

Well-Defined Nanostructures by Block Copolymers and Mass Transport Applications in Energy Conversion

Abstract

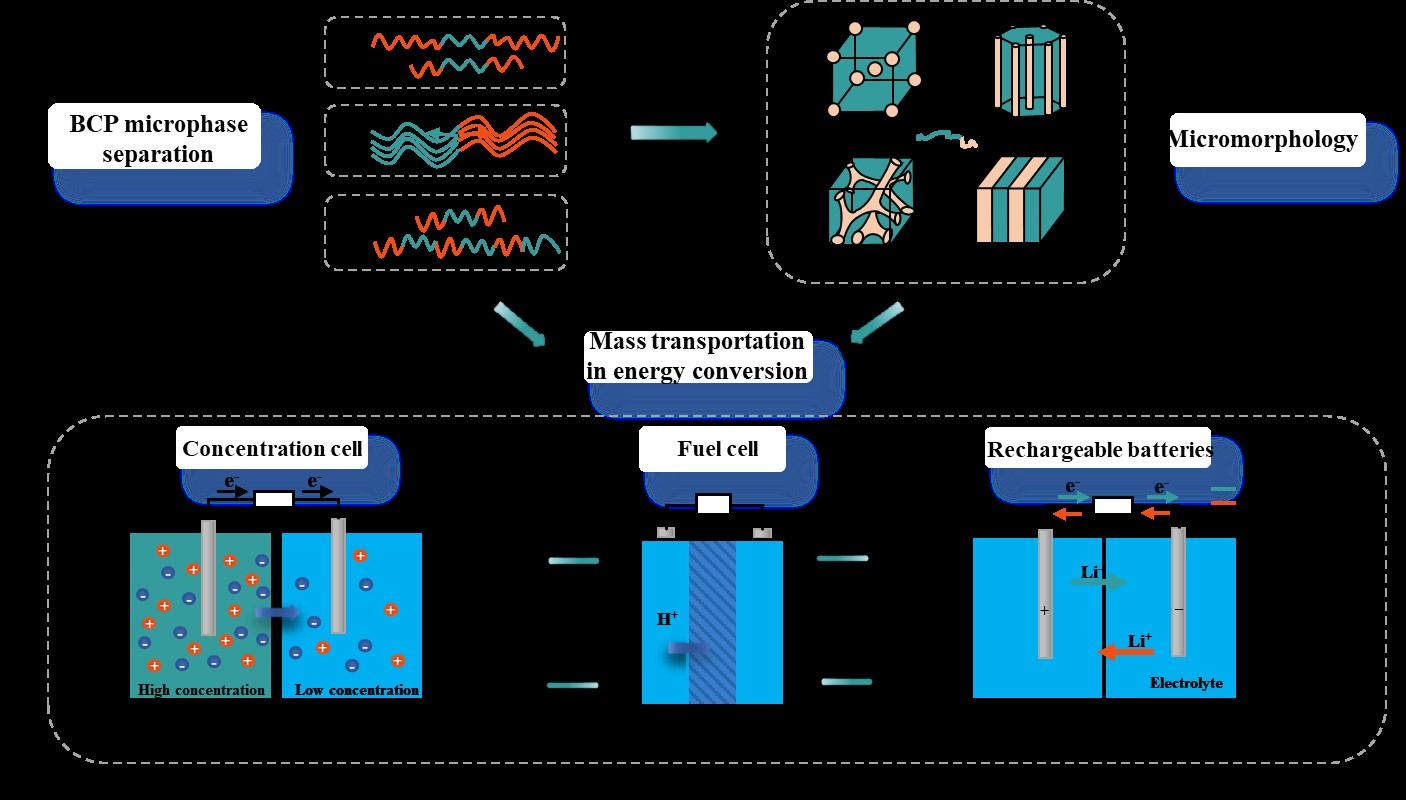{kind=link}
{kind=link}
{kind=link}
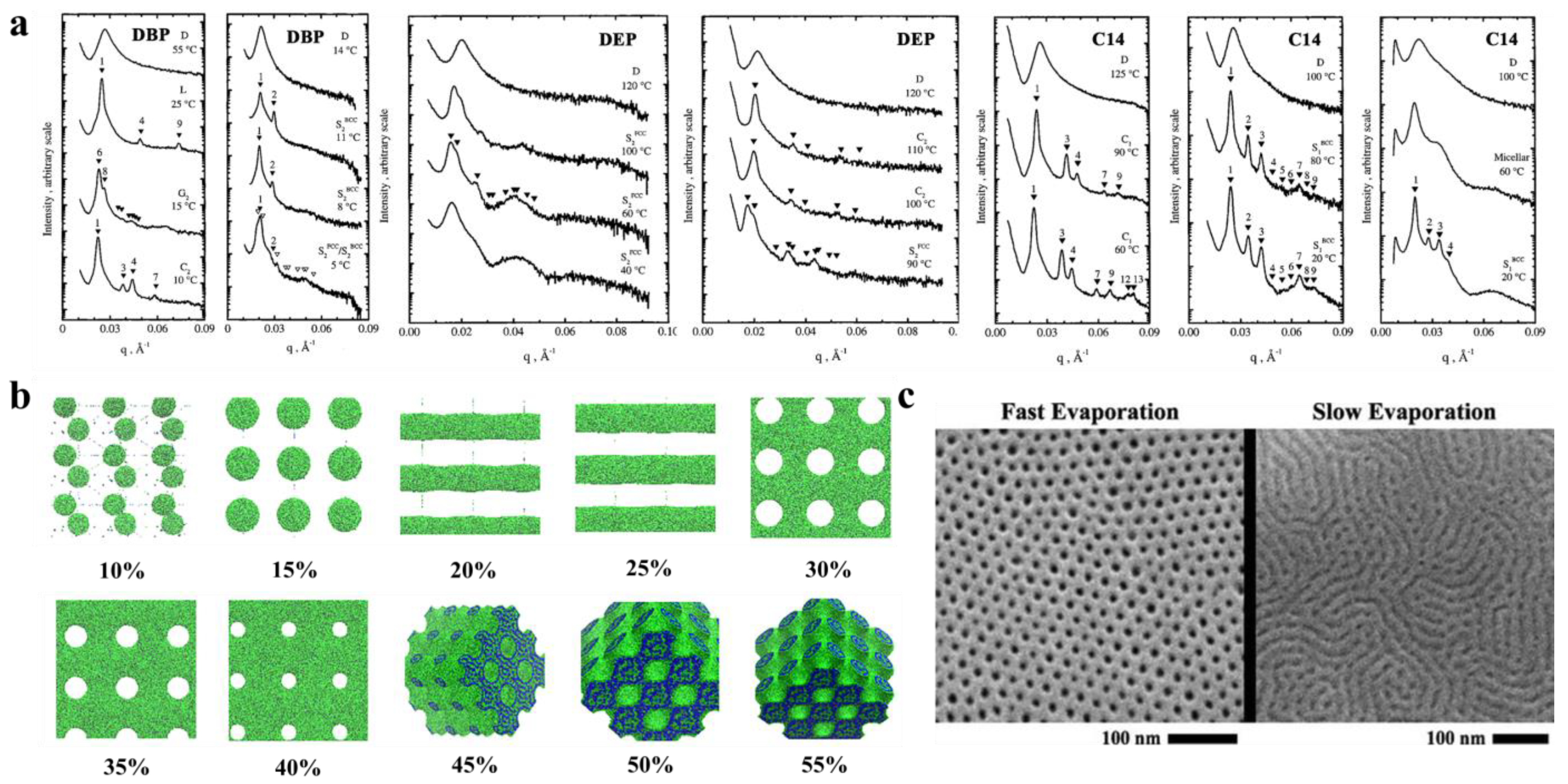{kind=link}
{kind=link}
{kind=link}
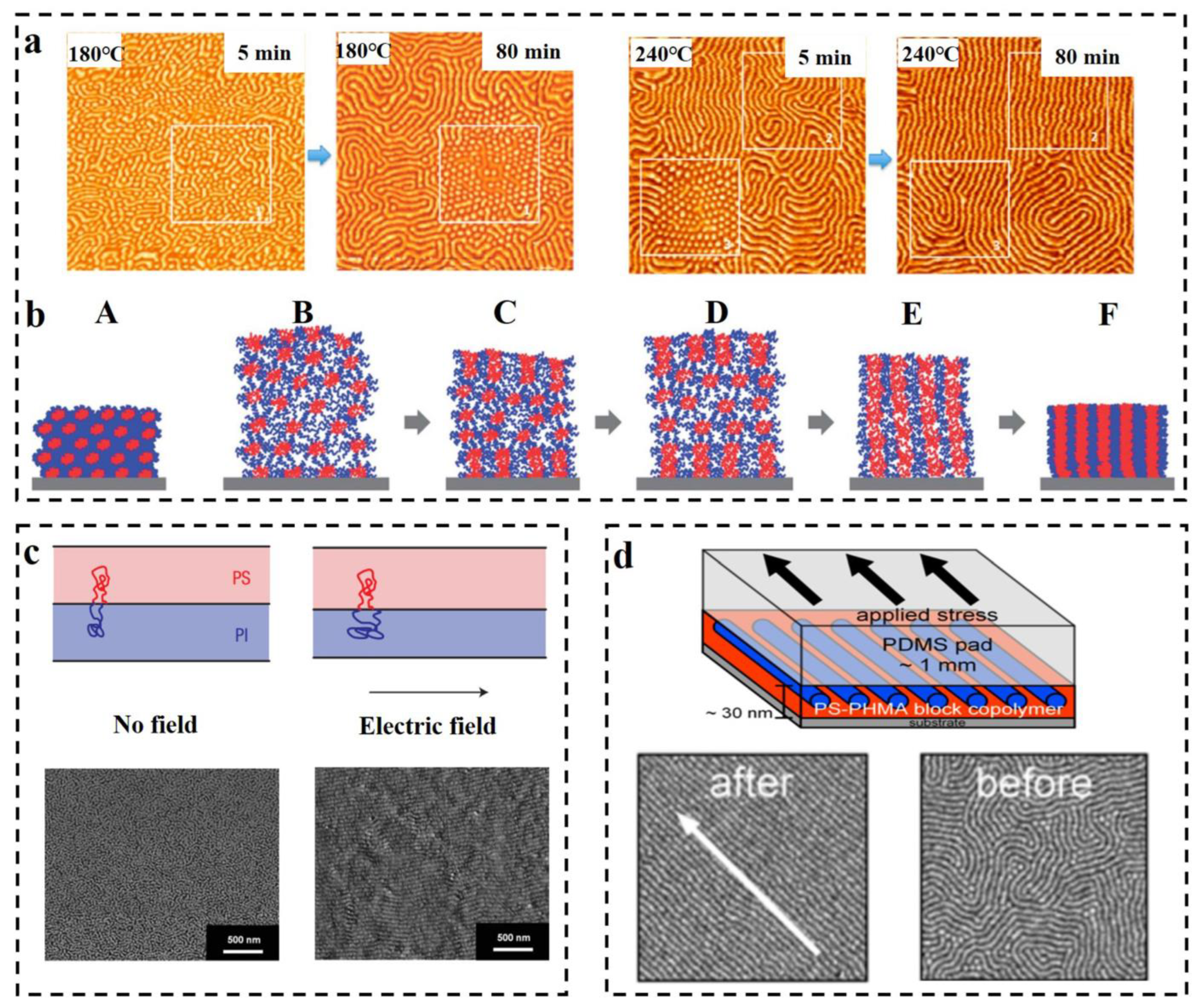{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
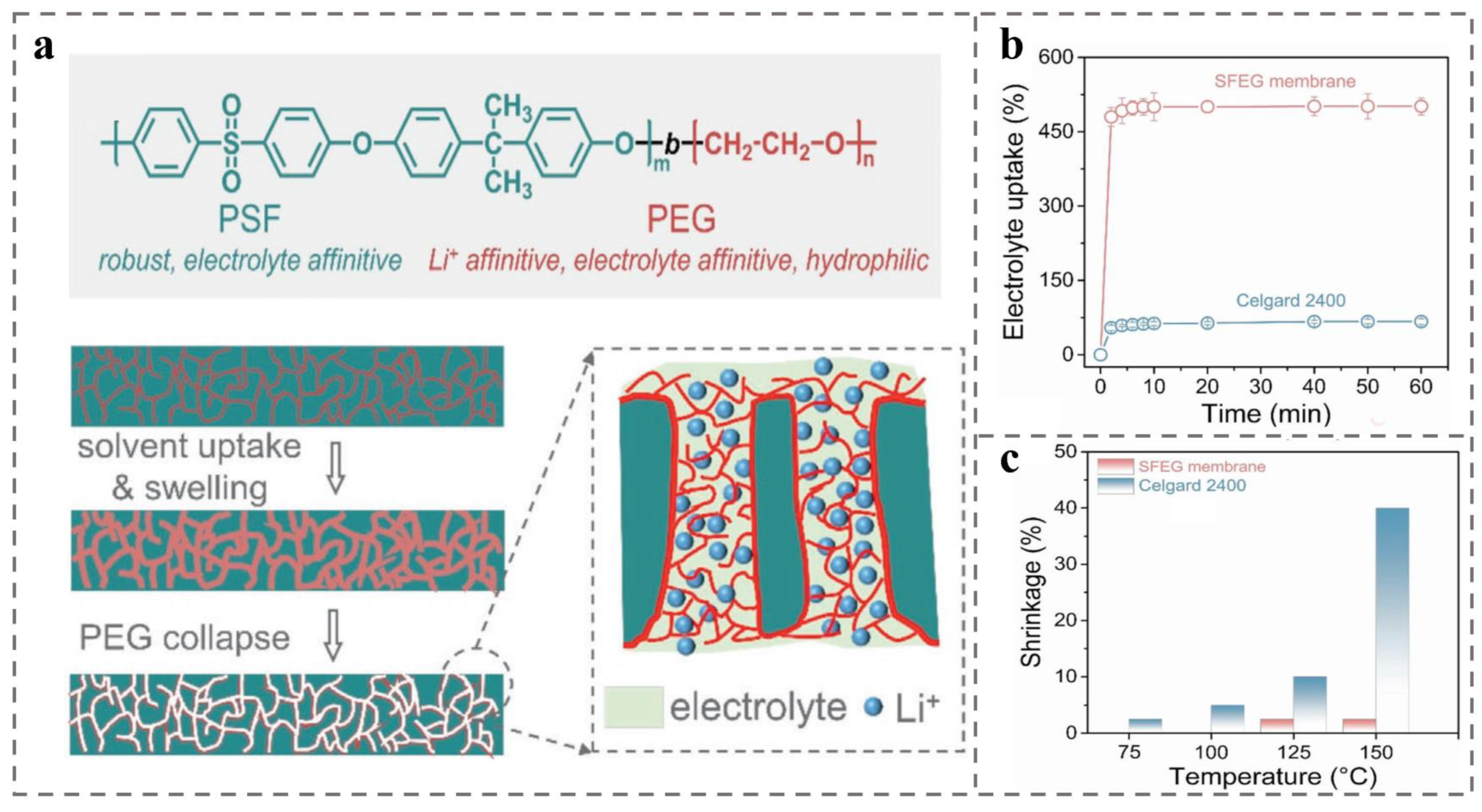{kind=link}
{kind=link}
1. Introduction
2. Microphase Separation of Block Copolymer

3. Regulation of the Self-Assembled Structure of the Block Copolymer
3.1. Influence of the Molecular Structure on the Self-Assembled Structure

3.2. Influence of Assembly Conditions on the Self-Assembled Structure
3.2.1. Solvents to Prepare the Membranes and Self-Assembly


3.2.2. Nonsolvent Approaches for Constructing Nanostructure

3.2.3. The Influence of External Field on Self-Assembled Structure of BCP

3.2.4. Additives for Regulating Self-Assembly Structure
4. Mass Transport Applications in Energy Conversion of Block Copolymers
4.1. Ion Selective Membranes for Concentration Cell


4.2. Ion Exchange Membranes for Fuel Cell


4.3. Battery Separator for Rechargeable Batteries (e.g., Lithium-Ion Batteries)

5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Schacher, F.H.; Rupar, P.A.; Manners, I. Functional Block Copolymers: Nanostructured Materials with Emerging Applications. Angew. Chem. Int. Ed. 2012, 51, 7898–7921. [Google Scholar] [CrossRef] [PubMed]
- Bates, C.M.; Bates, F.S. 50th Anniversary Perspective: Block Polymers-Pure Potential. Macromolecules 2017, 50, 3–22. [Google Scholar] [CrossRef]
- Mankowich, A.M. Micellar Molecular Weights of Selected Surface Active Agents. J. Phys. Chem. 1954, 58, 1027–1030. [Google Scholar] [CrossRef]
- Vaughn, T.H.; Suter, H.R.; Lundsted, L.G.; Kramer, M.G. Properties of some newly developed nonionic detergents. J. Am. Oil. Chem. Soc. 1951, 28, 294–299. [Google Scholar] [CrossRef]
- Szwarc, M.; Levy, M.; Milkovich, R. Polymerization initiated by electron transfer to monomer. A new method of formation of block polymers. J. Am. Chem. Soc. 1956, 78, 2656–2657. [Google Scholar] [CrossRef]
- Lanson, D.; Ariura, F.; Schappacher, M.; Borsali, R.; Deffieux, A. Application of living ionic polymerizations to the design of AB-type comb-like copolymers of various topologies and organizations. Macromol. Res. 2007, 15, 173–177. [Google Scholar] [CrossRef]
- Charleux, B.; Delaittre, G.; Rieger, J.; D’Agosto, F. Polymerization-Induced Self-Assembly: From Soluble Macromolecules to Block Copolymer Nano-Objects in One Step. Macromolecules 2012, 45, 6753–6765. [Google Scholar] [CrossRef]
- Nasrullah, M.J.; Vora, A.; Webster, D.C. Block Copolymer Synthesis via a Combination of ATRP and RAFT Using Click Chemistry. Macromol. Chem. Phys. 2011, 212, 539–549. [Google Scholar] [CrossRef]
- Siegwart, D.J.; Oh, J.K.; Matyjaszewski, K. ATRP in the design of functional materials for biomedical applications. Prog. Polym. Sci. 2012, 37, 18–37. [Google Scholar] [CrossRef] [PubMed]
- Matyjaszewski, K. Advanced Materials by Atom Transfer Radical Polymerization. Adv. Mater. 2018, 30, 1706441. [Google Scholar] [CrossRef] [PubMed]
- Keddie, D.J. A guide to the synthesis of block copolymers using reversible-addition fragmentation chain transfer (RAFT) polymerization. Chem. Soc. Rev. 2014, 43, 496–505. [Google Scholar] [CrossRef]
- Eggers, S.; Eckert, T.; Abetz, V. Double thermoresponsive block-random copolymers with adjustable phase transition temperatures: From block-like to gradient-like behavior. J. Polym. Sci. Pol. Chem. 2018, 56, 399–411. [Google Scholar] [CrossRef]
- Gyorgy, C.; Hunter, S.J.; Girou, C.; Derry, M.J.; Armes, S.P. Synthesis of poly(stearyl methacrylate)-poly(2-hydroxypropyl methacrylate) diblock copolymer nanoparticles via RAFT dispersion polymerization of 2-hydroxypropyl methacrylate in mineral oil. Polym. Chem. 2020, 11, 4579–4590. [Google Scholar] [CrossRef]
- Edwards, S.F. The statistical mechanics of polymers with excluded volume. Proc. Phys. Soc. 1965, 85, 613–624. [Google Scholar] [CrossRef]
- Matsen, M.W.; Schick, M. Stable and unstable phases of a diblock copolymer melt. Phys. Rev. Lett. 1994, 72, 2660–2663. [Google Scholar] [CrossRef] [PubMed]
- Matsen, M.W.; Bates, F.S. Unifying weak- and strong-segregation block copolymer theories. Macromolecules 1996, 29, 1091–1098. [Google Scholar] [CrossRef]
- Spencer, R.K.W.; Matsen, M.W. Fluctuation effects in blends of A plus B homopolymers with AB diblock copolymer. J. Chem. Phys. 2018, 148, 204907. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Kim, G.-H.; Park, S.-J. Conjugated Block Copolymers for Functional Nanostructures. Acc. Chem. Res. 2022, 55, 2224–2234. [Google Scholar] [CrossRef]
- Rosler, A.; Vandermeulen, G.W.M.; Klok, H.A. Advanced drug delivery devices via self-assembly of amphiphilic block copolymers. Adv. Drug Deliv. Rev. 2012, 64, 270–279. [Google Scholar] [CrossRef]
- Cummins, C.; Lundy, R.; Walsh, J.J.; Ponsinet, V.; Fleury, G.; Morris, M.A. Enabling future nanomanufacturing through block copolymer self-assembly: A review. Nano Today 2020, 35, 100936. [Google Scholar] [CrossRef]
