



Chiral Polymers from Norbornenes Based on Renewable Chemical Feedstocks

 , ,
, , 
Abstract
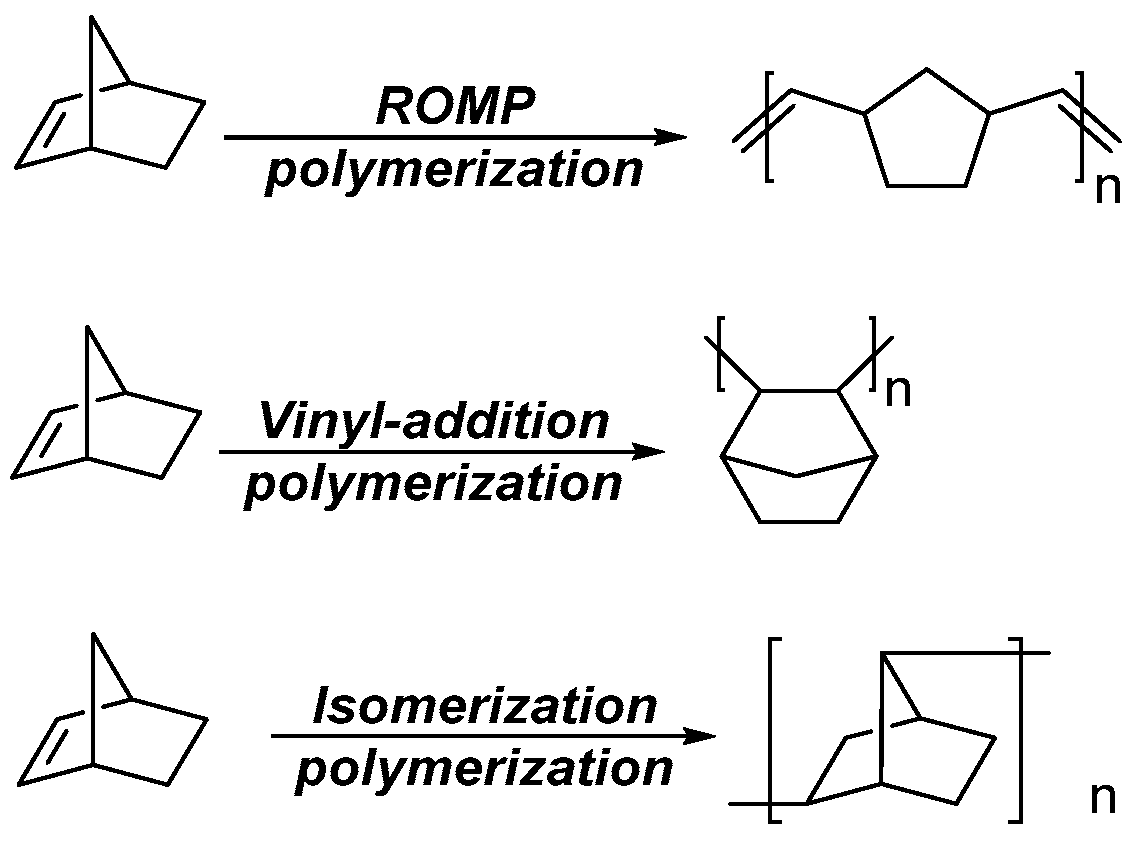
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Physico-Chemical Characterization
2.3. Film Preparation
2.4. Synthetic Part
- 1H NMR (300 MHz, CDCl3, δ, ppm): 1.50 (dd, 2H, RCH2, J = 10.20); 2.97 (s, 2H); 3.42 (s, 2H); 6.30 (s, 2H, R-CH=CH-R).
- 13C NMR (100 MHz, CDCl3, δ, ppm): 44.07; 46.82; 48.71; 137.91; 171.59.
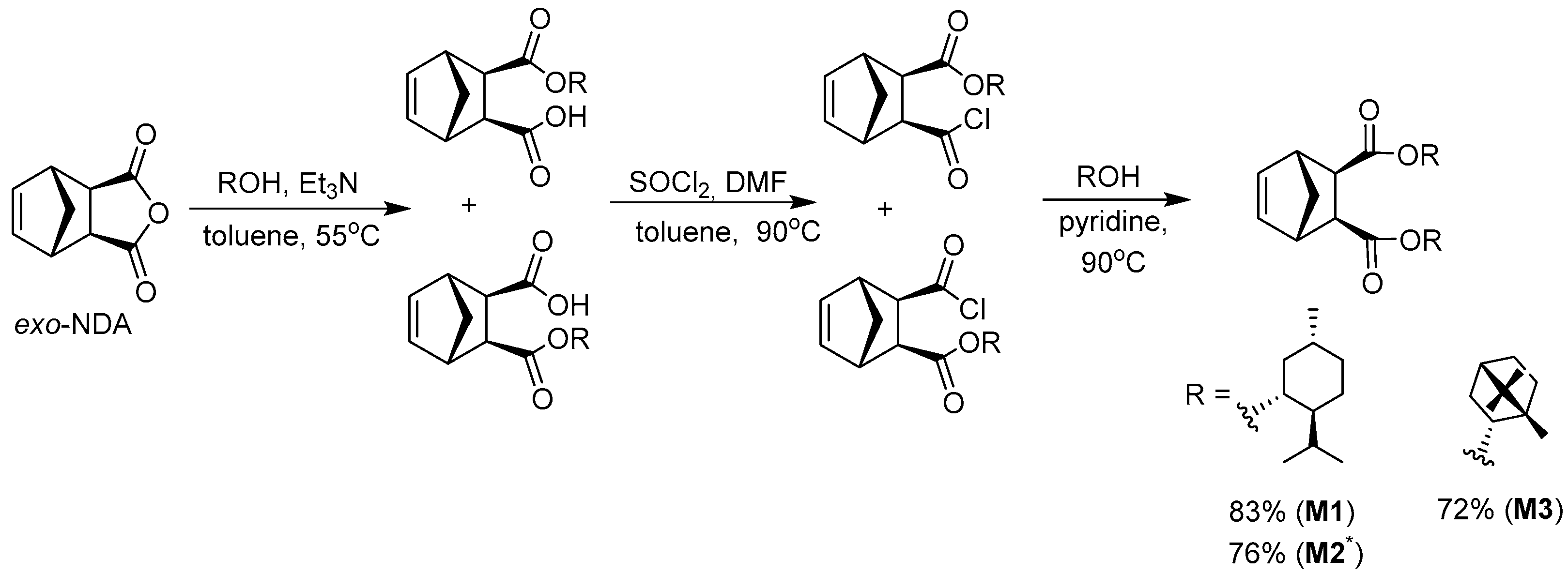
2.5. General Procedure for the Synthesis of Monomers M1–M3
- 1H NMR (400 MHz, CDCl3, δ, ppm): 6.26–6.24 (m, 2H, R-HC=CH-R), 4.61–4.76 (m, 2H), 3.15–3.14 (m, 1H), 3.05–3.04 (m, 1H), 2.63–2.62 (m, 1H), 2.55–2.54 (m, 1H), 2.13–2.04 (m, 3H), 2.03–1.86 (m, 2H), 1.76–1.60 (m, 4H), 1.56–1.30 (m, 5H), 1.62–0.76 (m, 24H).
- 13C NMR (100 MHz, CDCl3, δ, ppm): 172.93–172.78, 138.15–137.77, 74.52–74.24, 47.53, 47.34, 46.95, 46.88, 46.78, 45.68, 45.19, 40.68, 40.23, 34.33, 31.49–31.35, 26.19, 25.96, 23.42, 23.35, 22.07, 20.95, 16.57, 16.25.
- Monomer M2
- 1H NMR (400 MHz, CDCl3, δ, ppm): 6.28 (br. S., 1H), 6.20 (br.s., 1H), 4.68–4.53 (m, 2H), 3.25–3.15 (m, 4H), 2.05–1.96 (m, 2H), 1.95–1.82 (m, 2H), 1.68–1.62 (m, 4H), 1.47–1.23 (m, 7H), 1.10–0.96 (m, 2H), 0.94–0.79 (m, 17H), 0.78–0.70 (m, 6H).
