
Study on Mechanical Properties of Polyurethane Cross-Linked P(E-co-T)/PEG Blended Polyether Elastomer

Abstract
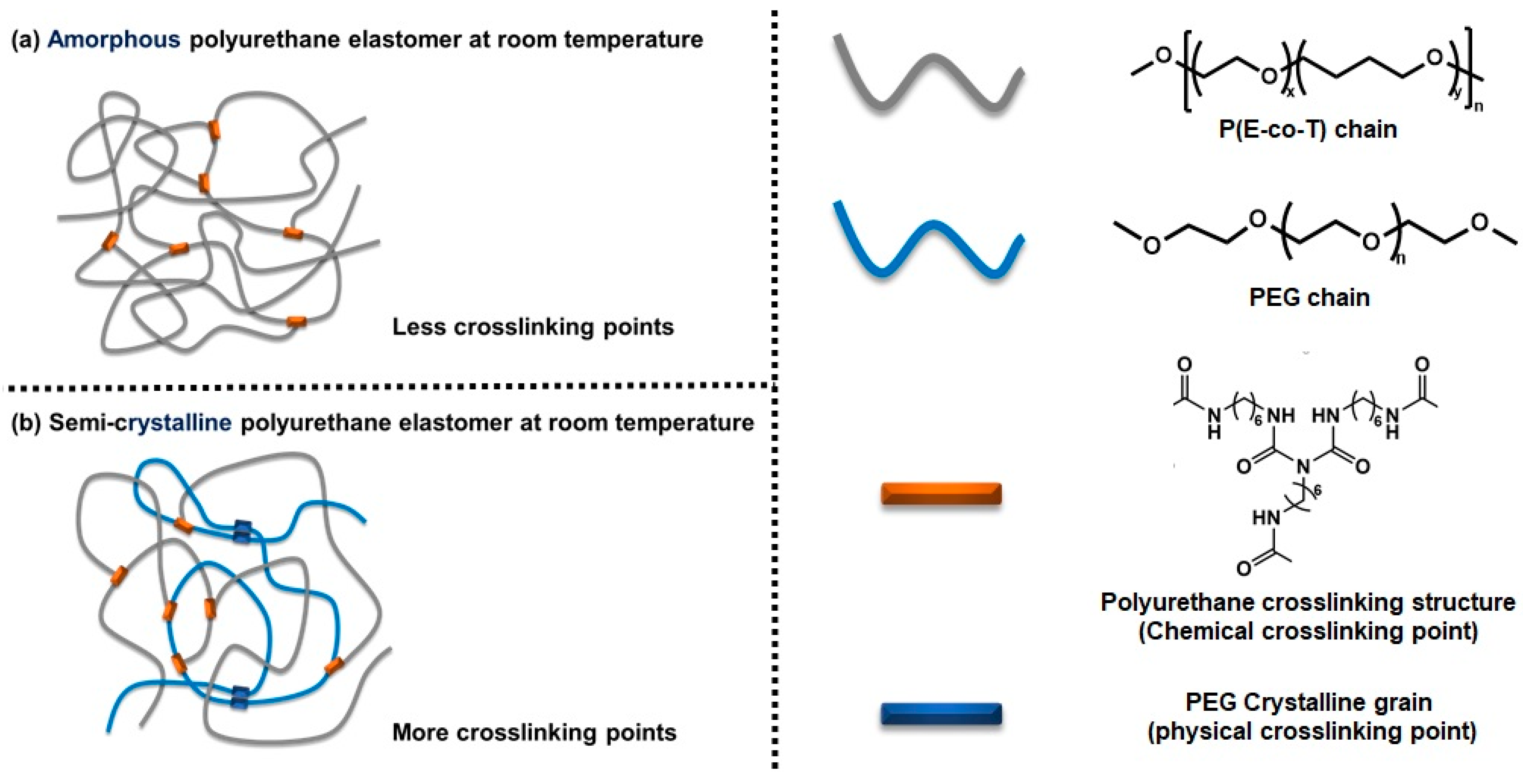
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Elastomers
2.3. Characterization
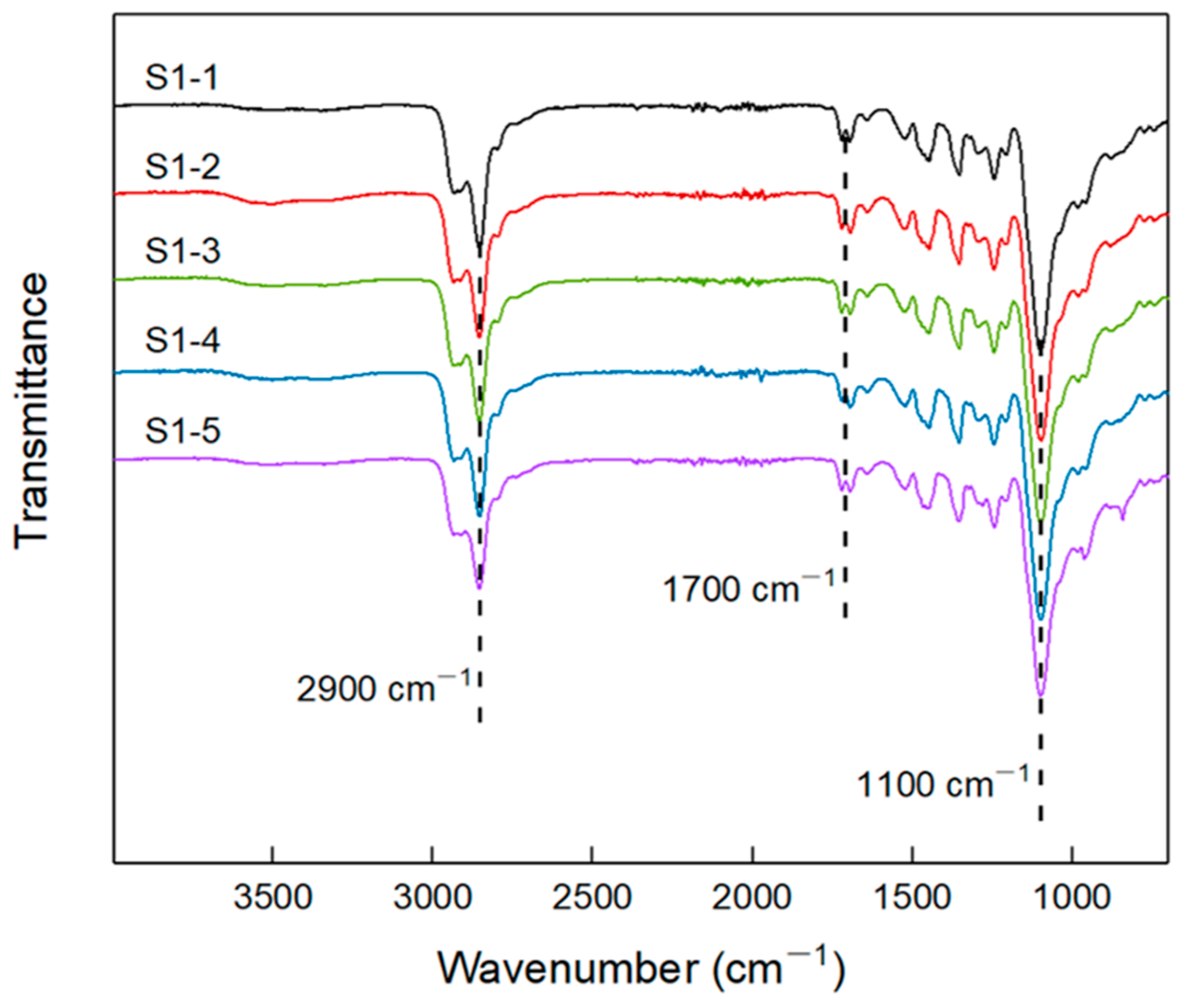
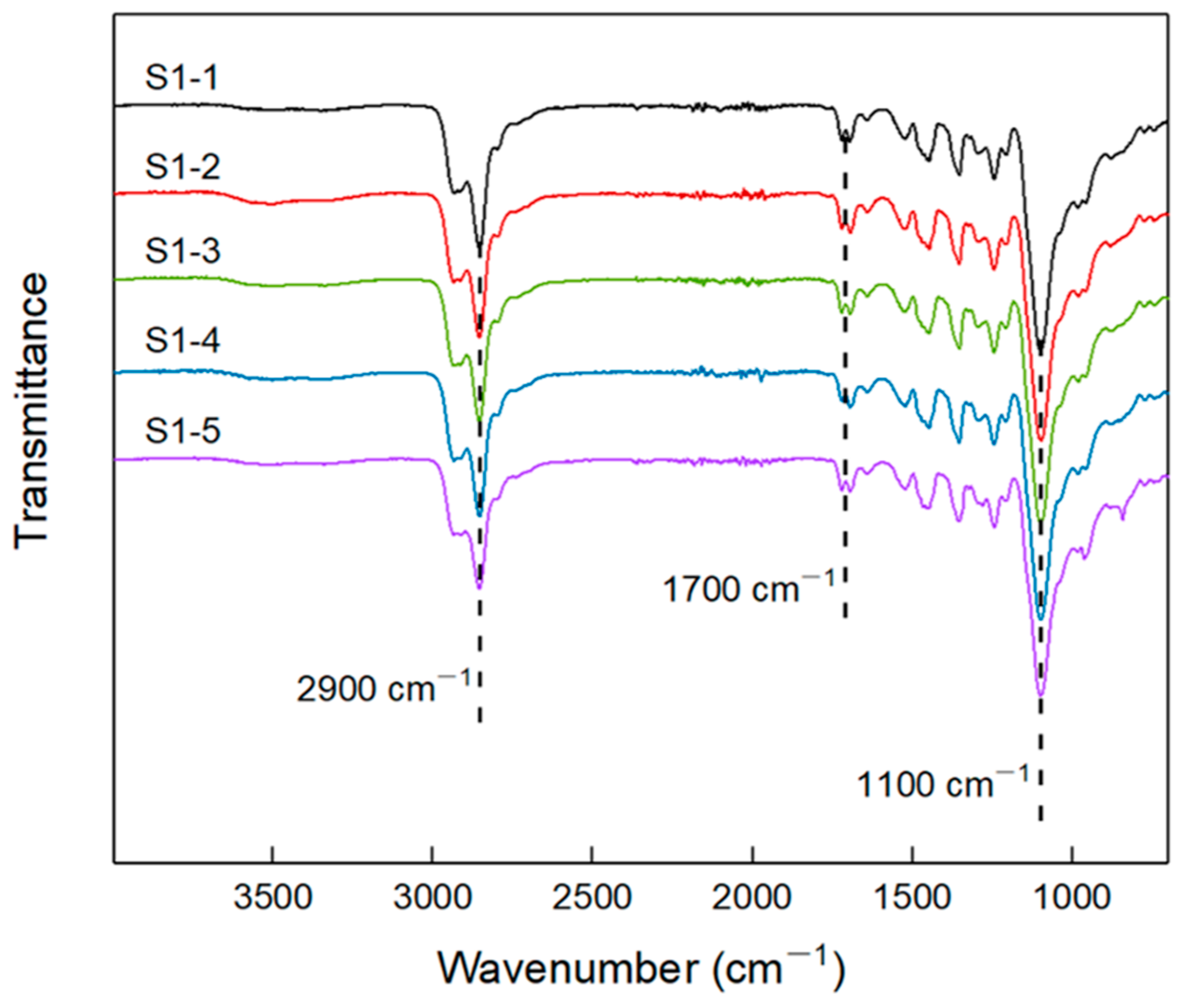
2.3.1. FTIR
2.3.2. Density Test
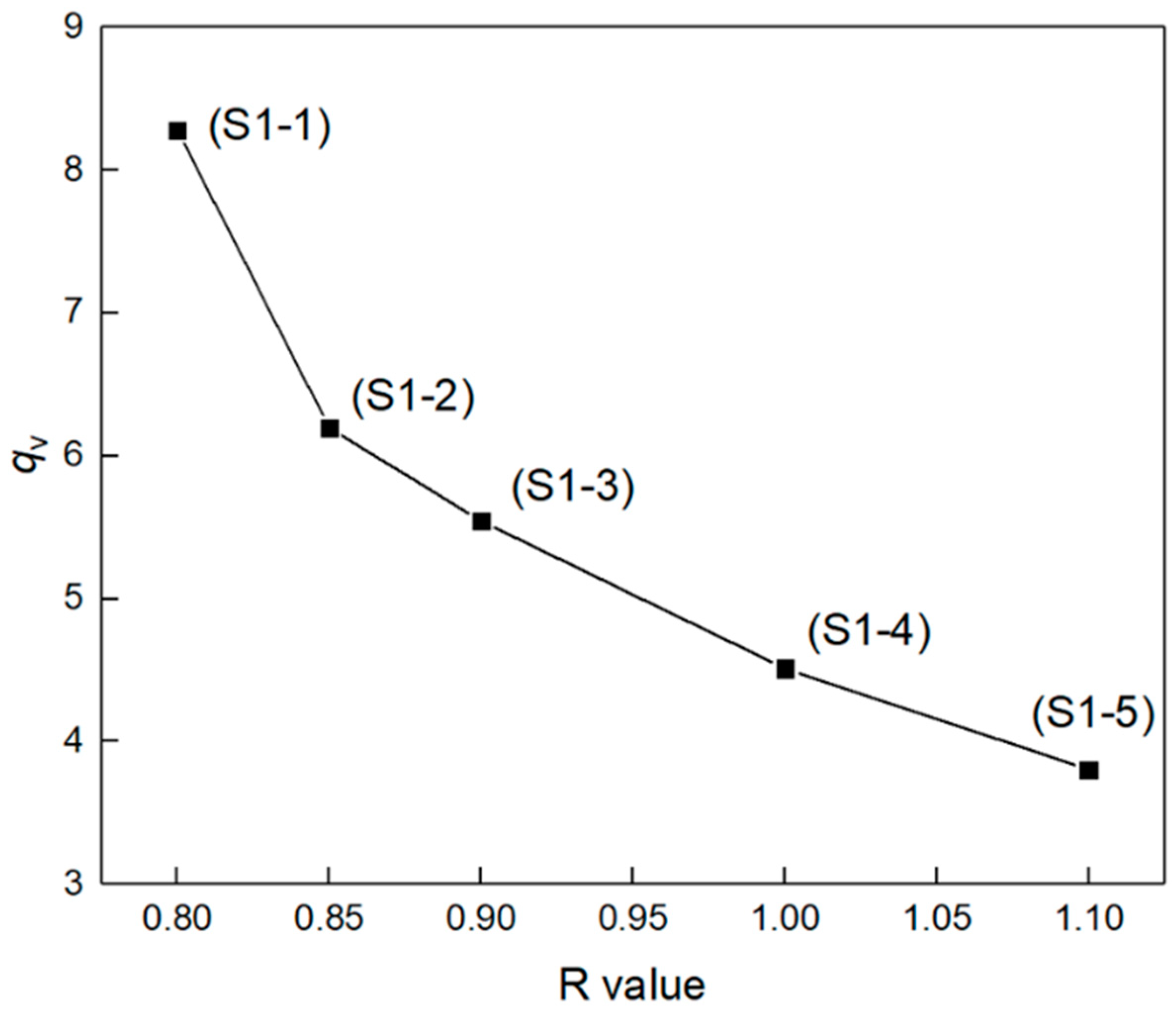
2.3.3. Equilibrium Swelling Measurement
2.3.4. DSC Test
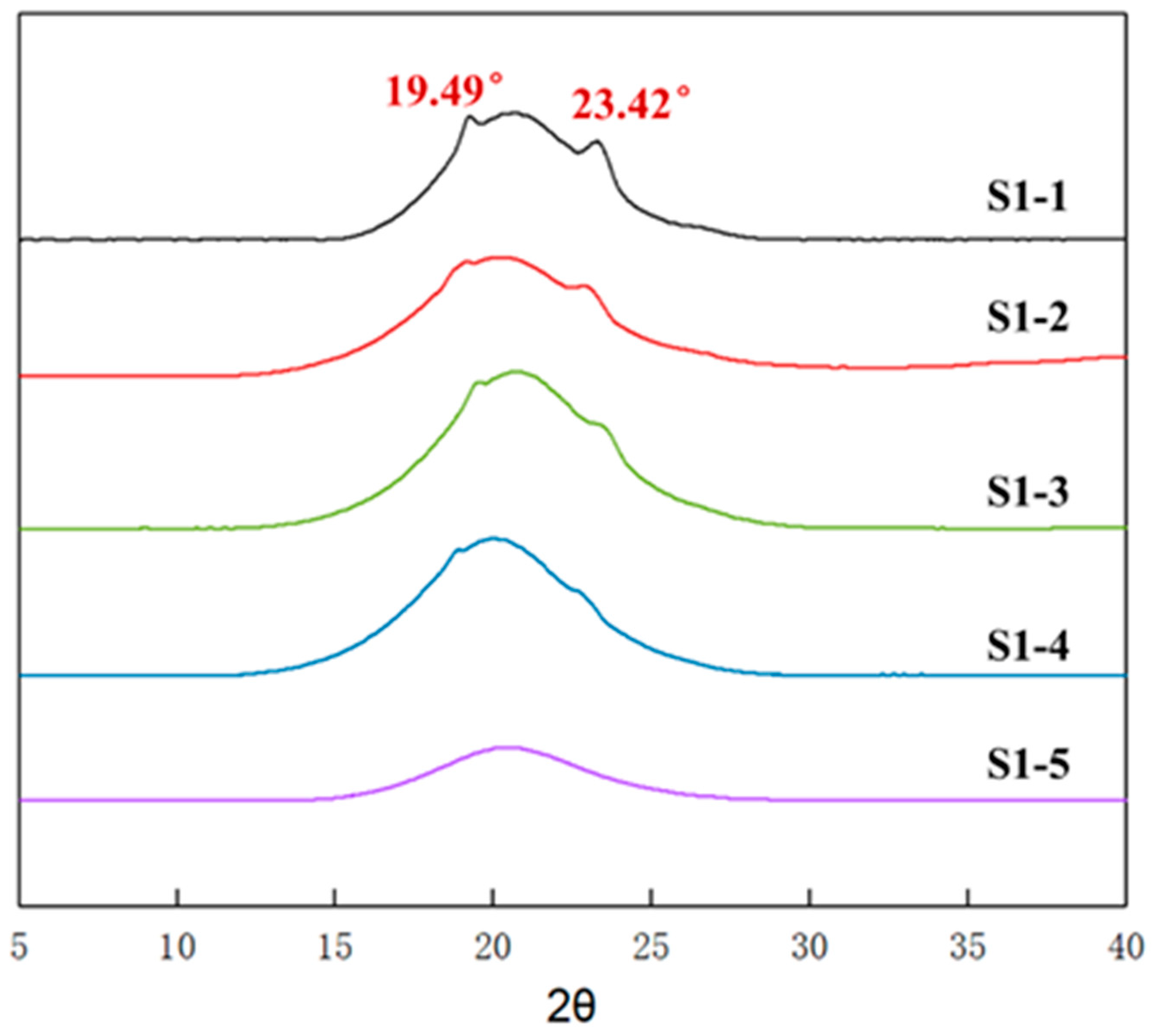
2.3.5. Wide-Angle X-ray Diffraction Test
2.3.6. Mechanical Properties Test
3. Result and Discussion
3.1. FT-IR of Elastomers with Different R Values
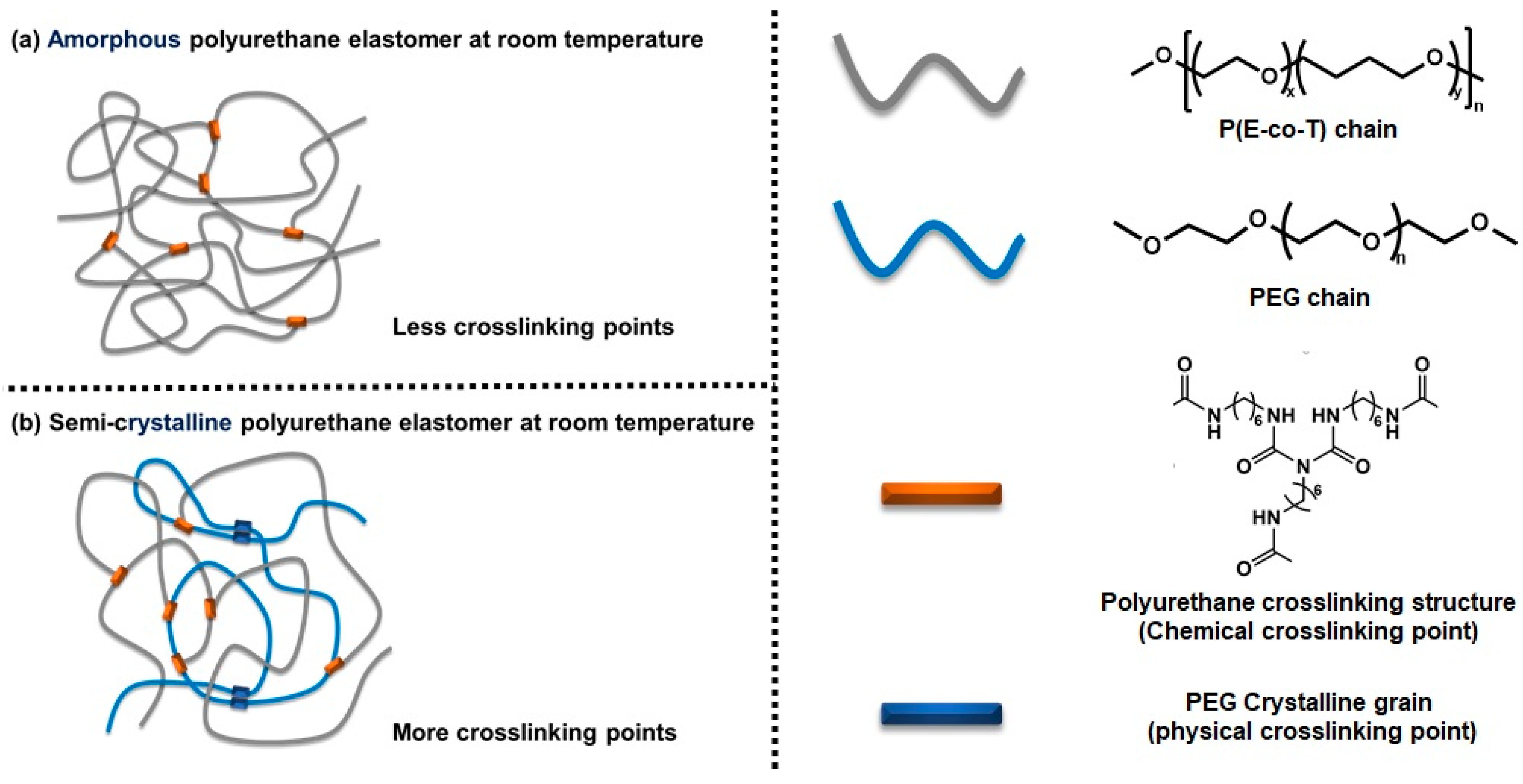
3.2. Chemical Crosslinking Networks of Elastomers with Different R Values
3.3. Aggregation of Elastomers with Different R Values
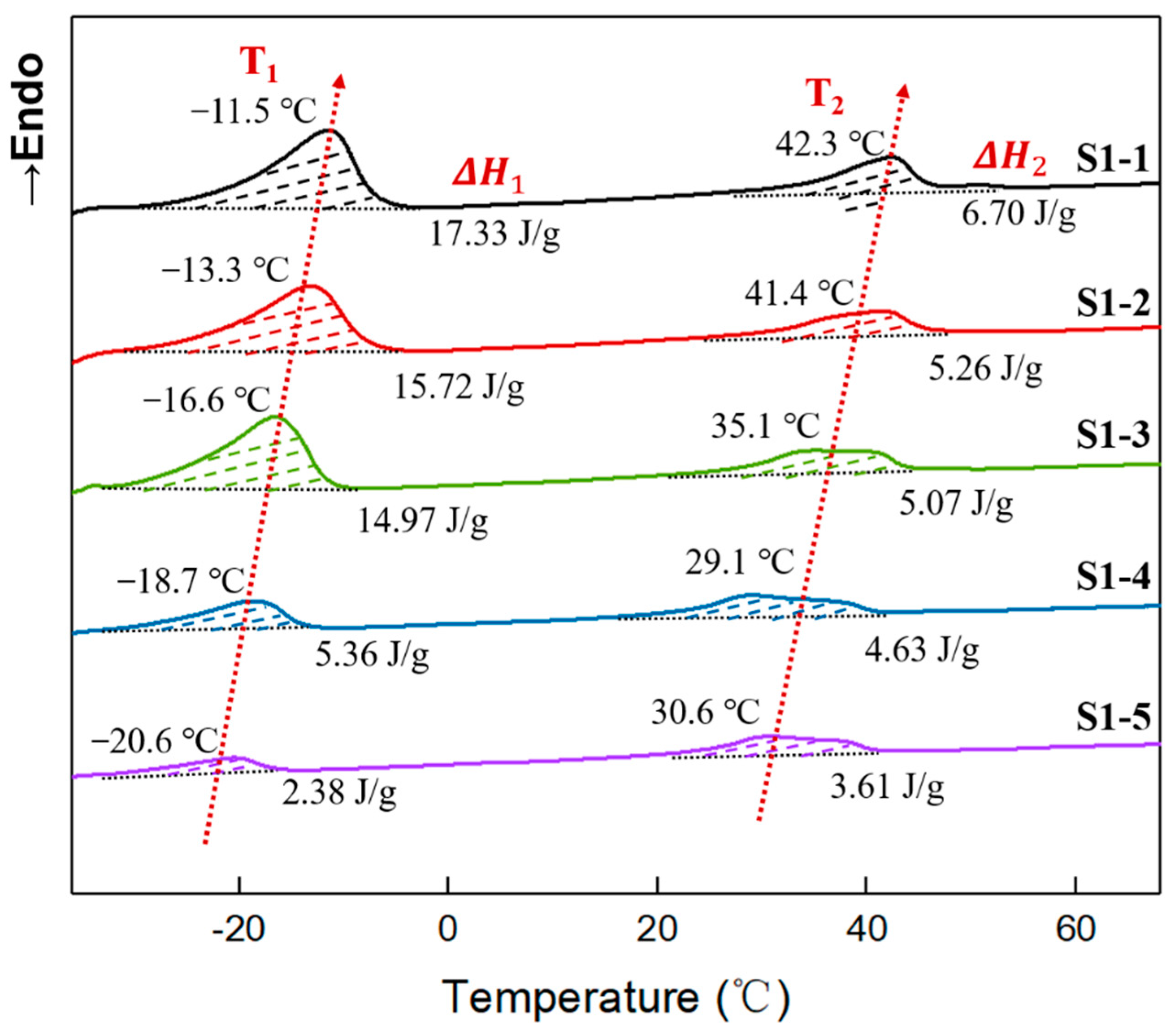
3.4. Crystallinity of Elastomers with Different R Values
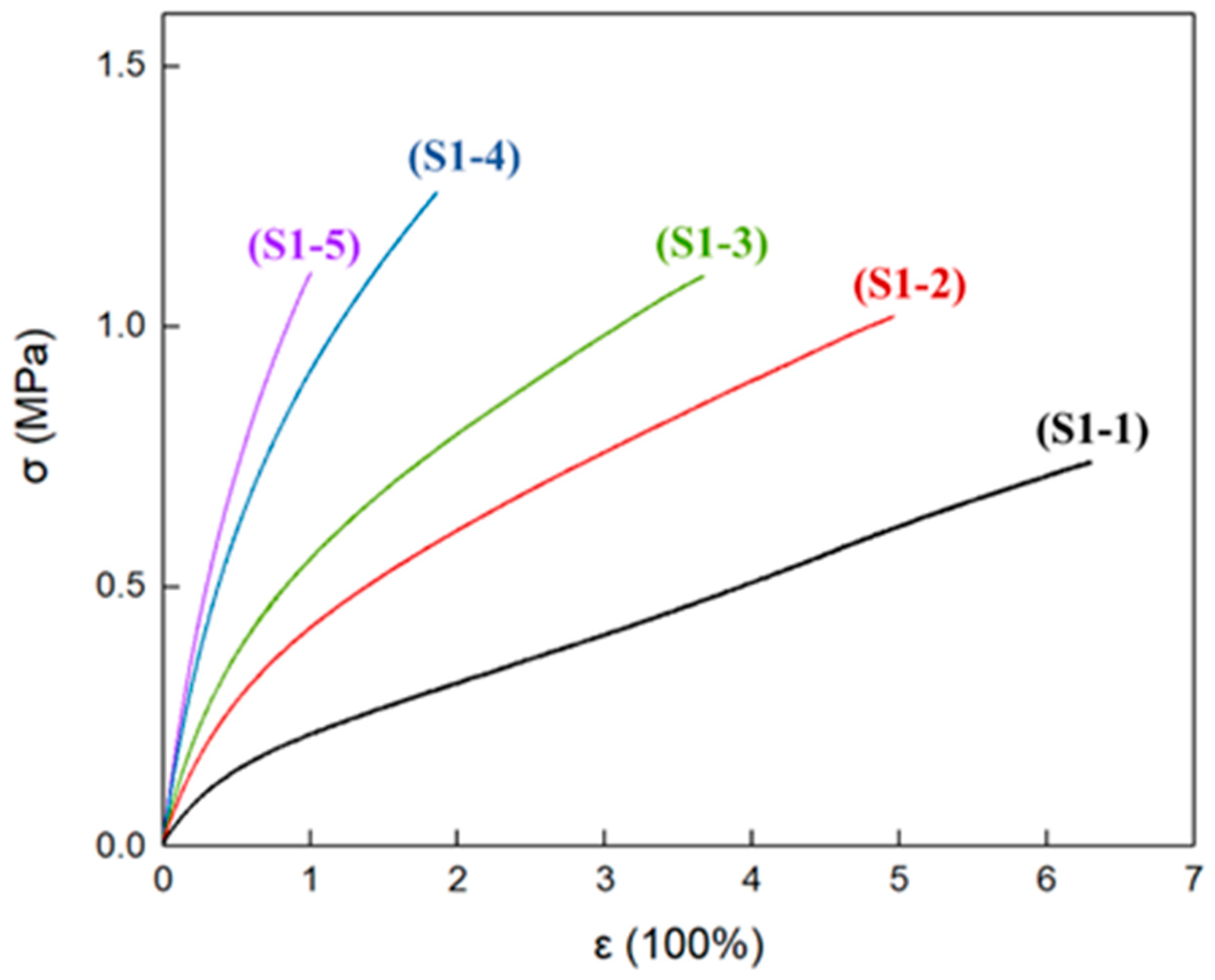
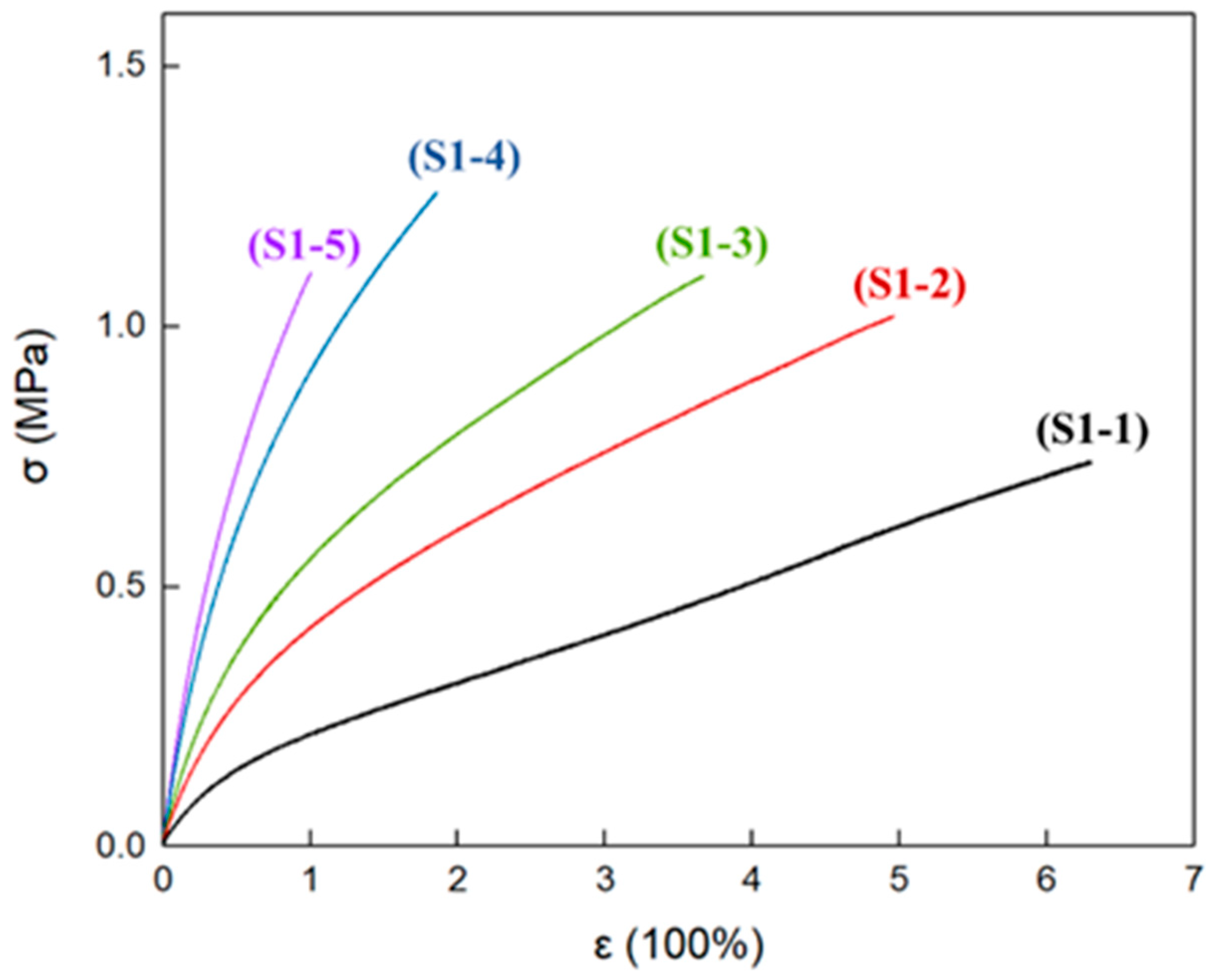
3.5. Mechanical Properties of Elastomers with Different R Values
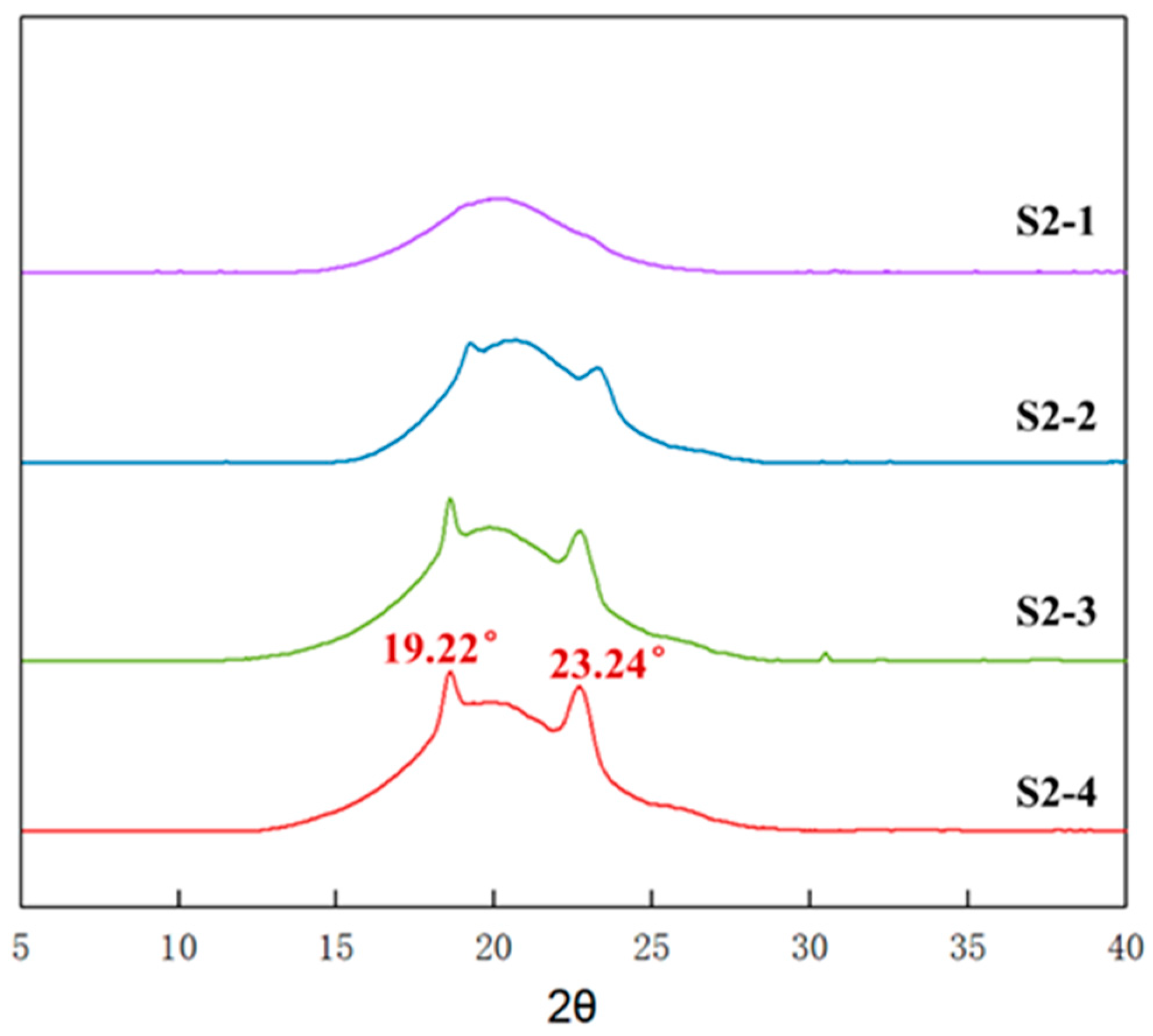
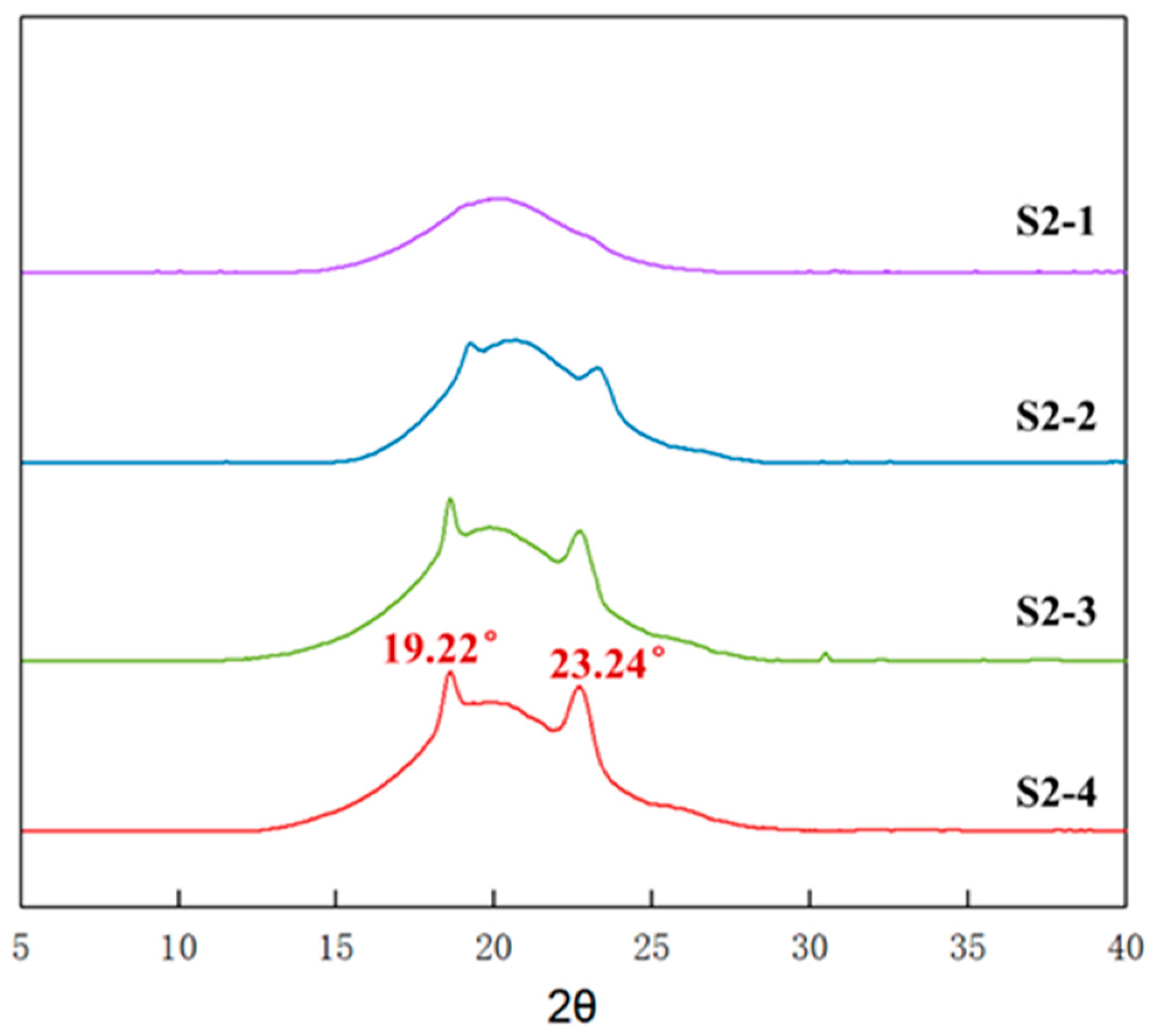
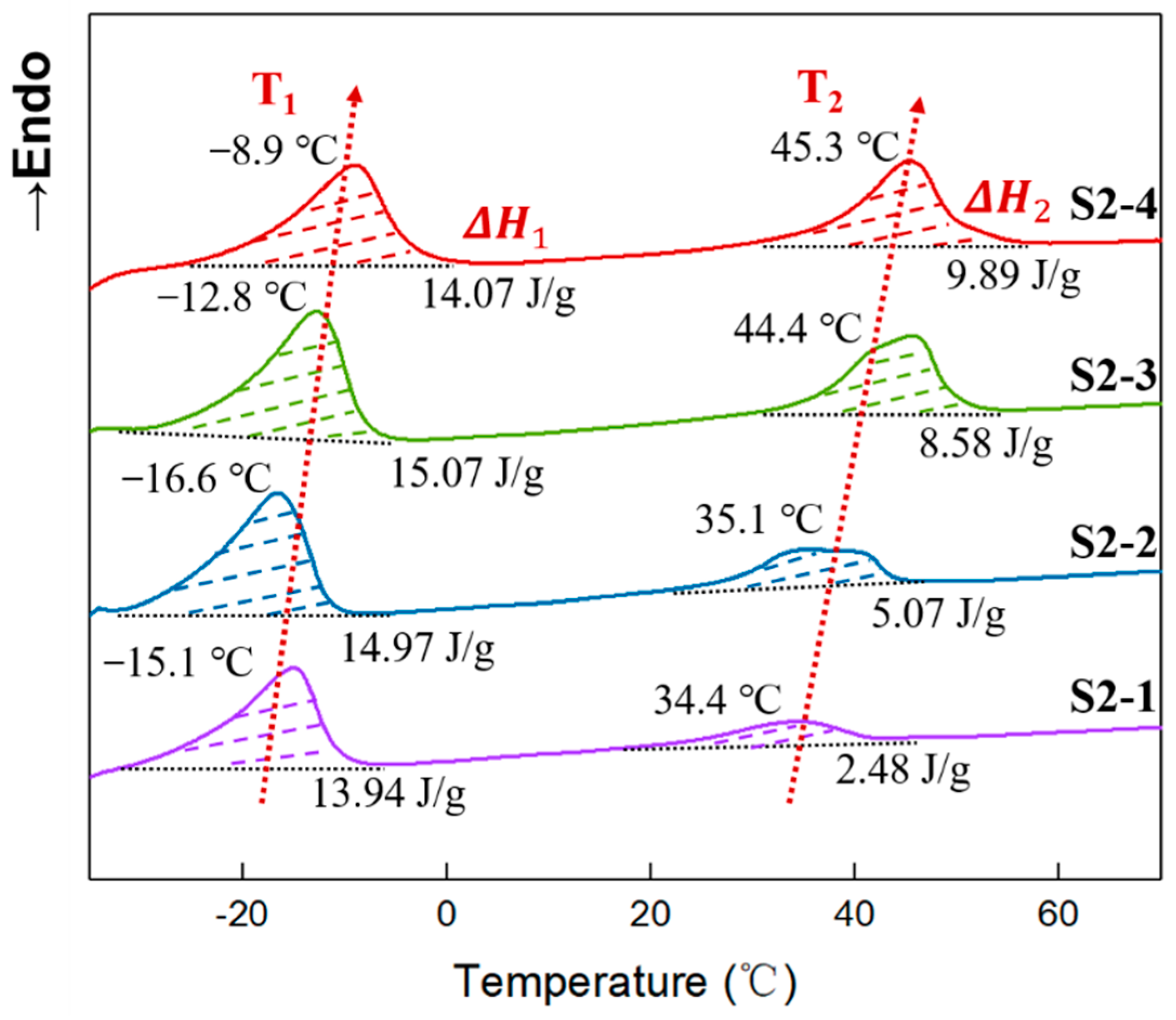
3.6. Aggregation of Elastomers with Different PEG Contents
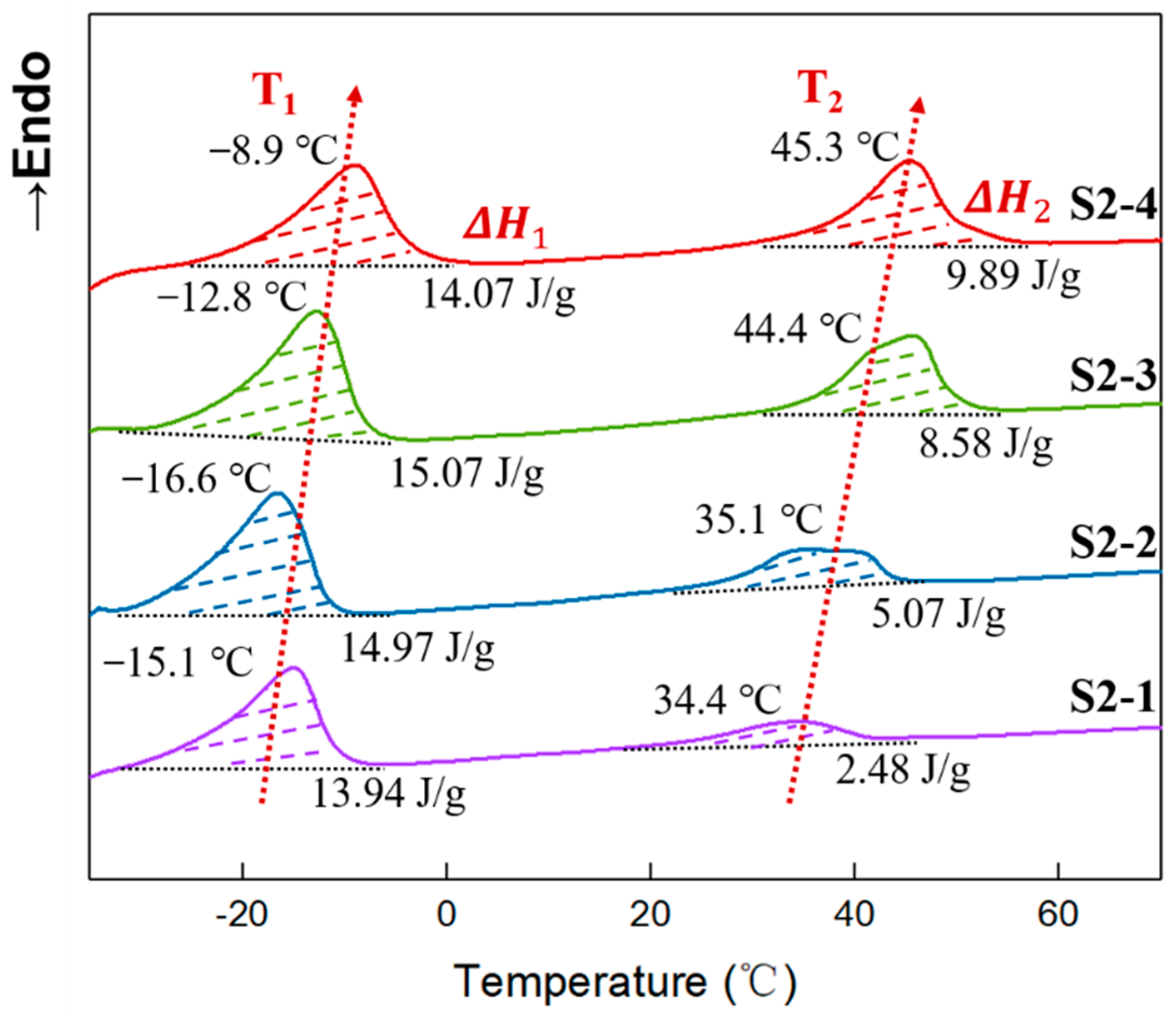
3.7. Crystallinity of Elastomers with Different PEG Contents
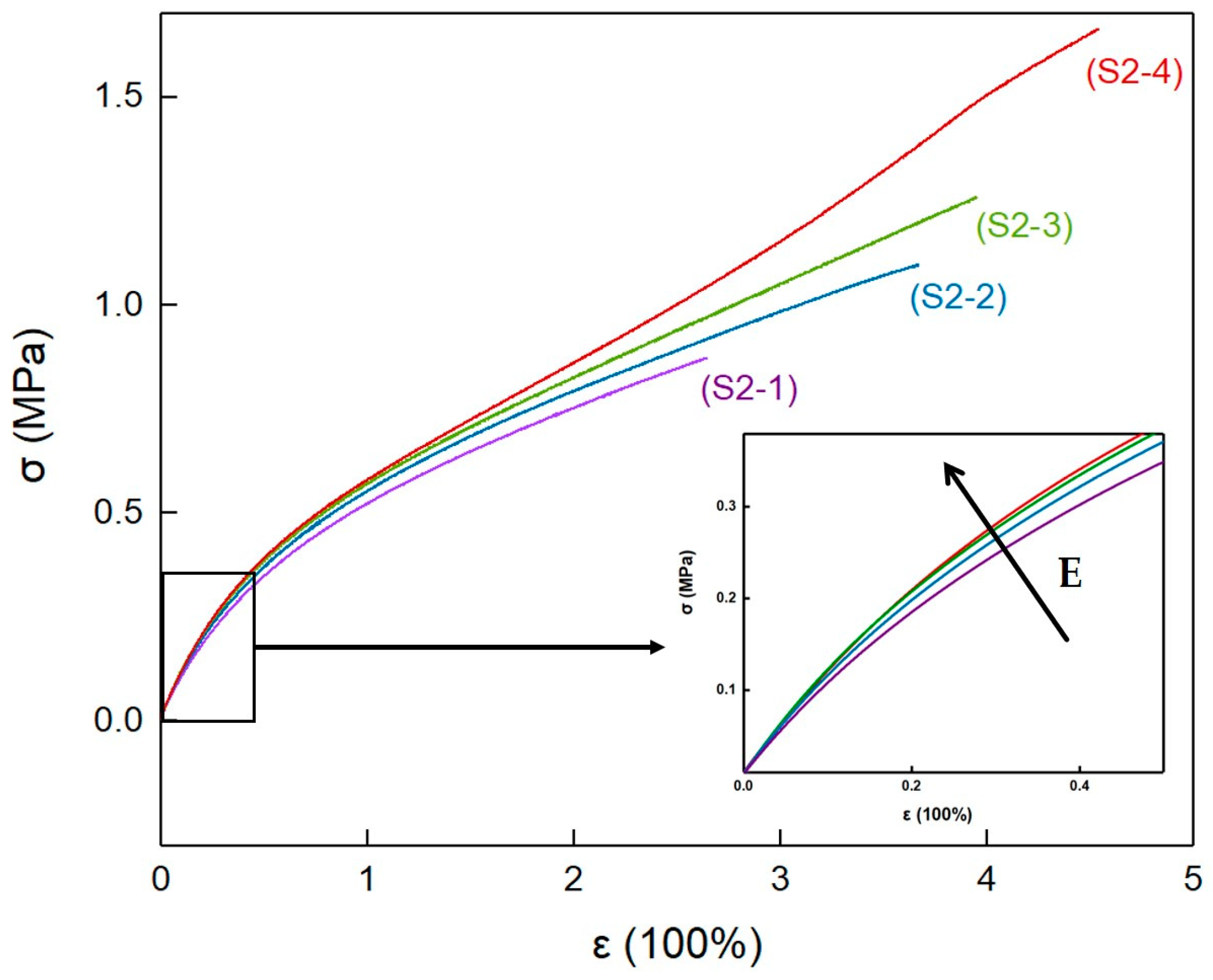
3.8. Mechanical Properties of Elastomers with Different PEG Contents
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Engels, H.W.; Pirkl, H.G.; Albers, R.; Albach, R.W.; Krause, J.; Hoffmann, A.; Casselmann, H.; Dormish, J. Polyurethanes: Versatile materials and sustainable problem solvers for today’s challenges. J. Ger. Chem. Soc. 2013, 52, 9422–9441. [Google Scholar] [CrossRef] [PubMed]
- Berezkin, Y.; Urick, M. Modern Polyurethanes: Overview of Structure Property Relationship. In Polymers for Personal Care and Cosmetics; ACS Publication: Washington, DC, USA, 2013; pp. 65–81. [Google Scholar] [CrossRef]