- Meng, L.; Fan, H.; Lane, J.M.D.; Qin, Y. Bottom-Up Approaches for Precisely Nanostructuring Hybrid Organic/Inorganic Multi-Component Composites for Organic Photovoltaics. MRS Adv. 2020, 5, 2055–2065. [Google Scholar] [CrossRef]
- Yu, H.Z.; Qiu, X.Y.; Moreno, N.; Ma, Z.W.; Calo, V.M.; Nunes, S.P.; Peinemann, K.V. Self-Assembled Asymmetric Block Copolymer Membranes: Bridging the Gap from Ultra- to Nanofiltration. Angew. Chem. Int. Ed. 2015, 54, 13937–13941. [Google Scholar] [CrossRef] [PubMed]
- Radjabian, M.; Abetz, V. Advanced porous polymer membranes from self-assembling block copolymers. Prog. Polym. Sci. 2020, 102, 101219. [Google Scholar] [CrossRef]
- Cheng, X.Q.; Wang, Z.X.; Jiang, X.; Li, T.X.; Lau, C.H.; Guo, Z.H.; Ma, J.; Shao, L. Towards sustainable ultrafast molecular-separation membranes: From conventional polymers to emerging materials. Prog. Mater. Sci. 2018, 92, 258–283. [Google Scholar] [CrossRef]
- Park, H.B.; Kamcev, J.; Robeson, L.M.; Elimelech, M.; Freeman, B.D. Maximizing the right stuff: The trade-off between membrane permeability and selectivity. Science 2017, 356, eaab0530. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Hou, F.J.; Le, H.P. The impact of natural resources, energy consumption, and population growth on environmental quality: Fresh evidence from the United States of America. Sci. Total Environ. 2021, 754, 142222. [Google Scholar] [CrossRef]
- Schiermeier, Q. Increased flood risk linked to global warming. Nature 2011, 470, 316. [Google Scholar] [CrossRef] [PubMed]
- Zandalinas, S.I.; Fritschi, F.B.; Mittler, R. Global Warming, Climate Change, and Environmental Pollution: Recipe for a Multifactorial Stress Combination Disaster. Trends Plant Sci. 2021, 26, 588–599. [Google Scholar] [CrossRef]
- Fan, E.S.; Li, L.; Wang, Z.P.; Lin, J.; Huang, Y.X.; Yao, Y.; Chen, R.J.; Wu, F. Sustainable Recycling Technology for Li-Ion Batteries and Beyond: Challenges and Future Prospects. Chem. Rev. 2020, 120, 7020–7063. [Google Scholar] [CrossRef] [PubMed]
- Orilall, M.C.; Wiesner, U. Block copolymer based composition and morphology control in nanostructured hybrid materials for energy conversion and storage: Solar cells, batteries, and fuel cells. Chem. Soc. Rev. 2011, 40, 520–535. [Google Scholar] [CrossRef]
- Bates, F.S. Polymer-polymer phase behavior. Sci.-New York 1991, 251, 898–905. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; Qiang, X.L.; Muller, A.H.E.; Groschel, A.H. Self-Assembly of block copolymers into internally ordered microparticles. Prog. Polym. Sci. 2020, 102, 101211. [Google Scholar] [CrossRef]
- Feng, H.B.; Lu, X.Y.; Wang, W.Y.; Kang, N.G.; Mays, J.W. Block Copolymers: Synthesis, Self-Assembly, and Applications. Polymers 2017, 9, 494. [Google Scholar] [CrossRef] [PubMed]
- Farrell, R.A.; Fitzgerald, T.G.; Borah, D.; Holmes, J.D.; Morris, M.A. Chemical Interactions and Their Role in the Microphase Separation of Block Copolymer Thin Films. Int. J. Mol. Sci. 2009, 10, 3671–3712. [Google Scholar] [CrossRef]
- Bates, C.M.; Seshimo, T.; Maher, M.J.; Durand, W.J.; Cushen, J.D.; Dean, L.M.; Blachut, G.; Ellison, C.J.; Willson, C.G. Polarity-Switching Top Coats Enable Orientation of Sub-10-nm Block Copolymer Domains. Science 2012, 338, 775–779. [Google Scholar] [CrossRef]
- Hagita, K.; Aoyagi, T.; Abe, Y.; Genda, S.; Honda, T. Deep learning-based estimation of Flory–Huggins parameter of A–B block copolymers from cross-sectional images of phase-separated structures. Sci. Rep. 2021, 11, 12322. [Google Scholar] [CrossRef]
- Yoshida, H.; Takenaka, M. 1-Physics of block copolymers from bulk to thin films. In Directed Self-Assembly of Block Co-Polymers for Nano-Manufacturing; Gronheid, R., Nealey, P., Eds.; Woodhead Publishing: Sawston, UK, 2015; pp. 3–26. [Google Scholar]
- Liu, M.J.; Qiang, Y.C.; Li, W.H.; Qiu, F.; Shi, A.C. Stabilizing the Frank-Kasper Phases via Binary Blends of AB Diblock Copolymers. Acs Macro Lett. 2016, 5, 1167–1171. [Google Scholar] [CrossRef]
- Bates, F.S.; Fredrickson, G.H. Block copolymers-Designer soft materials. Phys. Today 1999, 52, 32–38. [Google Scholar] [CrossRef]
- Groschel, A.H.; Muller, A.H.E. Self-assembly concepts for multicompartment nanostructures. Nanoscale 2015, 7, 11841–11876. [Google Scholar] [CrossRef]
- Wanka, G.; Hoffmann, H.; Ulbricht, W. Phase-Diagrams and aggregation behavior of poly(oxyethylene)-poly(oxypropylene)-poly(oxyethylene) triblock copolymers in aqueous-solutions. Macromolecules 1994, 27, 4145–4159. [Google Scholar] [CrossRef]
- Bates, F.S.; Hillmyer, M.A.; Lodge, T.P.; Bates, C.M.; Delaney, K.T.; Fredrickson, G.H. Multiblock Polymers: Panacea or Pandora’s Box? Science 2012, 336, 434–440. [Google Scholar] [CrossRef] [PubMed]
- Majewski, P.W.; Yager, K.G. Rapid ordering of block copolymer thin films. J. Phys.-Condes. Matter 2016, 28, 403002. [Google Scholar] [CrossRef] [PubMed]
- Abetz, V.; Kremer, K.; Muller, M.; Reiter, G. Functional Macromolecular Systems: Kinetic Pathways to Obtain Tailored Structures. Macromol. Chem. Phys. 2019, 220, 1800334. [Google Scholar] [CrossRef]
- Cui, H.G.; Chen, Z.Y.; Zhong, S.; Wooley, K.L.; Pochan, D.J. Block copolymer assembly via kinetic control. Science 2007, 317, 647–650. [Google Scholar] [CrossRef]
- Lynd, N.A.; Meuler, A.J.; Hillmyer, M.A. Polydispersity and block copolymer self-assembly. Prog. Polym. Sci. 2008, 33, 875–893. [Google Scholar] [CrossRef]