- 13C NMR (100 MHz, CDCl3, δ, ppm): 171.98, 171.79, 135.32, 134.52, 74.36, 74.00, 48.62, 48.18, 48.14, 47.48, 47.04, 46.78, 40.95, 40.84, 34.50, 34.45, 31.59, 31.45, 26.25, 25.96, 23.42, 23.37, 22.19, 21.09, 21.06, 16.46, 16.36.
- Monomer M3
- 1H NMR (400 MHz, CDCl3, δ, ppm): 6.24 (br. S., 2H), 3.11–3.07 (m, 2H), 2.68–2.63 (m, 2H), 2.43–2.24 (m, 2H), 2.21–2.14 (m, 1H), 2.00–1.85 (m, 2H), 1.81–1.63 (m, 4H), 1.55–1.46 (m, 1H), 1.36–1.12 (m, 5H), 1.12–0.96 (m, 2H), 0.92–0.82 (m, 20H).
- 13C NMR (100 MHz, CDCl3, δ, ppm): 173.62, 173.61, 137.99, 137.94, 80.40, 80.15, 48.96, 48.58, 47.82, 47.70, 47.38, 45.91, 45.35, 44.86, 44.84, 36.87, 36.14, 28.03, 27.24, 27.22, 19.71, 18.85, 13.65, 13.51.
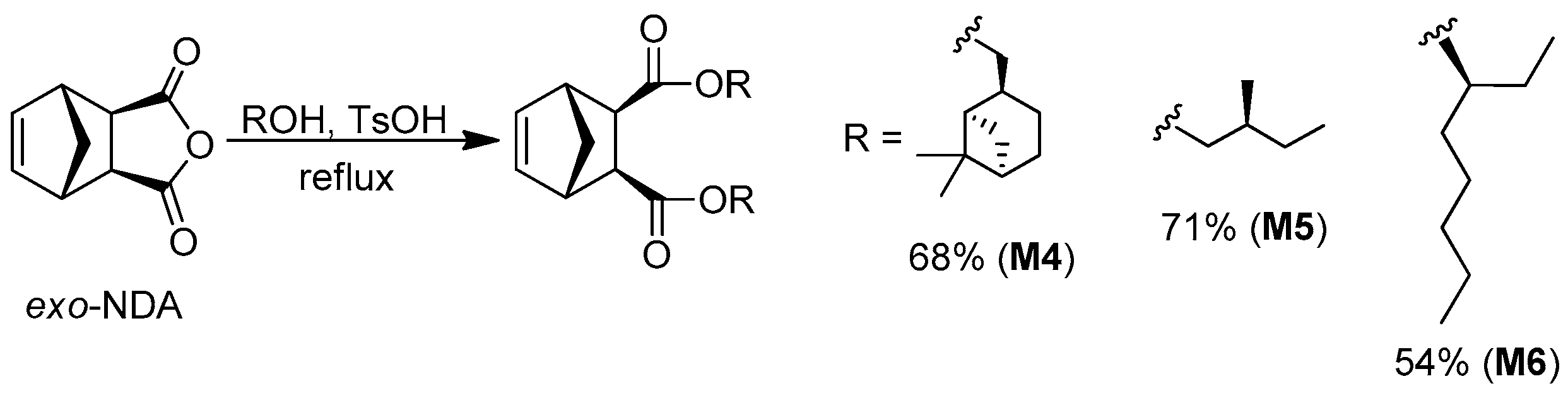
2.6. General Procedure for the Synthesis of Monomers M4–M6
- 1H NMR (300 MHz, CDCl3, δ, ppm): 6.15 (br. s, 2H, R-CH=CH-R), 3.96–3.67 (M, 4H), 3.02 (s, 2H), 2.57 (s, 2H), 2.07 (m, 2H), 1.67–1.55 (m, 2H), 1.45–1.29 (m, 3H), 1.23–1.03 (m, 2H), 0.92–0.77 (m, 12H).
- 13C NMR (100 MHz, CDCl3, δ, ppm): 172.99, 137.36, 68.82, 46.77, 45.13, 44.80, 33.50, 33.46, 25.48, 15.87, 15.83, 10.66, 10.61.
- Monomer M4
- 1H NMR (400 MHz, CDCl3, δ, ppm): 6.20 (s, 2H, R-CH=CH-R), 4.08–4.02 (m, 2H), 3.94–3.89 (m, 2H), 3.08–3.05 (m, 2H), 2.60–2.57 (m, 2H), 2.38–2.30 (m, 4H), 2.13–2.08 (m, 1H), 1.95–1.84 (m, 10H), 1.52–1.41 (m, 3H), 1.19–1.17 (m, 6H), 1.00–0.96 (m, 6H), 0.94–0.90 (m, 2H).
- 13C NMR (100 MHz, CDCl3, δ, ppm): 173.72, 173.67, 138.09, 138.07, 65.51, 69.30, 47.49, 47.47, 45.90, 45.82, 45.73, 45.54, 46.40, 43.18, 41.39, 41.37, 40.41, 40.30, 38.64, 33.12, 33.07, 27.99, 27.97, 25.98, 23.39, 23.34, 18.94, 18.81.
- Monomer M6
- 1H NMR (400 MHz, CDCl3, δ, ppm): 6.17 (s, 2H, R-CH=CH-R), 4.74–4.71 (m, 2H), 3.03–3.02 (m, 2H), 2.54–2.53 (m, 2H), 2.11–2.09 (m, 1H), 1.52–1.42 (m, 9H), 1.37–1.17 (m, 12H), 0.87–0.83 (m, 12H).
- 13C NMR (100 MHz, CDCl3, δ, ppm): 173.15, 138.03, 137.99, 75.84, 75.79, 47.45, 47.37, 46.19, 46.08, 45.33, 33.21, 33.13, 31.92, 31.82, 26.55, 26.54, 25.05, 24.93, 22.62, 14.09, 14.06, 9.63, 9.50.
- Epoxide based on M5
- 1H NMR (400 MHz, CDCl3, δ, ppm): 4.03–3.72 (M, 4H), 3.18 (s, 2H), 2.82 (s, 2H), 2.78 (s, 2H), 1.76–0.84 (m, 20H).
- 13C NMR (100 MHz, CDCl3, δ, ppm): 172.11, 69.68, 50.68, 47.08, 40.97, 34.02, 33.98, 26.03, 23.27, 16.43, 16.40, 11.23, 11.19.
- MS (EI, m/z (intensity, %)): 338 (2%, M+), 181 (60%, C9H9O4+).
