390/polym14050872
Chinnappan BA, Krishnaswamy M, Thanigachalam M, Xu H, Khan SI, Hoque ME. Fabrication, Characterization and In Vitro Assessment of Laevistrombus canarium-Derived Hydroxyapatite Particulate-Filled Polymer Composite for Implant Applications. Polymers. 2022; 14(5):872. https://doi.org/10.3390/polym14050872
Chicago/Turabian StyleChinnappan, Balaji Ayyanar, Marimuthu Krishnaswamy, Mugilan Thanigachalam, Huaizhong Xu, Saiful Islam Khan, and Md Enamul Hoque. 2022. "Fabrication, Characterization and In Vitro Assessment of Laevistrombus canarium-Derived Hydroxyapatite Particulate-Filled Polymer Composite for Implant Applications" Polymers 14, no. 5: 872. https://doi.org/10.3390/polym14050872
APA StyleChinnappan, B. A., Krishnaswamy, M., Thanigachalam, M., Xu, H., Khan, S. I., & Hoque, M. E. (2022). Fabrication, Characterization and In Vitro Assessment of Laevistrombus canarium-Derived Hydroxyapatite Particulate-Filled Polymer Composite for Implant Applications. Polymers, 14(5), 872. https://doi.org/10.3390/polym14050872