- Hadziioannou, G.; Skoulios, A. Molecular weight dependence of lamellar structure in styrene isoprene two- and three-block copolymers. Macromolecules 1982, 15, 258–262. [Google Scholar] [CrossRef]
- Carrot, C.; Guillet, J. From dynamic moduli to molecular weight distribution: A study of various polydisperse linear polymers. J. Rheol. 1997, 41, 1203–1220. [Google Scholar] [CrossRef]
- Llorens, J.; Rude, E.; Marcos, R.M. Polydispersity index from linear viscoelastic data: Unimodal and bimodal linear polymer melts. Polymer 2003, 44, 1741–1750. [Google Scholar] [CrossRef][Green Version]
- Levy, M. The impact of the concept of “Living Polymers” on material science. Polym. Adv. Technol. 2007, 18, 681–684. [Google Scholar] [CrossRef]
- Nguyen, H.T.; Tran, T.T.; Nguyen-Thai, N.U. Preparation of polydisperse polystyrene-block-poly(4-vinyl pyridine) synthesized by TEMPO-mediated radical polymerization and the facile nanostructure formation by self-assembly. J. Nanostruct. Chem. 2018, 8, 61–69. [Google Scholar] [CrossRef]
- Ruzette, A.V.; Tence-Girault, S.; Leibler, L.; Chauvin, F.; Bertin, D.; Guerret, O.; Gerard, P. Molecular disorder and mesoscopic order in polydisperse acrylic block copolymers prepared by controlled radical polymerization. Macromolecules 2006, 39, 5804–5814. [Google Scholar] [CrossRef]
- Lynd, N.A.; Hillmyer, M.A. Effects of polydispersity on the order-disorder transition in block copolymer melts. Macromolecules 2007, 40, 8050–8055. [Google Scholar] [CrossRef]
- Widin, J.M.; Schmitt, A.K.; Schmitt, A.L.; Im, K.; Mahanthappa, M.K. Unexpected Consequences of Block Polydispersity on the Self-Assembly of ABA Triblock Copolymers. J. Am. Chem. Soc. 2012, 134, 3834–3844. [Google Scholar] [CrossRef] [PubMed]
- Hanley, K.J.; Lodge, T.P.; Huang, C.-I. Phase Behavior of a Block Copolymer in Solvents of Varying Selectivity. Macromolecules 2000, 33, 5918–5931. [Google Scholar] [CrossRef][Green Version]
- Wang, Y. Nondestructive Creation of Ordered Nanopores by Selective Swelling of Block Copolymers: Toward Homoporous Membranes. Acc. Chem. Res. 2016, 49, 1401–1408. [Google Scholar] [CrossRef]
- Yi, Z.; Zhang, P.-B.; Liu, C.-J.; Zhu, L.-P. Symmetrical Permeable Membranes Consisting of Overlapped Block Copolymer Cylindrical Micelles for Nanoparticle Size Fractionation. Macromolecules 2016, 49, 3343–3351. [Google Scholar] [CrossRef]
- Gohil, J.M.; Choudhury, R.R. Chapter 2-Introduction to Nanostructured and Nano-enhanced Polymeric Membranes: Preparation, Function, and Application for Water Purification. In Nanoscale Materials in Water Purification; Thomas, S., Pasquini, D., Leu, S.-Y., Gopakumar, D.A., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 25–57. [Google Scholar]
- Jiang, H.; Chen, T.; Chen, Z.; Huo, J.; Zhang, L.; Zhou, J. Computer simulations on double hydrophobic PS-b-PMMA porous membrane by non-solvent induced phase separation. Fluid Phase Equilib. 2020, 523, 112784. [Google Scholar] [CrossRef]
- Paradiso, S.P.; Delaney, K.T.; Garcia-Cervera, C.J.; Ceniceros, H.D.; Fredrickson, G.H. Block Copolymer Self Assembly during Rapid Solvent Evaporation: Insights into Cylinder Growth and Stability. Acs Macro Lett. 2014, 3, 16–20. [Google Scholar] [CrossRef]
- Russell, T.P.; Coulon, G.; Deline, V.R.; Miller, D.C. Characteristics of the surface-induced orientation for symmetric diblock PS/PMMA copolymers. Macromolecules 1989, 22, 4600–4606. [Google Scholar] [CrossRef]
- Phillip, W.A.; O’Neill, B.; Rodwogin, M.; Hillmyer, M.A.; Cussler, E.L. Self-Assembled Block Copolymer Thin Films as Water Filtration Membranes. ACS Appl. Mater. Interfaces 2010, 2, 847–853. [Google Scholar] [CrossRef]
- Tseng, Y.H.; Lin, Y.L.; Ho, J.H.; Chang, C.T.; Fan, Y.C.; Shen, M.H.; Chen, J.T. Reversible and tunable morphologies of amphiphilic block copolymer nanorods confined in nanopores: Roles of annealing solvents. Polymer 2021, 228, 123859. [Google Scholar] [CrossRef]
- Lee, S.; Cheng, L.-C.; Gadelrab, K.R.; Ntetsikas, K.; Moschovas, D.; Yager, K.G.; Avgeropoulos, A.; Alexander-Katz, A.; Ross, C.A. Double-Layer Morphologies from a Silicon-Containing ABA Triblock Copolymer. ACS Nano 2018, 12, 6193–6202. [Google Scholar] [CrossRef] [PubMed]
- Chavis, M.A.; Smilgies, D.M.; Wiesner, U.B.; Ober, C.K. Widely Tunable Morphologies in Block Copolymer Thin Films Through Solvent Vapor Annealing Using Mixtures of Selective Solvents. Adv. Funct. Mater. 2015, 25, 3057–3065. [Google Scholar] [CrossRef]
- Peng, J.; Han, Y.C.; Knoll, W.; Kim, D.H. Development of nanodomain and fractal morphologies in solvent annealed block copolymer thin films. Macromol. Rapid Commun. 2007, 28, 1422–1428. [Google Scholar] [CrossRef]
- Stenbock-Fermor, A.; Rudov, A.A.; Gumerov, R.A.; Tsarkova, L.A.; Boker, A.; Moller, M.; Potemkin, I.I. Morphology-Controlled Kinetics of Solvent Uptake by Block Copolymer Films in Nonselective Solvent Vapors. Acs Macro Lett. 2014, 3, 803–807. [Google Scholar] [CrossRef] [PubMed]
- Tsarkova, L. Distortion of a Unit Cell versus Phase Transition to Nonbulk Morphology in Frustrated Films of Cylinder-Forming Polystyrene-b-polybutadiene Diblock Copolymers. Macromolecules 2012, 45, 7985–7994. [Google Scholar] [CrossRef]