- Tian, S. Recent Advances in Functional Polyurethane and Its Application in Leather Manufacture: A Review. Polymers 2020, 12, 1996. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.M.; Li, X.P.; Chen, J.; He, X.D.; Huang, W.M.; Zhu, K.; Yu, W.H.; Ni, H.L.; Zhao, K.Q.; Hu, P. Unified method to prepare thermoplastic/thermoset soft polyurethanes reshape-able around room temperature on-demand. J. Polym. Res. 2021, 28, 201. [Google Scholar] [CrossRef]
- Meiorin, C.; Calvo-Correas, T.; Mosiewicki, M.A.; Aranguren, M.I.; Corcuera, M.A.; Eceiza, A. Comparative effects of two different crosslinkers on the properties of vegetable oil-based polyurethanes. J. Appl. Polym. Sci. 2019, 137, 48741. [Google Scholar] [CrossRef]
- Li, B.; Zhao, Y.; Liu, G.; Li, X.; Luo, Y. Mechanical properties and thermal decomposition of PBAMO/GAP random block ETPE. J. Therm. Anal. Calorim. 2016, 126, 717–724. [Google Scholar] [CrossRef]
- Zhang, C.; Li, J.; Luo, Y. Synthesis and Characterization of 3,3′-Bisazidomethyl Oxetane-3-Azidomethyl-3′-Methyl Oxetane Alternative Block Energetic Thermoplastic Elastomer. Propellants Explos. Pyrotech. 2012, 37, 235–240. [Google Scholar] [CrossRef]
- Eroglu, M.S.; Guven, O. Characterization of network structure of poly(glycidyl azide) elastomers by swelling, solubility and mechanical measurements. Polymer 1998, 39, 1173–1176. [Google Scholar] [CrossRef]
- Li, Y.; Li, J.; Ma, S.; Luo, Y. Compatibility, mechanical and thermal properties of GAP/P(EO-co-THF) blends obtained upon a urethane-curing reaction. Polym. Bull. 2017, 74, 4607–4618. [Google Scholar] [CrossRef]
- Chen, K.; Yuan, S.; Wen, X.; Sang, C.; Luo, Y. Effect of Mixed Isocyanate Curing Agents on the Performance of In Situ-Prepared HTPE Binder Applied in Propellant. Propellants Explos. Pyrotech. 2021, 46, 428–439. [Google Scholar] [CrossRef]
- Jutrzenka Trzebiatowska, P.; Santamaria Echart, A.; Calvo Correas, T.; Eceiza, A.; Datta, J. The changes of crosslink density of polyurethanes synthesised with using recycled component. Chemical structure and mechanical properties investigations. Prog. Org. Coat. 2018, 115, 41–48. [Google Scholar] [CrossRef]