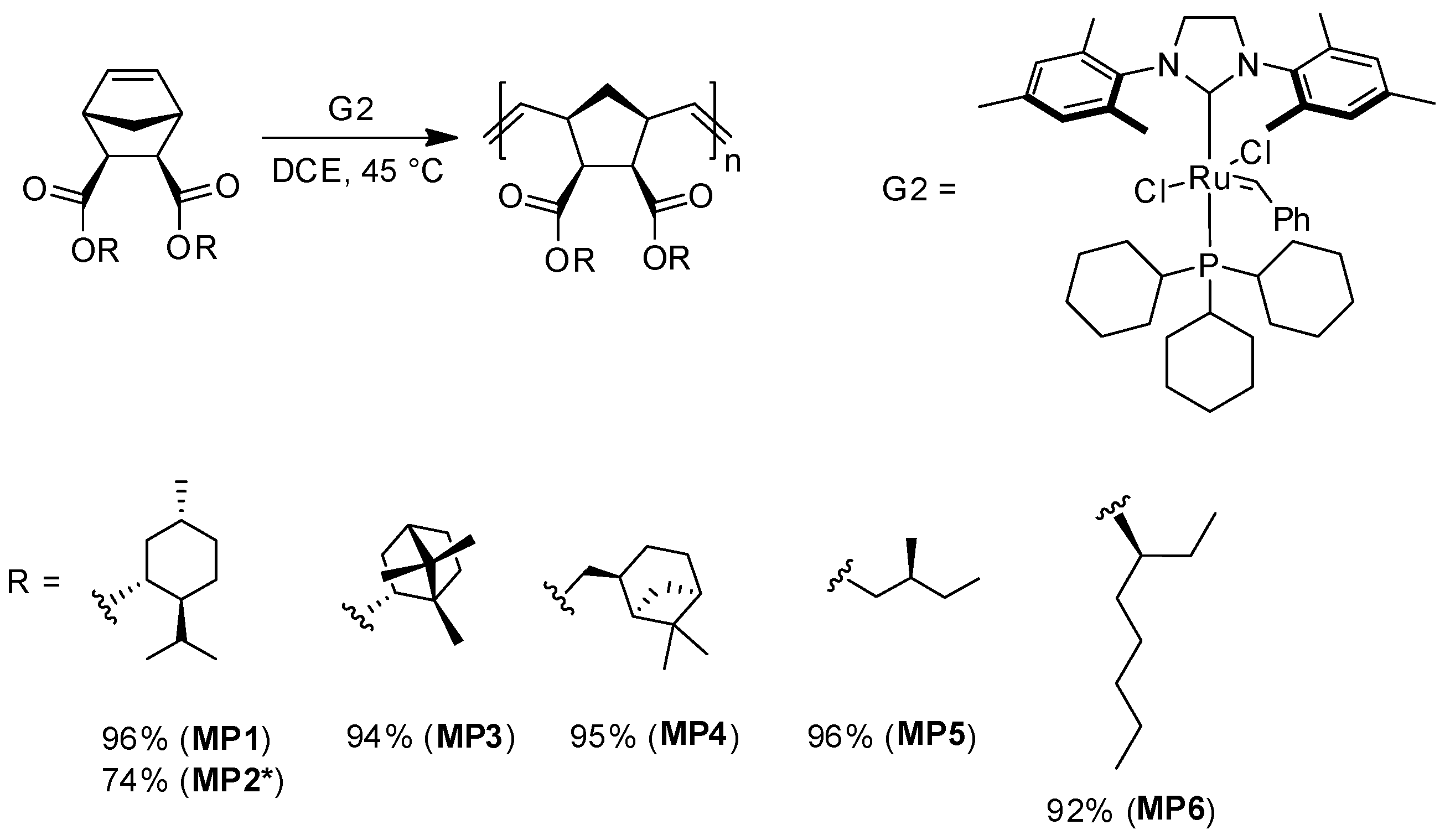
2.7. General Procedure of ROMP Polymerization (MP1–MP6)
- 1H NMR (400 MHz, CDCl3, δ, ppm): 5.51–5.23 (m, 2H, HRC=CRH), 3.94–0.72 (m, 31H).
- 13C NMR (100 MHz, CDCl3, δ, ppm): 178.14–178.10 (C=O), 133.37–131.80, 69.74–69.30, 53.91–52.62, 45.71–44.80, 41.25–39.45, 34.27–33.96, 26.31–26.05, 16.72–16.33, 11.41–11.23.
- Polymer MP1
- 1H NMR (400 MHz, CDCl3, δ, ppm): 5.55–4.51 (m, 4H), 3.56–0.56 (m, 42H).
- 13C NMR (100 MHz, CDCl3, δ, ppm): 172.87–171.44, 134.54–131.65, 74.50–74.00, 54.31–52.07, 47.55–44.51, 41.26–39.18, 34.72–34.13, 31.90–31.32, 26.62–25.70, 23.86–23.18, 22.61–21.82, 24.41–20.85, 16.97–16.10.
- Polymer MP2
- 1H NMR (400 MHz, CDCl3, δ, ppm): 5.74–5.28 (m, 2H), 4.81–4.55 (m, 2H), 3.31–0.45 (m, 42H).
- 13C NMR (100 MHz, CDCl3, δ, ppm): 171.94–170.91, 132.14–130.53, 75.12–73.81, 52.21–15.62.
- Polymer MP3
- 1H NMR (400 MHz, CDCl3, δ, ppm): 5.66–5.12 (m, 2H), 5.01–4.71 (m, 2H), 3.31–0.45 (m, 38H).
- 13C NMR (100 MHz, CDCl3, δ, ppm): 173.15–171.92, 133.42–131.48, 80.68–79.12, 53.62–13.52.
- Polymer MP4
- 1H NMR (400 MHz, CDCl3, δ, ppm): 5.50–5.11 (m, 2H, HRC=CRH), 4.12–0.77 (m, 40H).
- 13C NMR (100 MHz, CDCl3, δ, ppm): 172.94–172.52, 133.20–132.26, 69.29–69.27, 53.58–52.59, 45.25–38.60, 33.20–33.00, 28.12–27.95, 25.98–25.96, 23.42–23.41, 19.05–18.95
- Polymer MP6
- 1H NMR (400 MHz, CDCl3, δ, ppm): 5.54–5.13 (m, 2H), 4.81–4.64 (m, 2H), 3.53–0.76 (m, 38H).
- 13C NMR (100 MHz, CDCl3, δ, ppm): 172.93–171.85, 134.12–131.61, 76.16–75.43, 54.30–52.16, 46.07–44.27, 41.03–39.23, 33.73–32.86, 32.16–31.58, 27.12–26.25, 25.50–24.51, 22.95–22.43, 14.55–13.79, 9.97–9.39.
2.8. Synthesis of Polymer MP7
- 1H NMR (400 MHz, CDCl3, δ, ppm): 5.54–5.37 (m, 2H), 4.75–458 (m, 2H), 3.10–0.55 (m, 42H).