- Cavicchi, K.A.; Berthiaume, K.J.; Russell, T.P. Solvent annealing thin films of poly(isoprene-b-lactide). Polymer 2005, 46, 11635–11639. [Google Scholar] [CrossRef]
- Xuan, Y.; Peng, J.; Cui, L.; Wang, H.; Li, B.; Han, Y. Morphology Development of Ultrathin Symmetric Diblock Copolymer Film via Solvent Vapor Treatment. Macromolecules 2004, 37, 7301–7307. [Google Scholar] [CrossRef]
- Phillip, W.A.; Hillmyer, M.A.; Cussler, E.L. Cylinder Orientation Mechanism in Block Copolymer Thin Films Upon Solvent Evaporation. Macromolecules 2010, 43, 7763–7770. [Google Scholar] [CrossRef]
- Lin, Z.Q.; Kim, D.H.; Wu, X.D.; Boosahda, L.; Stone, D.; LaRose, L.; Russell, T.P. A rapid route to arrays of nanostructures in thin films. Adv. Mater. 2002, 14, 1373–1376. [Google Scholar] [CrossRef]
- Kim, S.H.; Misner, M.J.; Xu, T.; Kimura, M.; Russell, T.P. Highly oriented and ordered arrays from block copolymers via solvent evaporation. Adv. Mater. 2004, 16, 226–231. [Google Scholar] [CrossRef]
- Kim, K.; Park, S.; Kim, Y.; Bang, J.; Park, C.; Ryu, D.Y. Optimized Solvent Vapor Annealing for Long-Range Perpendicular Lamellae in PS-b-PMMA Films. Macromolecules 2016, 49, 1722–1730. [Google Scholar] [CrossRef]
- Li, X.; Peng, J.; Wen, Y.; Kim, D.H.; Knoll, W. Morphology change of asymmetric diblock copolymer micellar films during solvent annealing. Polymer 2007, 48, 2434–2443. [Google Scholar] [CrossRef]
- Park, S.; Kim, Y.; Lee, W.; Hur, S.-M.; Ryu, D.Y. Gyroid Structures in Solvent Annealed PS-b-PMMA Films: Controlled Orientation by Substrate Interactions. Macromolecules 2017, 50, 5033–5041. [Google Scholar] [CrossRef]
- Cheng, X.; Boker, A.; Tsarkova, L. Temperature-Controlled Solvent Vapor Annealing of Thin Block Copolymer Films. Polymers 2019, 11, 1312. [Google Scholar] [CrossRef] [PubMed]
- Gowd, E.B.; Koga, T.; Endoh, M.K.; Kumar, K.; Stamm, M. Pathways of cylindrical orientations in PS-b-P4VP diblock copolymer thin films upon solvent vapor annealing. Soft Matter 2014, 10, 7753–7761. [Google Scholar] [CrossRef] [PubMed]
- Bosworth, J.K.; Paik, M.Y.; Ruiz, R.; Schwartz, E.L.; Huang, J.Q.; Ko, A.W.; Smilgies, D.M.; Black, C.T.; Ober, C.K. Control of self-assembly of lithographically patternable block copolymer films. Acs Nano 2008, 2, 1396–1402. [Google Scholar] [CrossRef] [PubMed]
- Arias-Zapata, J.; Bohme, S.; Garnier, J.; Girardot, C.; Legrain, A.; Zelsmann, M. Ultrafast Assembly of PS-PDMS Block Copolymers on 300 mm Wafers by Blending with Plasticizers. Adv. Funct. Mater. 2016, 26, 5690–5700. [Google Scholar] [CrossRef]
- Guillen, G.R.; Ramon, G.Z.; Kavehpour, H.P.; Kaner, R.B.; Hoek, E.M.V. Direct microscopic observation of membrane formation by nonsolvent induced phase separation. J. Membr. Sci. 2013, 431, 212–220. [Google Scholar] [CrossRef]
- Wang, D.-M.; Lai, J.-Y. Recent advances in preparation and morphology control of polymeric membranes formed by nonsolvent induced phase separation. Curr. Opin. Chem. Eng. 2013, 2, 229–237. [Google Scholar] [CrossRef]
- Guillen, G.R.; Pan, Y.; Li, M.; Hoek, E.M.V. Preparation and Characterization of Membranes Formed by Nonsolvent Induced Phase Separation: A Review. Ind. Eng. Chem. Res. 2011, 50, 3798–3817. [Google Scholar] [CrossRef]
- Tan, X.; Li, K. Inorganic hollow fibre membranes in catalytic processing. Curr. Opin. Chem. Eng. 2011, 1, 69–76. [Google Scholar] [CrossRef]
- Plisko, T.V.; Penkova, A.V.; Burts, K.S.; Bildyukevich, A.V.; Dmitrenko, M.E.; Melnikova, G.B.; Atta, R.R.; Mazur, A.S.; Zolotarev, A.A.; Missyul, A.B. Effect of Pluronic F127 on porous and dense membrane structure formation via non-solvent induced and evaporation induced phase separation. J. Membr. Sci. 2019, 580, 336–349. [Google Scholar] [CrossRef]
- Gu, Y.; Wiesner, U. Tailoring Pore Size of Graded Mesoporous Block Copolymer Membranes: Moving from Ultrafiltration toward Nanofiltration. Macromolecules 2015, 48, 6153–6159. [Google Scholar] [CrossRef]
- Abed, M.R.M.; Kumbharkar, S.C.; Groth, A.M.; Li, K. Ultrafiltration PVDF hollow fibre membranes with interconnected bicontinuous structures produced via a single-step phase inversion technique. J. Membr. Sci. 2012, 407–408, 145–154. [Google Scholar] [CrossRef]
- Gin, D.L.; Noble, R.D. Designing the Next Generation of Chemical Separation Membranes. Science 2011, 332, 674–676. [Google Scholar] [CrossRef]
- Li, D.F.; Chung, T.S.; Ren, J.Z.; Wang, R. Thickness dependence of macrovoid evolution in wet phase-inversion asymmetric membranes. Ind. Eng. Chem. Res. 2004, 43, 1553–1556. [Google Scholar] [CrossRef]
- Widjojo, N.; Chung, T.S. Thickness and air gap dependence of macrovoid evolution in phase-inversion asymmetric hollow fiber membranes. Ind. Eng. Chem. Res. 2006, 45, 7618–7626. [Google Scholar] [CrossRef]
- Abetz, V. Isoporous Block Copolymer Membranes. Macromol. Rapid Commun. 2015, 36, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Foroutani, K.; Ghasemi, S.M.; Pourabbas, B. Molecular tailoring of polystyrene-block-poly (acrylic acid) block copolymer toward additive-free asymmetric isoporous membranes via SNIPS. J. Membr. Sci. 2021, 623, 119099. [Google Scholar] [CrossRef]
- Peinemann, K.V.; Abetz, V.; Simon, P.F.W. Asymmetric superstructure formed in a block copolymer via phase separation. Nat. Mater. 2007, 6, 992–996. [Google Scholar] [CrossRef]