- Zhai, J.X.; Pang, A.M.; Ding, T.F.; Liu, R.T.; Guo, X.Y.; Song, T.L. Effect of crosslinking point structures on properties of polyurethane end-crosslinked PBT elastomers. Iran. Polym. J. 2022, 31, 333–341. [Google Scholar] [CrossRef]
- Zheng, Q.; Wang, G.; Du, J.; Li, J.; Li, J.; Tang, Q.; Fan, X. Investigation of Hydroxyl-Terminated Polyether Cured with Different Isocyanates: Curing Process and Mechanical Property. Propellants Explos. Pyrotech. 2020, 45, 1972–1979. [Google Scholar] [CrossRef]
- Liu, Q.; Liu, Y.; Zheng, H.; Li, C.; Zhang, Y.; Zhang, Q. Design and development of self-repairable and recyclable crosslinked poly(thiourethane-urethane) via enhanced aliphatic disulfide chemistry. J. Polym. Sci. 2020, 58, 1092–1104. [Google Scholar] [CrossRef]
- Kojio, K.; Fukumaru, T.; Furukawa, M. Highly Softened Polyurethane Elastomer Synthesized with Novel 1,2-Bis(isocyanate)ethoxyethane. Macromolecules 2004, 37, 3287–3291. [Google Scholar] [CrossRef]
- Geng, Z.; Pang, A.; Ding, T.; Guo, X.; Yang, R.; Luo, Y.; Zhai, J. Overlooked Impact of Interchain H-Bonding between Cross-Links on the Mechanical Properties of Thermoset Polyurethane Elastomers. Macromolecules 2022, 55, 8749–8756. [Google Scholar] [CrossRef]
- de Keer, L.; Kilic, K.I.; Van Steenberge, P.H.M.; Daelemans, L.; Kodura, D.; Frisch, H.; De Clerck, K.; Reyniers, M.F.; Barner-Kowollik, C.; Dauskardt, R.H.; et al. Computational prediction of the molecular configuration of three-dimensional network polymers. Nat. Mater. 2021, 20, 1422–1430. [Google Scholar] [CrossRef] [PubMed]
- de Keer, L.; van Steenberge, P.H.M.; Reyniers, M.F.; D’Hooge, D.R. Going Beyond the Carothers, Flory and Stockmayer Equation by Including Cyclization Reactions and Mobility Constraints. Polymers 2021, 13, 2410. [Google Scholar] [CrossRef] [PubMed]
- Nofar, M.; Mohammadi, M.; Carreau, P.J. Effect of TPU hard segment content on the rheological and mechanical properties of PLA/TPU blends. J. Appl. Polym. Sci. 2020, 137, 49387. [Google Scholar] [CrossRef]