3. Results and Discussion
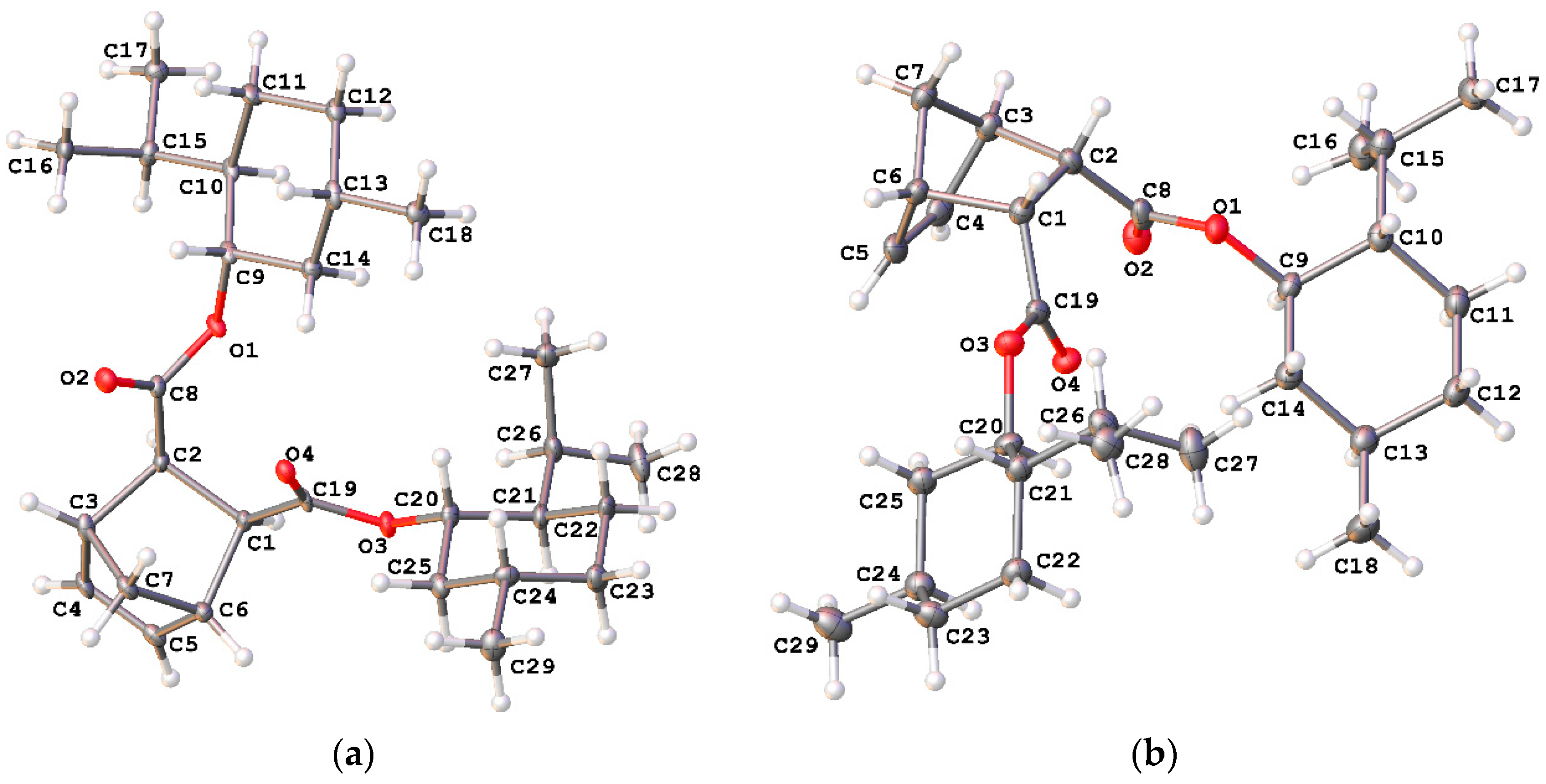
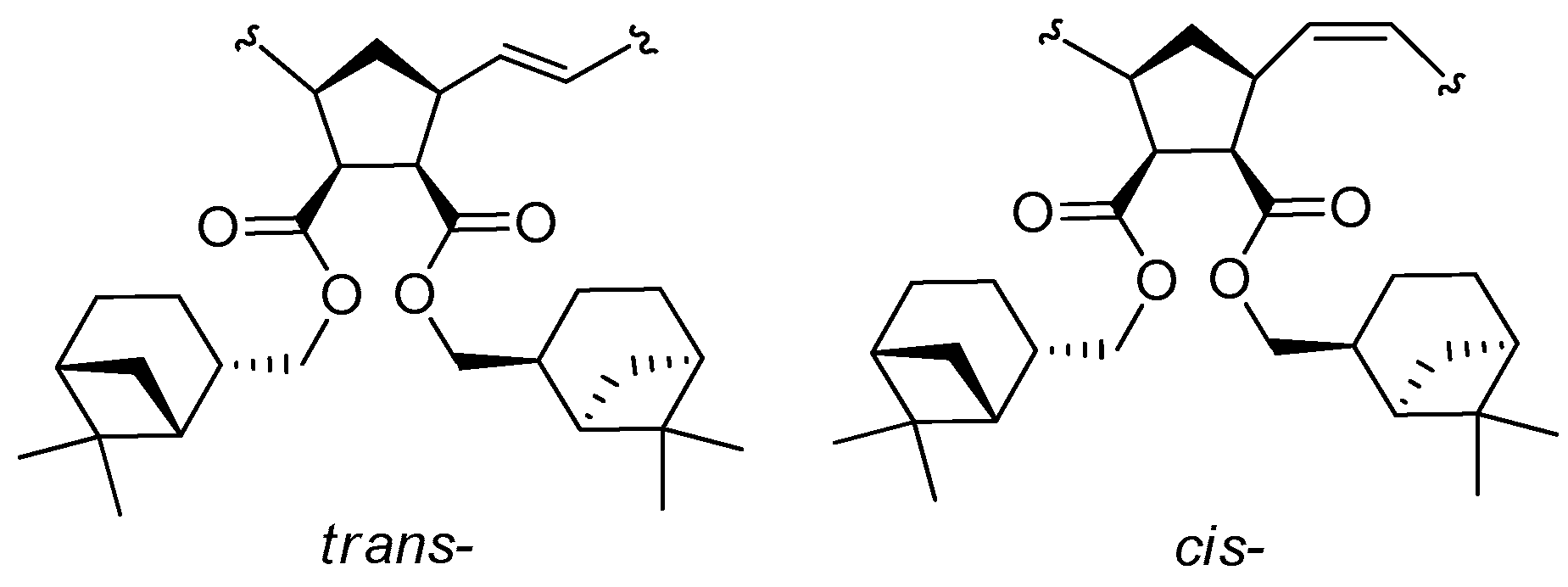
3.1. Synthesis of Monomers
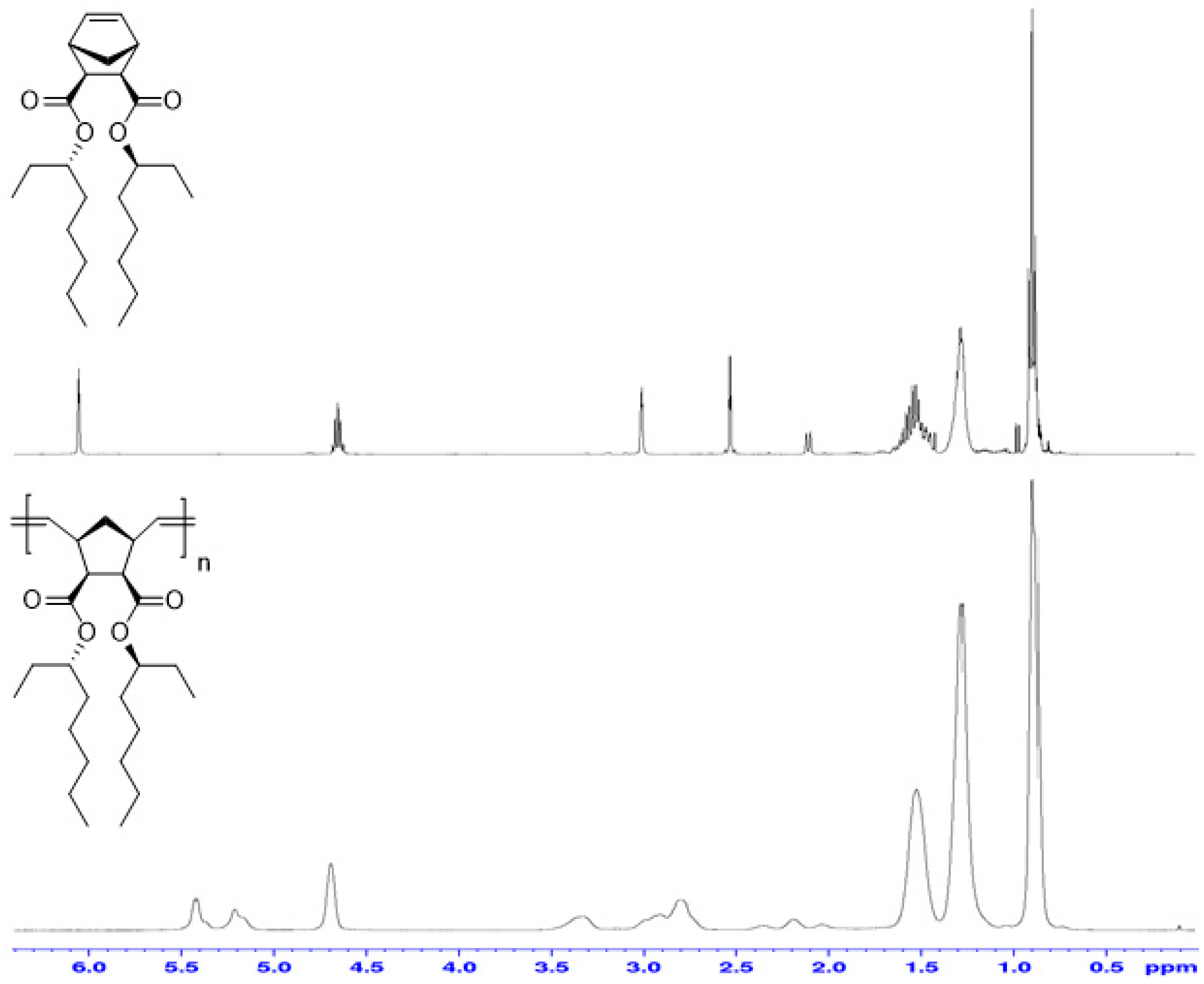
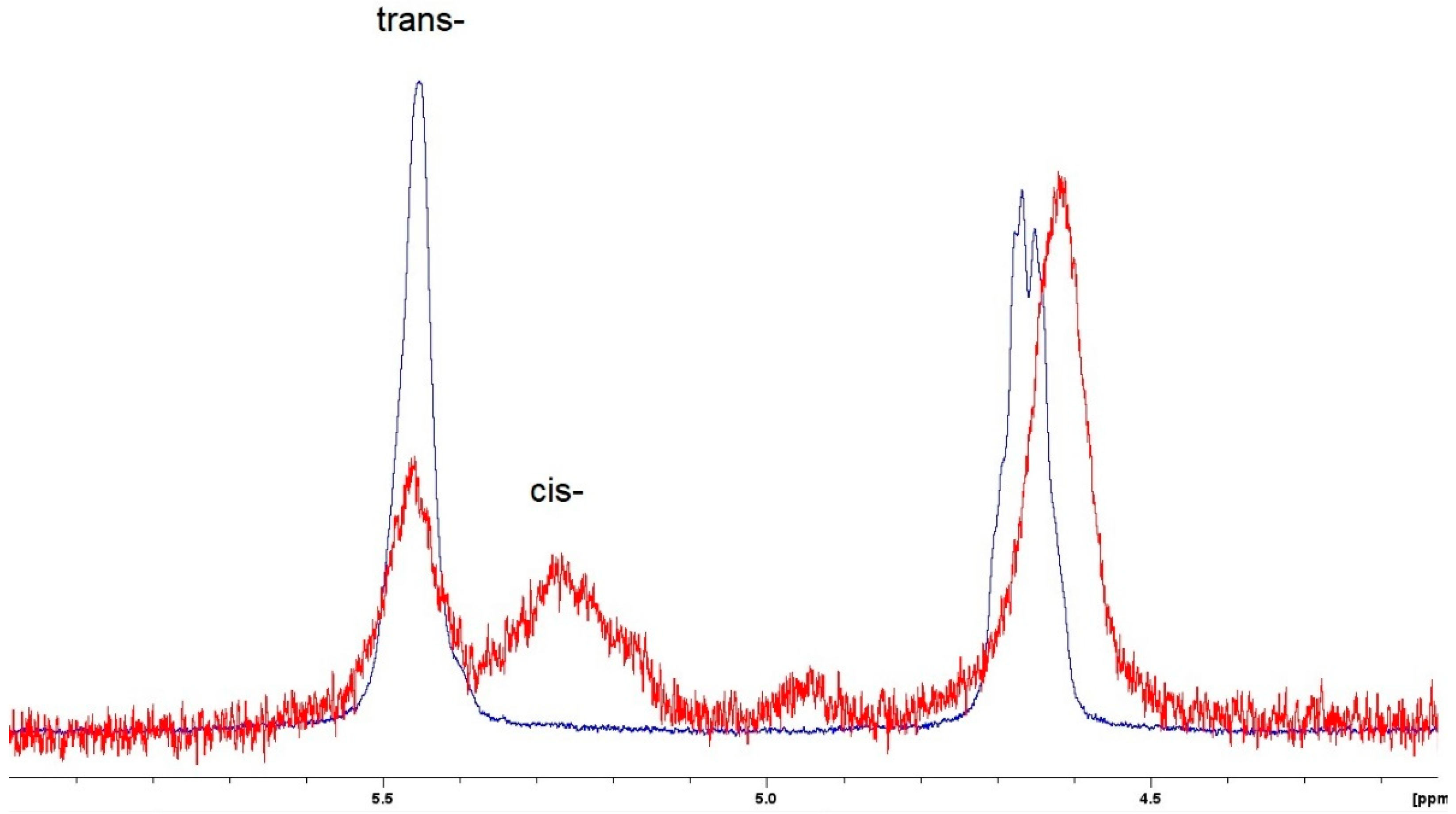
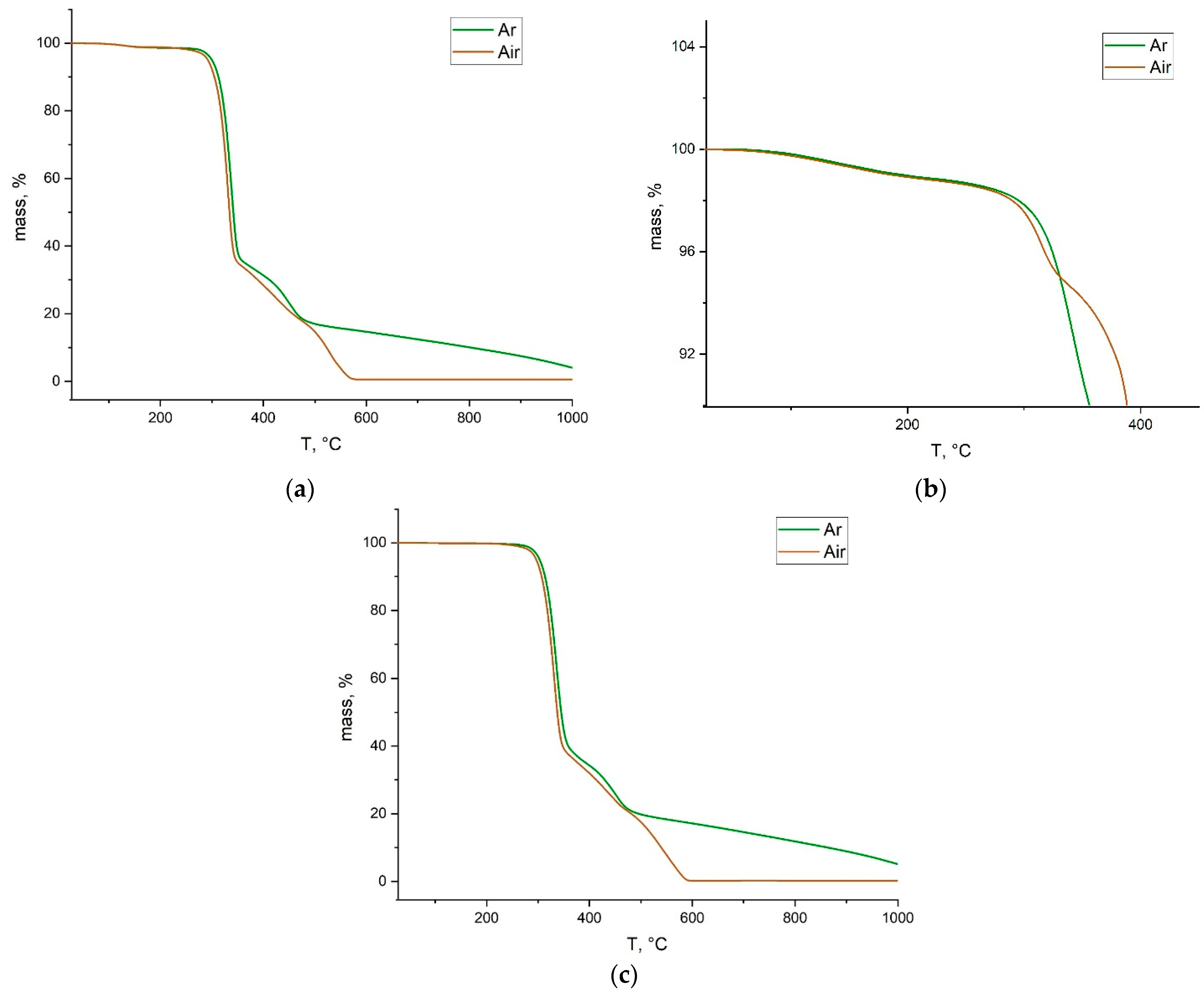
3.2. Ring-Opening Metathesis Polymerization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bolm, C.; Tanyeli, C.; Grenz, A.; Dinter, C.L. ROMP-Polymers in Asymmetric Catalysis: The Role of the Polymer Backbone. Adv. Synth. Catal. 2002, 344, 649–656. [Google Scholar] [CrossRef]
- Abdelkawy, M.A.; Davin, C.; Aly, E.-S.A.; El-Badawi, M.A.; Itsuno, S. Chiral Polyureas Derived Cinchona Alkaloids: Highly Efficient Bifunctional Organocatalysts for the Asymmetric Michael Addition Reaction. ChemistrySelect 2021, 6, 11971–11979. [Google Scholar] [CrossRef]
- Wali Ullah, M.; Haraguchi, N. Asymmetric Diels-Alder Reaction Catalyzed by Facile Recoverable Ionically Core-Corona Polymer Microsphere-Immobilized MacMillan Catalyst. ChemistrySelect 2022, 7, e202202568. [Google Scholar] [CrossRef]
- Han, H.; Liu, W.; Xiao, Y.; Ma, X.; Wang, Y. Advances of enantioselective solid membranes. New J. Chem. 2021, 45, 6586–6599. [Google Scholar] [CrossRef]
- Vedovello, P.; Marcio Paranhos, C.; Fernandes, C.; Elizabeth Tiritan, M. Chiral polymeric membranes: Recent applications and trends. Sep. Purif. Technol. 2022, 280, 119800. [Google Scholar] [CrossRef]
- Choi, H.-J.; Ahn, Y.-H.; Koh, D.-Y. Enantioselective Mixed Matrix Membranes for Chiral Resolution. Membranes 2021, 11, 279. [Google Scholar] [CrossRef]
- Guntari, S.N.; Nam, E.; Pranata, N.N.; Chia, K.; Wong, E.H.H.; Blencowe, A.; Goh, T.K.; Caruso, F.; Qiao, G.G. Fabrication of Chiral Stationary Phases via Continuous Assembly of Polymers for Resolution of Enantiomers by Liquid Chromatography. Macromol. Mater. Eng. 2014, 299, 1285–1291. [Google Scholar] [CrossRef]
- Teixeira, J.; Tiritan, M.E.; Pinto, M.M.M.; Fernandes, C. Chiral Stationary Phases for Liquid Chromatography: Recent Developments. Molecules 2019, 24, 865. [Google Scholar] [CrossRef]
- Betzenbichler, G.; Huber, L.; Kräh, S.; Morkos, M.-L.K.; Siegle, A.F.; Trapp, O. Chiral stationary phases and applications in gas chromatography. Chirality 2022, 34, 732–759. [Google Scholar] [CrossRef]
- Wang, L.; Lu, N.; Huang, S.; Wang, M.; Chen, X.-M.; Yang, H. Optically Active Nucleobase-Functionalized Polynorbornenes Mimicking Double-Helix DNA. CCS Chem. 2021, 3, 1787–1796. [Google Scholar] [CrossRef]
- Dana, D.M.; Yitzhak, M. Biomimetic Polymers for Chiral Resolution and Antifreeze Applications. In On Biomimetics; Lilyana, D.P., Ed.; IntechOpen: Rijeka, Croatia, 2011. [Google Scholar]