- Karunakaran, M.; Nunes, S.P.; Qiu, X.Y.; Yu, H.Z.; Peinemann, K.V. Isoporous PS-b-PEO ultrafiltration membranes via self-assembly and water-induced phase separation. J. Membr. Sci. 2014, 453, 471–477. [Google Scholar] [CrossRef]
- Stegelmeier, C.; Filiz, V.; Abetz, V.; Perlich, J.; Fery, A.; Ruckdeschel, P.; Rosenfeldt, S.; Forster, S. Topological Paths and Transient Morphologies during Formation of Mesoporous Block Copolymer Membranes. Macromolecules 2014, 47, 5566–5577. [Google Scholar] [CrossRef]
- Jung, A.; Rangou, S.; Abetz, C.; Filiz, V.; Abetz, V. Structure Formation of Integral Asymmetric Composite Membranes of Polystyrene-block-Poly(2-vinylpyridine) on a Nonwoven. Macromol. Mater. Eng. 2012, 297, 790–798. [Google Scholar] [CrossRef]
- Rahman, M.M. Selective Swelling and Functionalization of Integral Asymmetric Isoporous Block Copolymer Membranes. Macromol. Rapid Commun. 2021, 42, 2100235. [Google Scholar] [CrossRef]
- Dorin, R.M.; Phillip, W.A.; Sai, H.; Werner, J.; Elimelech, M.; Wiesner, U. Designing block copolymer architectures for targeted membrane performance. Polymer 2014, 55, 347–353. [Google Scholar] [CrossRef]
- Rangou, S.; Buhr, K.; Filiz, V.; Clodt, J.I.; Lademann, B.; Hahn, J.; Jung, A.; Abetz, V. Self-organized isoporous membranes with tailored pore sizes. J. Membr. Sci. 2014, 451, 266–275. [Google Scholar] [CrossRef]
- Radjabian, M.; Abetz, C.; Fischer, B.; Meyer, A.; Abetz, V. Influence of Solvent on the Structure of an Amphiphilic Block Copolymer in Solution and in Formation of an Integral Asymmetric Membrane. Acs Appl. Mater. Interfaces 2017, 9, 31224–31234. [Google Scholar] [CrossRef]
- Tan, K.W.; Jung, B.; Werner, J.G.; Rhoades, E.R.; Thompson, M.O.; Wiesner, U. Transient laser heating induced hierarchical porous structures from block copolymer-directed self-assembly. Science 2015, 349, 54–58. [Google Scholar] [CrossRef]
- Seshimo, T.; Maeda, R.; Odashima, R.; Takenaka, Y.; Kawana, D.; Ohmori, K.; Hayakawa, T. Perpendicularly oriented sub-10-nm block copolymer lamellae by atmospheric thermal annealing for one minute. Sci. Rep. 2016, 6, 19481. [Google Scholar] [CrossRef]
- Albalak, R.J.; Thomas, E.L.; Capel, M.S. Thermal annealing of roll-cast triblock copolymer films. Polymer 1997, 38, 3819–3825. [Google Scholar] [CrossRef]
- Tong, Q.Q.; Zheng, Q.; Sibener, S.J. Alignment and Structural Evolution of Cylinder-Forming Diblock Copolymer Thin Films in Patterned Tapered-Width Nanochannels. Macromolecules 2014, 47, 4236–4242. [Google Scholar] [CrossRef]
- Sepe, A.; Hoppe, E.T.; Jaksch, S.; Magerl, D.; Zhong, Q.; Perlich, J.; Posselt, D.; Smilgies, D.M.; Papadakis, C.M. The effect of heat treatment on the internal structure of nanostructured block copolymer films. J. Phys.-Condes. Matter 2011, 23, 254213. [Google Scholar] [CrossRef]
- Shi, L.-Y.; Yin, C.; Zhou, B.; Xia, W.; Weng, L.; Ross, C.A. Annealing Process Dependence of the Self-Assembly of Rod–Coil Block Copolymer Thin Films. Macromolecules 2021, 54, 1657–1664. [Google Scholar] [CrossRef]
- Majewski, P.W.; Yager, K.G. Latent Alignment in Pathway-Dependent Ordering of Block Copolymer Thin Films. Nano Lett. 2015, 15, 5221–5228. [Google Scholar] [CrossRef]
- Wang, H.S.; Kim, K.H.; Bang, J. Thermal Approaches to Perpendicular Block Copolymer Microdomains in Thin Films: A Review and Appraisal. Macromol. Rapid Commun. 2019, 40, 1800728. [Google Scholar] [CrossRef] [PubMed]
- Stehlin, F.; Diot, F.; Gwiazda, A.; Dirani, A.; Salaun, M.; Zelsmann, M.; Soppera, O. Local Reorganization of Diblock Copolymer Domains in Directed Self-Assembly Monitored by in Situ High-Temperature AFM. Langmuir 2013, 29, 12796–12803. [Google Scholar] [CrossRef]
- Majewski, P.W.; Yager, K.G. Reordering transitions during annealing of block copolymer cylinder phases. Soft Matter 2016, 12, 281–294. [Google Scholar] [CrossRef]
- Zhou, Z.P.; Liu, G.L. Controlling the Pore Size of Mesoporous Carbon Thin Films through Thermal and Solvent Annealing. Small 2017, 13, 1603107. [Google Scholar] [CrossRef]
- Kim, S.; Jeon, G.; Heo, S.W.; Kim, H.J.; Kim, S.B.; Chang, T.; Kim, J.K. High aspect ratio cylindrical microdomains oriented vertically on the substrate using block copolymer micelles and temperature-programmed solvent vapor annealing. Soft Matter 2013, 9, 5550–5556. [Google Scholar] [CrossRef]
- Kim, E.; Ahn, H.; Park, S.; Lee, H.; Lee, M.; Lee, S.; Kim, T.; Kwak, E.-A.; Lee, J.H.; Lei, X.; et al. Directed Assembly of High Molecular Weight Block Copolymers: Highly Ordered Line Patterns of Perpendicularly Oriented Lamellae with Large Periods. ACS Nano 2013, 7, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Gurovich, E. On Microphase Separation of Block Copolymers In an Electric Field: Four Universal Classes. Macromolecules 1994, 27, 7339–7362. [Google Scholar] [CrossRef]
- Boker, A.; Elbs, H.; Hansel, H.; Knoll, A.; Ludwigs, S.; Zettl, H.; Urban, V.; Abetz, V.; Muller, A.H.E.; Krausch, G. Microscopic mechanisms of electric-field-induced alignment of block copolymer microdomains. Phys. Rev. Lett. 2002, 89, 135502. [Google Scholar] [CrossRef]