- Shen, Z.; Zheng, L.; Li, C.; Liu, G.; Xiao, Y.; Wu, S.; Liu, J.; Zhang, B. A comparison of non-isocyanate and HDI-based poly(ether urethane): Structure and properties. Polymer 2019, 175, 186–194. [Google Scholar] [CrossRef]
- Schimpf, V.; Max, J.B.; Stolz, B.; Heck, B.; Mülhaupt, R. Semicrystalline Non-Isocyanate Polyhydroxyurethanes as Thermoplastics and Thermoplastic Elastomers and Their Use in 3D Printing by Fused Filament Fabrication. Macromolecules 2018, 52, 320–331. [Google Scholar] [CrossRef]
- Eceiza, A.; Martin, M.D.; de la Caba, K.; Kortaberria, G.; Gabilondo, N.; Corcuera, M.A.; Mondragon, I. Thermoplastic polyurethane elastomers based on polycarbonate diols with different soft segment molecular weight and chemical structure: Mechanical and thermal properties. Polym. Eng. Sci. 2008, 48, 297–306. [Google Scholar] [CrossRef]
- Anokhin, D.V.; Gorbunova, M.A.; Abukaev, A.F.; Ivanov, D.A. Multiblock Thermoplastic Polyurethanes: In Situ Studies of Structural and Morphological Evolution under Strain. Materials 2021, 14, 3009. [Google Scholar] [CrossRef] [PubMed]
- Gorbunova, M.A.; Komov, E.V.; Grunin, L.Y.; Ivanova, M.S.; Abukaev, A.F.; Imamutdinova, A.M.; Ivanov, D.A.; Anokhin, D.V. The effect of separation of blocks on the crystallization kinetics and phase composition of poly(butylene adipate) in multi-block thermoplastic polyurethanes. Phys. Chem. Chem. Phys. 2022, 24, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gao, J.; Wang, Y.; Zhou, J.; Cao, L.; He, Z.; Zhang, Y.; Tang, C.; Zhong, L. Enhanced Temperature Stability of High Energy Density Ferroelectric Polymer Blends: The Spatial Confinement Effect. Macromol. Rapid Commun. 2019, 40, e1900406. [Google Scholar] [CrossRef]
- Alvarado, N.; Alegría, L.; Sandoval, C.; Kortaberría, G.; Leiva, A.; Gargallo, L.; Radic, D. Synthesis and Characterization of a Branched Poly(Methacrylamide): Thermal Stability and Molecular Simulation Studies of Their Blends With Vinylic Polymers. J. Macromol. Sci. Part A 2014, 51, 864–872. [Google Scholar] [CrossRef]