- Green, J.J.; Elisseeff, J.H. Mimicking biological functionality with polymers for biomedical applications. Nature 2016, 540, 386–394. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, C.; Di Girolamo, R.; Talarico, G. Expanding the Origin of Stereocontrol in Propene Polymerization Catalysis. ACS Catal. 2016, 6, 3767–3770. [Google Scholar] [CrossRef]
- Emmerich, A.; Daniliuc, C.G.; Studer, A. Synthesis of Polymers Bearing a Chiral Backbone via Stereospecific Ionic Ring-Opening Polymerization of Chiral Donor-Acceptor Cyclopropanes. Macromol. Rapid Commun. 2021, 42, 2100030. [Google Scholar] [CrossRef]
- Mizuta, K.; Fukutomi, S.; Yamabuki, K.; Onimura, K.; Oishi, T. Ring-opening metathesis polymerization of N-substituted-5-norbornene-2,3-dicarboximides in the presence of chiral additives. Polym. J. 2010, 42, 534–539. [Google Scholar] [CrossRef]
- Zang, Y.; Aoki, T.; Teraguchi, M.; Kaneko, T.; Ma, L.; Jia, H.; Miao, F. Synthesis of Well-Defined Chiral Oligopinanylsiloxane Graft Copoly(phenylacetylene)s Using the Macromonomer Method and Their Enantioselective Permeability. ACS Appl. Polym. Mater. 2020, 2, 853–861. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, C.; Zhou, Z.; Zhu, R.; Liu, L.; Bai, J.; Dong, H.; Satoh, T.; Okamoto, Y. Influence of different sequences of l-proline dipeptide derivatives in the pendants on the helix of poly(phenylacetylene)s and their enantioseparation properties. Polym. Chem. 2019, 10, 4810–4817. [Google Scholar] [CrossRef]
- Daeffler, C.S.; Miyake, G.M.; Li, J.; Grubbs, R.H. Partial Kinetic Resolution of Oxanorbornenes by Ring-Opening Metathesis Polymerization with a Chiral Ruthenium Initiator. ACS Macro Lett. 2014, 3, 102–104. [Google Scholar] [CrossRef]
- Ogba, O.M.; Warner, N.C.; O'Leary, D.J.; Grubbs, R.H. Recent advances in ruthenium-based olefin metathesis. Chem. Soc. Rev. 2018, 47, 4510–4544. [Google Scholar] [CrossRef]
- Alentiev, D.A.; Bermeshev, M.V. Design and Synthesis of Porous Organic Polymeric Materials from Norbornene Derivatives. Polym. Rev. 2021, 62, 400–437. [Google Scholar] [CrossRef]
- Ponkratov, D.O.; Shaplov, A.S.; Vygodskii, Y.S. Metathesis Polymerization in Ionic Media. Polym. Sci. Ser. C 2019, 61, 2–16. [Google Scholar] [CrossRef]
- Petrov, V.A.; Vasil'ev, N.V. Synthetic chemistry of quadricyclane. Curr. Org. Synth. 2006, 3, 215–259. [Google Scholar] [CrossRef]
- Houk, K.N.; Liu, F.; Yang, Z.; Seeman, J.I. Evolution of the Diels–Alder Reaction Mechanism since the 1930s: Woodward, Houk with Woodward, and the Influence of Computational Chemistry on Understanding Cycloadditions. Angew. Chem. Int. Ed. 2021, 60, 12660–12681. [Google Scholar] [CrossRef] [PubMed]