- Schmidt, K.; Schoberth, H.G.; Ruppel, M.; Zettl, H.; Hansel, H.; Weiss, T.M.; Urban, V.; Krausch, G.; Boker, A. Reversible tuning of a block-copolymer nanostructure via electric fields. Nat. Mater. 2008, 7, 142–145. [Google Scholar] [CrossRef] [PubMed]
- Schoberth, H.G.; Olszowka, V.; Schmidt, K.; Boker, A. Effects of Electric Fields on Block Copolymer Nanostructures. In Complex Macromolecular Systems I; Muller, A.H.E., Schmidt, H.W., Eds.; Springer: Amsterdam, The Netherlands, 2010; Volume 227, pp. 1–31. [Google Scholar]
- Olszowka, V.; Hund, M.; Kuntermann, V.; Scherdel, S.; Tsarkova, L.; Boker, A. Electric Field Alignment of a Block Copolymer Nanopattern: Direct Observation of the Microscopic Mechanism. Acs Nano 2009, 3, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Schoberth, H.G.; Pester, C.W.; Ruppe, M.; Urban, V.S.; Boker, A. Orientation-Dependent Order-Disorder Transition of Block Copolymer Lamellae in Electric Fields. Acs Macro Lett. 2013, 2, 469–473. [Google Scholar] [CrossRef]
- Marencic, A.P.; Chaikin, P.M.; Register, R.A. Orientational order in cylinder-forming block copolymer thin films. Phys. Rev. E 2012, 86, 021507. [Google Scholar] [CrossRef]
- Morrison, F.; Le Bourvellec, G.; Winter, H.H. Flow-induced structure and rheology of a triblock copolymer. J. Appl. Polym. Sci. 1987, 33, 1585–1600. [Google Scholar] [CrossRef]
- Angelescu, D.E.; Waller, J.H.; Adamson, D.H.; Deshpande, P.; Chou, S.Y.; Register, R.A.; Chaikin, P.M. Macroscopic orientation of block copolymer cylinders in single-layer films by shearing. Adv. Mater. 2004, 16, 1736–1740. [Google Scholar] [CrossRef]
- Angelescu, D.E.; Waller, J.H.; Register, R.A.; Chaikin, P.M. Shear-induced alignment in thin films of spherical nanodomains. Adv. Mater. 2005, 17, 1878–1881. [Google Scholar] [CrossRef]
- Pujari, S.; Keaton, M.A.; Chaikin, P.M.; Register, R.A. Alignment of perpendicular lamellae in block copolymer thin films by shearing. Soft Matter 2012, 8, 5358–5363. [Google Scholar] [CrossRef]
- Davis, R.L.; Chaikin, P.M.; Register, R.A. Cylinder Orientation and Shear Alignment in Thin Films of Polystyrene-Poly(n-hexyl methacrylate) Diblock Copolymers. Macromolecules 2014, 47, 5277–5285. [Google Scholar] [CrossRef]
- Marencic, A.P.; Adamson, D.H.; Chaikin, P.M.; Register, R.A. Shear alignment and realignment of sphere-forming and cylinder-forming block-copolymer thin films. Phys. Rev. E 2010, 81, 011503. [Google Scholar] [CrossRef]
- Kim, S.Y.; Nunns, A.; Gwyther, J.; Davis, R.L.; Manners, I.; Chaikin, P.M.; Register, R.A. Large-Area Nanosquare Arrays from Shear-Aligned Block Copolymer Thin Films. Nano Lett. 2014, 14, 5698–5705. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Goswami, M.; Kumar, R.; Sumpter, B.G.; Mays, J. Morphologies of block copolymers composed of charged and neutral blocks. Soft Matter 2012, 8, 3036–3052. [Google Scholar] [CrossRef]
- Sing, C.E.; Zwanikken, J.W.; de la Cruz, M.O. Electrostatic control of block copolymer morphology. Nat. Mater. 2014, 13, 694–698. [Google Scholar] [CrossRef]
- Goswami, M.; Sumpter, B.G.; Mays, J. Controllable stacked disk morphologies of charged diblock copolymers. Chem. Phys. Lett. 2010, 487, 272–278. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, C.G.; Gao, C.Q.; Qu, Y.Q.; Shi, K.Y.; Zhang, W.Q. Polymerization-Induced Self-Assembly of Block Copolymer Through Dispersion RAFT Polymerization in Ionic Liquid. J. Polym. Sci. Pol. Chem. 2016, 54, 1517–1525. [Google Scholar] [CrossRef]
- Kim, S.Y.; Yoon, E.; Joo, T.; Park, M.J. Morphology and Conductivity in Ionic Liquid Incorporated Sulfonated Block Copolymers. Macromolecules 2011, 44, 5289–5298. [Google Scholar] [CrossRef]
- Bennett, T.M.; Jack, K.S.; Thurecht, K.J.; Blakey, I. Perturbation of the Experimental Phase Diagram of a Diblock Copolymer by Blending with an Ionic Liquid. Macromolecules 2016, 49, 205–214. [Google Scholar] [CrossRef]
- Cui, T.; Li, X.; Dong, B.; Li, X.; Guo, M.; Wu, L.; Li, B.; Li, H. Janus onions of block copolymers via confined self-assembly. Polymer 2019, 174, 70–76. [Google Scholar] [CrossRef]
- He, Q.B.; Zhang, Y.J.; Li, H.L.; Chen, Q. Rheological Properties of ABA-Type Copolymers Physically End-Cross-Linked by Polyoxometalate. Macromolecules 2020, 53, 10927–10941. [Google Scholar] [CrossRef]
- Zhang, L.Y.; Liu, C.; Shang, H.Y.; Cao, X.; Chai, S.C.; Chen, Q.; Wu, L.X.; Li, H.L. Electrostatic tuning of block copolymer morphologies by inorganic macroions. Polymer 2016, 106, 53–61. [Google Scholar] [CrossRef]
- Guo, W.; Cao, L.X.; Xia, J.C.; Nie, F.Q.; Ma, W.; Xue, J.M.; Song, Y.L.; Zhu, D.B.; Wang, Y.G.; Jiang, L. Energy Harvesting with Single-Ion-Selective Nanopores: A Concentration-Gradient-Driven Nanofluidic Power Source. Adv. Funct. Mater. 2010, 20, 1339–1344. [Google Scholar] [CrossRef]
- Logan, B.E.; Elimelech, M. Membrane-based processes for sustainable power generation using water. Nature 2012, 488, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Hao, J.; Sun, Q.; Zhao, M.; Liu, H.; Li, C.; Sui, X. Carbon nanofibers membrane bridged with graphene nanosheet and hyperbranched polymer for high-performance osmotic energy harvesting. Nano Res. 2022. [Google Scholar] [CrossRef]
- Siria, A.; Bocquet, M.L.; Bocquet, L. New avenues for the large-scale harvesting of blue energy. Nat. Rev. Chem. 2017, 1, 0091. [Google Scholar] [CrossRef]