- Karim, A.; Liu, D.W.; Douglas, J.F.; Nakatani, A.I.; Amis, E.J. Modification of the phase stability of polymer blends by fillers. Polymer 2000, 41, 8455–8458. [Google Scholar] [CrossRef]
- Zou, Y.C.; Yang, R.J.; Zhai, J.X. Polytriazole polyether elastomers with widely tunable mechanical properties: The role of network structure and crystallization behavior. J. Appl. Polym. Sci. 2017, 134, 45298. [Google Scholar] [CrossRef]
- Kambe, Y. Thermal-Behavior of Poly(Ethylene Oxide) as Revealed by Differential Scanning Calorimetry. Polymer 1980, 21, 352–355. [Google Scholar] [CrossRef]
- van Krevelen, D.W. Properties of Polymers: Their Correlation with Chemical Structure; Their Numerical Estimation and Prediction from Additive Group Contributions, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 1–1004. [Google Scholar]
- Qu, Z.; Zhai, J.; Yang, R. Comparison between properties of polyether polytriazole elastomers and polyether polyurethane elastomers. Polym. Adv. Technol. 2014, 25, 314–321. [Google Scholar] [CrossRef]
- Flory, P.J.; Rehner, J. Statistical Mechanics of Cross-Linked Polymer Networks II. Swelling. J. Chem. Phys. 1943, 11, 521–526. [Google Scholar] [CrossRef]
- Bristow, G.M.; Watson, W.F. Cohesive Energy Densities of Polymers Part 1—Cohesive Energy Densities of Rubbers by Swelling Measurements. Trans. Faraday Soc. 1958, 54, 1731–1741. [Google Scholar] [CrossRef]
- Ozdemir, C.; Guner, A. Solubility profiles of poly(ethylene glycol)/solvent systems, I: Qualitative comparison of solubility parameter approaches. Eur. Polym. J. 2007, 43, 3068–3093. [Google Scholar] [CrossRef]
- Zhang, L.; Shi, H.; Li, W.; Han, X.; Zhang, X. Thermal performance and crystallization behavior of poly(ethylene glycol) hexadecyl ether in confined environment. Polym. Int. 2014, 63, 982–988. [Google Scholar] [CrossRef]