- Guseva, M.A.; Alentiev, D.A.; Bermesheva, E.V.; Zamilatskov, I.A.; Bermeshev, M.V. The selective hydrosilylation of norbornadiene-2,5 by monohydrosiloxanes. RSC Adv. 2019, 9, 33029–33037. [Google Scholar] [CrossRef] [PubMed]
- Sundell, B.J.; Lawrence, J.A.; Harrigan, D.J.; Lin, S.; Headrick, T.P.; O’Brien, J.T.; Penniman, W.F.; Sandler, N. Exo-selective, Reductive Heck Derived Polynorbornenes with Enhanced Molecular Weights, Yields, and Hydrocarbon Gas Transport Properties. ACS Macro Lett. 2020, 9, 1363–1368. [Google Scholar] [CrossRef]
- Bermeshev, M.V.; Chapala, P.P. Addition polymerization of functionalized norbornenes as a powerful tool for assembling molecular moieties of new polymers with versatile properties. Prog. Polym. Sci. 2018, 84, 1–46. [Google Scholar] [CrossRef]
- Bermesheva, E.V.; Wozniak, A.I.; Andreyanov, F.A.; Karpov, G.O.; Nechaev, M.S.; Asachenko, A.F.; Topchiy, M.A.; Melnikova, E.K.; Nelyubina, Y.V.; Gribanov, P.S.; et al. Polymerization of 5-Alkylidene-2-norbornenes with Highly Active Pd–N-Heterocyclic Carbene Complex Catalysts: Catalyst Structure–Activity Relationships. ACS Catal. 2020, 10, 1663–1678. [Google Scholar] [CrossRef]
- Bermeshev, M.V.; Bulgakov, B.A.; Genaev, A.M.; Kostina, J.V.; Bondarenko, G.N.; Finkelshtein, E.S. Cationic Polymerization of Norbornene Derivatives in the Presence of Boranes. Macromolecules 2014, 47, 5470–5483. [Google Scholar] [CrossRef]
- García-Loma, R.; Albéniz, A.C. Vinylic Addition Polynorbornene in Catalysis. Asian J. Org. Chem. 2019, 8, 304–315. [Google Scholar] [CrossRef]
- Coles, M.P.; Gibson, V.C.; Mazzariol, L.; North, M.; Teasdale, W.G.; Williams, C.M.; Zamuner, D. Amino acid derived homochiral polymers via ring-opening metathesis polymerisation. J. Chem. Soc. Chem. Commun. 1994, 21, 2505–2506. [Google Scholar] [CrossRef]
- Sutthasupa, S.; Terada, K.; Sanda, F.; Masuda, T. Ring-opening metathesis polymerization of amino acid-functionalized norbornene diester monomers. Polymer 2007, 48, 3026–3032. [Google Scholar] [CrossRef]
- Rosebrugh, L.E.; Marx, V.M.; Keitz, B.K.; Grubbs, R.H. Synthesis of Highly Cis, Syndiotactic Polymers via Ring-Opening Metathesis Polymerization Using Ruthenium Metathesis Catalysts. J. Am. Chem. Soc. 2013, 135, 10032–10035. [Google Scholar] [CrossRef] [PubMed]
- Choinopoulos, I.; Koinis, S.; Pitsikalis, M. Synthesis and characterization of chiral poly(l-lactide-b-hexyl isocyanate) macromonomers with norbornenyl end groups and their homopolymerization through ring opening metathesis polymerization to afford polymer brushes. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 1102–1112. [Google Scholar] [CrossRef]