- Mei, Y.; Tang, C.Y.Y. Recent developments and future perspectives of reverse electrodialysis technology: A review. Desalination 2018, 425, 156–174. [Google Scholar] [CrossRef]
- Kingsbury, R.S.; Chu, K.; Coronell, O. Energy storage by reversible electrodialysis: The concentration battery. J. Membr. Sci. 2015, 495, 502–516. [Google Scholar] [CrossRef]
- van Egmond, W.J.; Saakes, M.; Porada, S.; Meuwissen, T.; Buisman, C.J.N.; Hamelers, H.V.M. The concentration gradient flow battery as electricity storage system: Technology potential and energy dissipation. J. Power Sources 2016, 325, 129–139. [Google Scholar] [CrossRef]
- Zaffora, A.; Culcasi, A.; Gurreri, L.; Cosenza, A.; Tamburini, A.; Santamaria, M.; Micale, G. Energy Harvesting by Waste Acid/Base Neutralization via Bipolar Membrane Reverse Electrodialysis. Energies 2020, 13, 5510. [Google Scholar] [CrossRef]
- Tedesco, M.; Cipollina, A.; Tamburini, A.; Micale, G. Towards 1 kW power production in a reverse electrodialysis pilot plant with saline waters and concentrated brines. J. Membr. Sci. 2017, 522, 226–236. [Google Scholar] [CrossRef]
- Siria, A.; Poncharal, P.; Biance, A.L.; Fulcrand, R.; Blase, X.; Purcell, S.T.; Bocquet, L. Giant osmotic energy conversion measured in a single transmembrane boron nitride nanotube. Nature 2013, 494, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Wang, S.; Yu, B.; Sui, X.; Shen, Y.; Cong, H. Bioinspired nanochannels based on polymeric membranes. Sci. China Mater. 2021, 64, 1320–1342. [Google Scholar] [CrossRef]
- Li, C.; Jiang, H.; Liu, P.; Zhai, Y.; Yang, X.; Gao, L.; Jiang, L. One Porphyrin Per Chain Self-Assembled Helical Ion-Exchange Channels for Ultrahigh Osmotic Energy Conversion. J. Am. Chem. Soc. 2022, 144, 9472–9478. [Google Scholar] [CrossRef]
- Xiao, K.; Wen, L.P.; Jiang, L. Biomimetic Solid-State Nanochannels: From Fundamental Research to Practical Applications. Small 2016, 12, 2810–2831. [Google Scholar] [CrossRef]
- Laucirica, G.; Albesa, A.G.; Toimil-Molares, M.E.; Trautmann, C.; Marmisolle, W.A.; Azzaroni, O. Shape matters: Enhanced osmotic energy harvesting in bullet-shaped nanochannels. Nano Energy 2020, 71, 104621. [Google Scholar] [CrossRef]
- Chen, W.; Xiang, Y.; Kong, X.-Y.; Wen, L. Polymer-based membranes for promoting osmotic energy conversion. Giant 2022, 10, 100094. [Google Scholar] [CrossRef]
- Emmerich, T.; Vasu, K.S.; Nigues, A.; Keerthi, A.; Radha, B.; Siria, A.; Bocquet, L. Enhanced nanofluidic transport in activated carbon nanoconduits. Nat. Mater. 2022, 26, 696–702. [Google Scholar] [CrossRef]
- Lin, C.Y.; Combs, C.; Su, Y.S.; Yeh, L.H.; Siwy, Z.S. Rectification of Concentration Polarization in Mesopores Leads To High Conductance Ionic Diodes and High Performance Osmotic Power. J. Am. Chem. Soc. 2019, 141, 3691–3698. [Google Scholar] [CrossRef]
- Wu, Y.D.; Qian, Y.C.; Niu, B.; Chen, J.J.; He, X.F.; Yang, L.S.; Kong, X.Y.; Zhao, Y.F.; Lin, X.B.; Zhou, T.; et al. Surface Charge Regulated Asymmetric Ion Transport in Nanoconfined Space. Small 2021, 17, 2101199. [Google Scholar] [CrossRef] [PubMed]
- Sharon, D.; Bennington, P.; Dolejsi, M.; Webb, M.A.; Dong, B.X.; de Pablo, J.J.; Nealey, P.F.; Patel, S.N. Intrinsic Ion Transport Properties of Block Copolymer Electrolytes. Acs Nano 2020, 14, 8902–8914. [Google Scholar] [CrossRef] [PubMed]
- DuChanois, R.M.; Porter, C.J.; Violet, C.; Verduzco, R.; Elimelech, M. Membrane Materials for Selective Ion Separations at the Water-Energy Nexus. Adv. Mater. 2021, 33, 2101312. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hou, J.; Zhang, H.C.; Tian, Y.; Jiang, L. Single Nanochannel-Aptamer-Based Biosensor for Ultrasensitive and Selective Cocaine Detection. Acs Appl. Mater. Interfaces 2018, 10, 2033–2039. [Google Scholar] [CrossRef]
- Apel, P.Y. Fabrication of functional micro- and nanoporous materials from polymers modified by swift heavy ions. Radiat. Phys. Chem. 2019, 159, 25–34. [Google Scholar] [CrossRef]
- Zhang, Z.; Kong, X.Y.; Xiao, K.; Liu, Q.; Xie, G.H.; Li, P.; Ma, J.; Tian, Y.; Wen, L.P.; Jiang, L. Engineered Asymmetric Heterogeneous Membrane: A Concentration-Gradient-Driven Energy Harvesting Device. J. Am. Chem. Soc. 2015, 137, 14765–14772. [Google Scholar] [CrossRef]
- Wang, J.; Liu, L.; Yan, G.L.; Li, Y.C.; Gao, Y.; Tian, Y.; Jiang, L. Ionic Transport and Robust Switching Properties of the Confined Self-Assembled Block Copolymer/Homopolymer in Asymmetric Nanochannels. ACS Appl. Mater. Interfaces 2021, 13, 14520–14530. [Google Scholar] [CrossRef]
- Yang, L.S.; Liu, P.; Zhu, C.C.; Zhao, Y.Y.; Yuan, M.M.; Kong, X.Y.; Wen, L.P.; Jiang, L. Ion transport regulation through triblock copolymer/PET asymmetric nanochannel membrane: Model system establishment and rectification mapping. Chin. Chem. Lett. 2021, 32, 822–825. [Google Scholar] [CrossRef]
- Wu, Y.F.; Yang, G.; Lin, M.C.; Kong, X.Y.; Mi, L.; Liu, S.Q.; Chen, G.S.; Tian, Y.; Jiang, L. Continuously Tunable Ion Rectification and Conductance in Submicrochannels Stemming from Thermoresponsive Polymer Self-Assembly. Angew. Chem. Int. Ed. 2019, 58, 12481–12485. [Google Scholar] [CrossRef]
- Macha, M.; Marion, S.; Nandigana, V.V.R.; Radenovic, A. 2D materials as an emerging platform for nanopore-based power generation. Nat. Rev. Mater. 2019, 4, 588–605. [Google Scholar] [CrossRef]