- Rachmawati, R.; Woortman, A.J.; Loos, K. Facile preparation method for inclusion complexes between amylose and polytetrahydrofurans. Biomacromolecules 2013, 14, 575–583. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, T.; Cui, J.; Liu, L.; Zhou, Y.; Li, G.; Zhou, E.; Chen, X. Nonisothermal crystallization behavior of the poly(ethylene glycol) block in poly(L-lactide)–poly(ethylene glycol) diblock copolymers: Effect of the poly(L-lactide) block length. J. Polym. Sci. Part B Polym. Phys. 2006, 44, 3215–3226. [Google Scholar] [CrossRef]
- Buckley, C.P.; Kovacs, A.J. Melting behavior of low-molecular weight poly (ethylene-oxide) fractions. 2. folded chain crystals. Colloid Polym. Sci. 1976, 254, 695–715. [Google Scholar] [CrossRef]
- Liu, M.; Zhao, Q.; Wang, Y.; Zhang, C.; Mo, Z.; Cao, S. Melting behaviors, isothermal and non-isothermal crystallization kinetics of nylon 1212. Polymer 2003, 44, 2537–2545. [Google Scholar] [CrossRef]
- Run, M.; Wu, S.; Zhang, D.; Wu, G. Melting behaviors and isothermal crystallization kinetics of poly(ethylene terephthalate)/mesoporous molecular sieve composite. Polymer 2005, 46, 5308–5316. [Google Scholar] [CrossRef]
- Jenkins, M.J.; Cao, Y.; Kukureka, S.N. The effect of molecular weight on the crystallization kinetics and equilibrium melting temperature of poly(tetramethylene ether glycol). Polym. Adv. Technol. 2006, 17, 1–5. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | R Value | P(E-co-T) (g) | PEG (g) | N100 (g) | T12 (g) |
|---|---|---|---|---|---|
| S1-1 | 0.80 | 17.6 | 2.4 | 1.409 | 0.001 |
| S1-2 | 0.85 | 17.6 | 2.4 | 1.494 | 0.001 |
| S1-3 | 0.90 | 17.6 | 2.4 | 1.582 | 0.001 |
| S1-4 | 1.00 | 17.6 | 2.4 | 1.757 | 0.001 |
| S1-5 | 1.10 | 17.6 | 2.4 | 1.933 | 0.001 |
| Samples | PEG (wt%) | P(E-co-T) (g) | PEG (g) | N100 (g) | T12 (g) |
|---|---|---|---|---|---|
| S2-1 | 8 | 18.4 | 1.6 | 1.577 | 0.001 |
| S2-2 | 12 | 17.6 | 2.4 | 1.582 | 0.001 |
| S2-3 | 16 | 16.8 | 3.2 | 1.586 | 0.001 |
| S2-4 | 20 | 16.0 | 4.0 | 1.590 | 0.001 |
| Samples | (g cm−3) | (g mol−1) | (mmol cm−3) | ||||
|---|---|---|---|---|---|---|---|