- Fadlallah, S.; Peru, A.A.M.; Flourat, A.L.; Allais, F. A straightforward access to functionalizable polymers through ring-opening metathesis polymerization of levoglucosenone-derived monomers. Eur. Polym. J. 2020, 138, 109980. [Google Scholar] [CrossRef]
- Fadlallah, S.; Peru, A.A.M.; Longé, L.; Allais, F. Chemo-enzymatic synthesis of a levoglucosenone-derived bi-functional monomer and its ring-opening metathesis polymerization in the green solvent Cyrene™. Polym. Chem. 2020, 11, 7471–7475. [Google Scholar] [CrossRef]
- Souto, J.A.; Stockman, R.A.; Ley, S.V. Development of a flow method for the hydroboration/oxidation of olefins. Org. Biomol. Chem. 2015, 13, 3871–3877. [Google Scholar] [CrossRef]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Bolm, C.; Atodiresei, I.; Schiffers, I. Asymmetric alcoholysis of meso-anhydrides mediated by alkaloids. Org. Synth. 2005, 82, 120–125. [Google Scholar] [CrossRef]
- Bermesheva, E.V.; Nazarov, I.V.; Kataranova, K.D.; Khrychikova, A.P.; Zarezin, D.P.; Melnikova, E.K.; Asachenko, A.F.; Topchiy, M.A.; Rzhevskiy, S.A.; Bermeshev, M.V. Cocatalyst versus precatalyst impact on the vinyl-addition polymerization of norbornenes with polar groups: Looking at the other side of the coin. Polym. Chem. 2021, 12, 6355–6362. [Google Scholar] [CrossRef]
- Yushkin, A.; Grekhov, A.; Matson, S.; Bermeshev, M.; Khotimsky, V.; Finkelstein, E.; Budd, P.M.; Volkov, V.; Vlugt, T.J.H.; Volkov, A. Study of glassy polymers fractional accessible volume (FAV) by extended method of hydrostatic weighing: Effect of porous structure on liquid transport. React. Funct. Polym. 2015, 86, 269–281. [Google Scholar] [CrossRef]
- Askadskii, A.A. Computational Materials Science of Polymers; Cambridge International Science Publishing Ltd.: Cambridge, UK, 2003; p. 16. [Google Scholar]
- Wilks, B.R.; Chung, W.J.; Ludovice, P.J.; Rezac, M.R.; Meakin, P.; Hill, A.J. Impact of average free-volume element size on transport in stereoisomers of polynorbornene. I. Properties at 35 °C. J. Polym. Sci. Part B Polym. Phys. 2003, 41, 2185–2199. [Google Scholar] [CrossRef]
- Wilks, B.R.; Chung, W.J.; Ludovice, P.J.; Rezac, M.E.; Meakin, P.; Hill, A.J. Structural and free-volume analysis for alkyl-substituted palladium-catalyzed poly(norbornene): A combined experimental and Monte Carlo investigation. J. Polym. Sci. Part B Polym. Phys. 2006, 44, 215–233. [Google Scholar] [CrossRef]
- Gringolts, M.; Bermeshev, M.; Yampolskii, Y.; Starannikova, L.; Shantarovich, V.; Finkelshtein, E. New High Permeable Addition Poly(tricyclononenes) with Si(CH3)3 Side Groups. Synthesis, Gas Permeation Parameters, and Free Volume. Macromolecules 2010, 43, 7165–7172. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
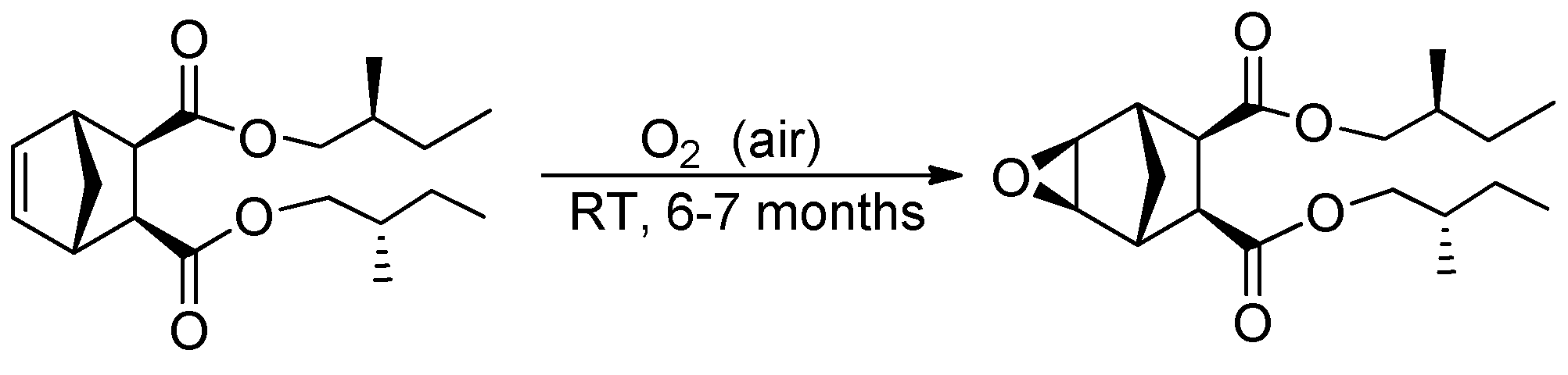{kind=link}
{kind=link}
| Monomer | Reaction Conditions a | Yield, % | [α]D b, deg·mL·g−1·dm−1 | Tm, °C |
|---|---|---|---|---|
| M1 | i | 83 | −75 (T = 25 °C, CHCl3) | 86–89 |
| M2 | i | 76 | −125 (T = 20 °C, CHCl3) | 97–100 |
| M3 | i | 72 | −42 (T = 25 °C, CHCl3) | 96–98 |
| M4 | ii | 68 | −11 (T = 20 °C, CHCl3) | Colorless oil |
| M5 | ii | 71 | +7 (T = 20 °C, THF) | Colorless oil |
| M6 | ii | 54 | +4 (T = 20 °C, CHCl3) | Colorless oil |
| Polymer | Monomer/[Ru] Molar Ratio | C, M b | Yield, % | Mw × 10−3 | Mw/Mn c | Tg, °C d | [α]D e, deg·mL·g−1·dm−1 |
|---|---|---|---|---|---|---|---|
 | 1000/1 | 0.5 | 96 | 488 | 2.1 | 80 | −59 |
 | 750/1 | 0.5 | 74 | 190 | 1.6 | 136 | −64 |
 | 500/1 | 0.5 | 94 | 469 | 2.1 | 139 | −34 |
 | 1000/1 | 0.5 | 95 | 582 | 2.9 | 68 | −11 |
 | 500/1 | 0.5 | 96 | 457 | 2.2 | −9 | +10 f |
 | 500/1 | 0.5 | 92 | 269 | 2.2 | −30 | +4 |
| Polymer | CH3CN | Acetone | DMA | DMSO | n-Hexane | PhCH3 | THF | CHCl3 |
|---|---|---|---|---|---|---|---|---|
 | ± | ± | ± | – | + | ± | + | + |
 | – | ± | ± | – | ± | + | + | + |
 | ± | ± | ± | ± | ± | + | + | + |
 | ± | ± | ± | ± | ± | + | + | + |
 | – | + | ± | ± | ± | + | + | + |
| Polymer | Density, g/cm3 | FFV, % |
|---|---|---|
 | 1.033 | 16 |
 | 1.064 | 17 |
 | 1.110 | 13 |
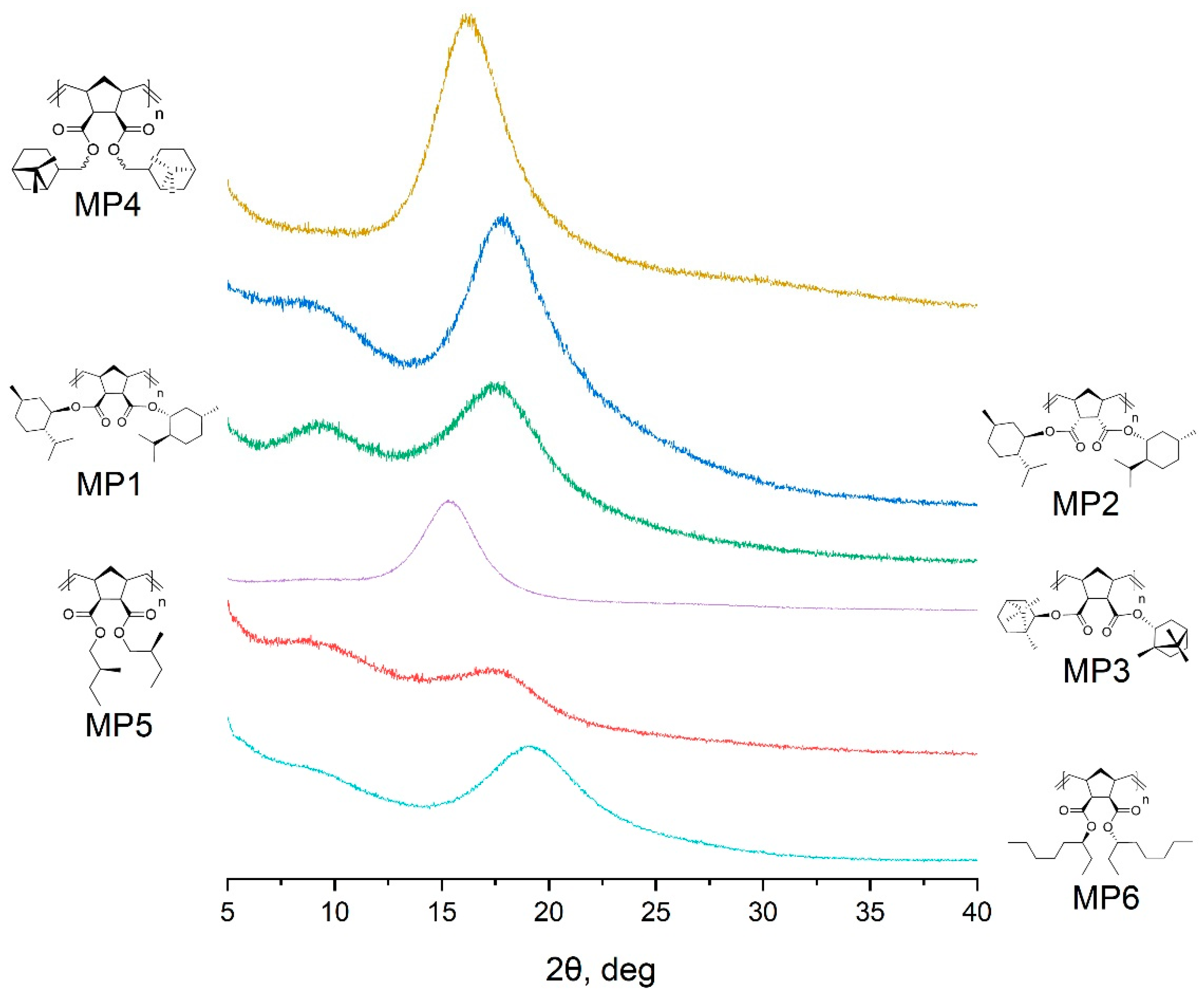
| Polymer | 2θ1,° | d1, Å | 2θ2,° | d2, Å |
|---|---|---|---|---|
| MP1 | 9.2 | 9.6 | 17.6 | 5.0 |
| MP2 | 9.1 | 9.7 | 17.8 | 5.0 |
| MP3 | 15.2 | 5.8 | - | - |
| MP4 | 15.9 | 5.6 | - | - |
| MP5 | 9.3 | 9.5 | 17.6 | 5.0 |
| MP6 | 9.1 | 9.7 | 19.0 | 4.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nazarov, I.V.; Zarezin, D.P.; Solomatov, I.A.; Danshina, A.A.; Nelyubina, Y.V.; Ilyasov, I.R.; Bermeshev, M.V. Chiral Polymers from Norbornenes Based on Renewable Chemical Feedstocks. Polymers 2022, 14, 5453. https://doi.org/10.3390/polym14245453
Nazarov IV, Zarezin DP, Solomatov IA, Danshina AA, Nelyubina YV, Ilyasov IR, Bermeshev MV. Chiral Polymers from Norbornenes Based on Renewable Chemical Feedstocks. Polymers. 2022; 14(24):5453. https://doi.org/10.3390/polym14245453
Chicago/Turabian StyleNazarov, Ivan V., Danil P. Zarezin, Ivan A. Solomatov, Anastasya A. Danshina, Yulia V. Nelyubina, Igor R. Ilyasov, and Maxim V. Bermeshev. 2022. "Chiral Polymers from Norbornenes Based on Renewable Chemical Feedstocks" Polymers 14, no. 24: 5453. https://doi.org/10.3390/polym14245453
APA StyleNazarov, I. V., Zarezin, D. P., Solomatov, I. A., Danshina, A. A., Nelyubina, Y. V., Ilyasov, I. R., & Bermeshev, M. V. (2022). Chiral Polymers from Norbornenes Based on Renewable Chemical Feedstocks. Polymers, 14(24), 5453. https://doi.org/10.3390/polym14245453