| S1-1 | 1.0594 | 18.371 | 0.341 | 8.2806 | 0.1208 | 16,416 | 0.0645 |
| S1-2 | 1.0609 | 18.371 | 0.341 | 6.1976 | 0.1614 | 9087 | 0.1168 |
| S1-3 | 1.0623 | 18.371 | 0.341 | 5.5454 | 0.1803 | 7178 | 0.1478 |
| S1-4 | 1.0577 | 18.371 | 0.341 | 4.5106 | 0.2217 | 4549 | 0.2325 |
| S1-5 | 1.0604 | 18.371 | 0.341 | 3.8050 | 0.2628 | 3086 | 0.3437 |
| Samples | T1 (°C) | (J/g) | (%) | T2 (°C) | (J/g) | (%) |
|---|---|---|---|---|---|---|
| S1-1 | −11.5 | 17.33 | 10.08 | 42.3 | 6.70 | 4.27 |
| S1-2 | −13.3 | 15.72 | 9.14 | 41.4 | 5.26 | 3.35 |
| S1-3 | −16.6 | 14.97 | 8.70 | 35.1 | 5.07 | 3.23 |
| S1-4 | −18.7 | 5.36 | 3.12 | 29.1 | 4.63 | 2.95 |
| S1-5 | −20.6 | 2.38 | 1.38 | 30.6 | 3.61 | 2.30 |
| Samples | R Value | εb (%) | σb (MPa) | E (MPa) |
|---|---|---|---|---|
| S1-1 | 0.80 | 630 | 0.738 | 0.356 |
| 617 | 0.738 | 0.348 | ||
| 588 | 0.727 | 0.362 | ||
| 612 ± 5 | 0.73 ± 0.01 | 0.36 ± 0.01 | ||
| S1-2 | 0.85 | 408 | 0.972 | 0.663 |
| 448 | 0.972 | 0.675 | ||
| 496 | 1.018 | 0.614 | ||
| 451 ± 7 | 0.99 ± 0.03 | 0.65 ± 0.03 | ||
| S1-3 | 0.90 | 312 | 0.980 | 0.809 |
| 367 | 1.096 | 0.863 | ||
| 370 | 1.111 | 0.880 | ||
| 349 ± 6 | 1.06 ± 0.07 | 0.85 ± 0.03 | ||
| S1-4 | 1.00 | 169 | 1.173 | 1.411 |
| 177 | 1.185 | 1.376 | ||
| 185 | 1.258 | 1.453 | ||
| 177 ± 3 | 1.20 ± 0.04 | 1.41 ± 0.03 | ||
| S1-5 | 1.10 | 81 | 0.980 | 1.700 |
| 88 | 1.018 | 1.616 | ||
| 100 | 1.101 | 1.717 | ||
| 90 ± 3 | 1.03 ± 0.06 | 1.68 ± 0.05 |
| Sample | R Value | εb (%) | σb (MPa) | E (MPa) |
|---|---|---|---|---|
| S1-1 | 0.80 | 635 | 0.730 | 0.350 |
| 622 | 0.731 | 0.346 | ||
| 614 | 0.721 | 0.339 | ||
| 624 ± 4 | 0.73 ± 0.01 | 0.35 ± 0.01 | ||
| S1-2 | 0.85 | 459 | 0.988 | 0.664 |
| 497 | 0.963 | 0.657 | ||
| 489 | 0.964 | 0.652 | ||
| 482 ± 5 | 0.97 ± 0.02 | 0.66 ± 0.01 | ||
| S1-3 | 0.90 | 331 | 0.999 | 0.787 |
| 359 | 0.981 | 0.851 | ||
| 376 | 1.055 | 0.780 | ||
| 355 ± 5 | 1.01 ± 0.03 | 0.81 ± 0.04 | ||
| S1-4 | 1.00 | 180 | 1.214 | 1.411 |
| 192 | 1.198 | 1.379 | ||
| 176 | 1.186 | 1.396 | ||
| 183 ± 3 | 1.20 ± 0.01 | 1.40 ± 0.02 | ||
| S1-5 | 1.10 | 114 | 1.034 | 1.680 |
| 97 | 0.991 | 1.608 | ||
| 86 | 1.011 | 1.721 | ||
| 99 ± 3 | 1.01 ± 0.02 | 1.66 ± 0.05 |
| Samples | T1 (°C) | (J/g) | (%) | T2 (°C) | (J/g) | (%) |
|---|---|---|---|---|---|---|
| S2-1 | −15.1 | 13.94 | 8.10 | 34.4 | 2.48 | 1.58 |
| S2-2 | −16.6 | 14.97 | 8.70 | 35.1 | 5.07 | 3.23 |
| S2-3 | −12.8 | 15.07 | 8.76 | 45.7 | 8.58 | 5.47 |
| S2-4 | −8.9 | 14.07 | 8.18 | 45.3 | 9.89 | 6.30 |
| Samples | ε (%) | σ (MPa) | E (MPa) |
|---|---|---|---|
| S2-1 | 264 | 0.872 | 0.820 |
| 224 | 0.771 | 0.777 | |
| 223 | 0.746 | 0.735 | |
| 237 ± 5 | 0.80 ± 0.07 | 0.78 ± 0.04 | |
| S2-2 | 312 | 0.980 | 0.809 |
| 367 | 1.096 | 0.863 | |
| 370 | 1.111 | 0.880 | |
| 331 ± 7 | 0.84 ± 0.02 | 1.03 ± 0.03 | |
| S2-3 | 339 | 1.170 | 0.962 |
| 349 | 1.157 | 0.931 | |
| 394 | 1.259 | 0.881 | |
| 361 ± 5 | 1.20 ± 0.06 | 0.92 ± 0.04 | |
| S2-4 | 427 | 1.428 | 0.918 |
| 449 | 1.467 | 0.921 | |
| 454 | 1.563 | 0.943 | |
| 443 ± 4 | 1.49 ± 0.06 | 0.93 ± 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, P.; Pang, A.; Yang, R.; Guo, X.; He, J.; Zhai, J. Study on Mechanical Properties of Polyurethane Cross-Linked P(E-co-T)/PEG Blended Polyether Elastomer. Polymers 2022, 14, 5419. https://doi.org/10.3390/polym14245419
Jin P, Pang A, Yang R, Guo X, He J, Zhai J. Study on Mechanical Properties of Polyurethane Cross-Linked P(E-co-T)/PEG Blended Polyether Elastomer. Polymers. 2022; 14(24):5419. https://doi.org/10.3390/polym14245419
Chicago/Turabian StyleJin, Puyu, Aimin Pang, Rongjie Yang, Xiaoyan Guo, Jiyu He, and Jinxian Zhai. 2022. "Study on Mechanical Properties of Polyurethane Cross-Linked P(E-co-T)/PEG Blended Polyether Elastomer" Polymers 14, no. 24: 5419. https://doi.org/10.3390/polym14245419
APA StyleJin, P., Pang, A., Yang, R., Guo, X., He, J., & Zhai, J. (2022). Study on Mechanical Properties of Polyurethane Cross-Linked P(E-co-T)/PEG Blended Polyether Elastomer. Polymers, 14(24), 5419. https://doi.org/10.3390/polym14245419