- Hao, J.; Wang, W.; Zhao, J.; Che, H.; Chen, L.; Sui, X. Construction and application of bioinspired nanochannels based on two-dimensional materials. Chin. Chem. Lett. 2022, 33, 2291–2300. [Google Scholar] [CrossRef]
- Lin, X.B.; Liu, P.; Xin, W.W.; Teng, Y.F.; Chen, J.J.; Wu, Y.D.; Zhao, Y.F.; Kong, X.Y.; Jiang, L.; Wen, L.P. Heterogeneous MXene/PS-b-P2VP Nanofluidic Membranes with Controllable Ion Transport for Osmotic Energy Conversion. Adv. Funct. Mater. 2021, 31, 2105013. [Google Scholar] [CrossRef]
- Sui, X.; Zhang, Z.; Zhang, Z.Y.; Wang, Z.W.; Li, C.; Yuan, H.; Gao, L.C.; Wen, L.P.; Fan, X.; Yang, L.J.; et al. Biomimetic Nanofluidic Diode Composed of Dual Amphoteric Channels Maintains Rectification Direction over a Wide pH Range. Angew. Chem. Int. Ed. 2016, 55, 13056–13060. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Zhang, Z.; Li, C.; Gao, L.C.; Zhao, Y.; Yang, L.J.; Wen, L.P.; Jiang, L. Engineered Nanochannel Membranes with Diode-like Behavior for Energy Conversion over a Wide pH Range. Acs Appl. Mater. Interfaces 2019, 11, 23815–23821. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sui, X.; Li, P.; Xie, G.H.; Kong, X.Y.; Xiao, K.; Gao, L.C.; Wen, L.P.; Jiang, L. Ultrathin and Ion-Selective Janus Membranes for High-Performance Osmotic Energy Conversion. J. Am. Chem. Soc. 2017, 139, 8905–8914. [Google Scholar] [CrossRef]
- Hao, J.L.; Yang, T.; He, X.L.; Tang, H.Y.; Sui, X. Hierarchical nanochannels based on rod-coil block copolymer for ion transport and energy conversion. Giant 2021, 5, 100049. [Google Scholar] [CrossRef]
- Li, C.; Wen, L.P.; Sui, X.; Cheng, Y.R.; Gao, L.C.; Jiang, L. Large-scale, robust mushroom-shaped nanochannel array membrane for ultrahigh osmotic energy conversion. Sci. Adv. 2021, 7, eabg2183. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.M.; Park, C.H.; Yoo, S.; Lee, G.W.; Yang, S.Y.; Kim, J.H.; Yoo, S.I. Selective ion transport through three-dimensionally interconnected nanopores of quaternized block copolymer membranes for energy harvesting application. Soft Matter 2021, 17, 3700–3708. [Google Scholar] [CrossRef]
- Xie, L.; Zhou, S.; Liu, J.R.; Qiu, B.L.; Liu, T.Y.; Liang, Q.R.; Zheng, X.Z.; Li, B.; Zeng, J.; Yan, M.; et al. Sequential Superassembly of Nanofiber Arrays to Carbonaceous Ordered Mesoporous Nanowires and Their Heterostructure Membranes for Osmotic Energy Conversion. J. Am. Chem. Soc. 2021, 143, 6922–6932. [Google Scholar] [CrossRef]
- Sazali, N.; Salleh, W.N.W.; Jamaludin, A.S.; Razali, M.N.M. New Perspectives on Fuel Cell Technology: A Brief Review. Membranes 2020, 10, 99. [Google Scholar] [CrossRef]
- Sharaf, O.Z.; Orhan, M.F. An overview of fuel cell technology: Fundamentals and applications. Renew. Sust. Energ. Rev. 2014, 32, 810–853. [Google Scholar] [CrossRef]
- Zaman, S.; Huang, L.; Douka, A.I.; Yang, H.; You, B.; Xia, B.Y. Oxygen Reduction Electrocatalysts toward Practical Fuel Cells: Progress and Perspectives. Angew. Chem. Int. Ed. 2021, 60, 17832–17852. [Google Scholar] [CrossRef] [PubMed]
- Cullen, D.A.; Neyerlin, K.C.; Ahluwalia, R.K.; Mukundan, R.; More, K.L.; Borup, R.L.; Weber, A.Z.; Myers, D.J.; Kusoglu, A. New roads and challenges for fuel cells in heavy-duty transportation. Nat. Energy 2021, 6, 462–474. [Google Scholar] [CrossRef]
- He, G.W.; Li, Z.; Zhao, J.; Wang, S.F.; Wu, H.; Guiver, M.D.; Jiang, Z.Y. Nanostructured Ion-Exchange Membranes for Fuel Cells: Recent Advances and Perspectives. Adv. Mater. 2015, 27, 5280–5295. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, J.; Zhao, D.Y. Mesoporous materials for energy conversion and storage devices. Nat. Rev. Mater. 2016, 1, 16023. [Google Scholar] [CrossRef]
- Ogungbemi, E.; Ijaodola, O.; Khatib, F.N.; Wilberforce, T.; El Hassan, Z.; Thompson, J.; Ramadan, M.; Olabi, A.G. Fuel cell membranes-Pros and cons. Energy 2019, 172, 155–172. [Google Scholar] [CrossRef]
- Adamski, M.; Skalski, T.J.G.; Britton, B.; Peckham, T.J.; Metzler, L.; Holdcroft, S. Highly Stable, Low Gas Crossover, Proton-Conducting Phenylated Polyphenylenes. Angew. Chem. Int. Ed. 2017, 56, 9058–9061. [Google Scholar] [CrossRef]
- Cheng, J.; He, G.H.; Zhang, F.X. A mini-review on anion exchange membranes for fuel cell applications: Stability issue and addressing strategies. Int. J. Hydrog. Energy 2015, 40, 7348–7360. [Google Scholar] [CrossRef]
- Luo, T.; Abdu, S.; Wessling, M. Selectivity of ion exchange membranes: A review. J. Membr. Sci. 2018, 555, 429–454. [Google Scholar] [CrossRef]
- Wang, Y.; Ruiz Diaz, D.F.; Chen, K.S.; Wang, Z.; Adroher, X.C. Materials, technological status, and fundamentals of PEM fuel cells—A review. Mater. Today 2020, 32, 178–203. [Google Scholar] [CrossRef]
- Scofield, M.E.; Liu, H.Q.; Wong, S.S. A concise guide to sustainable PEMFCs: Recent advances in improving both oxygen reduction catalysts and proton exchange membranes. Chem. Soc. Rev. 2015, 44, 5836–5860. [Google Scholar] [CrossRef]
- Xing, L.; Shi, W.; Su, H.; Xu, Q.; Das, P.K.; Mao, B.; Scott, K. Membrane electrode assemblies for PEM fuel cells: A review of functional graded design and optimization. Energy 2019, 177, 445–464. [Google Scholar] [CrossRef]
- Kim, D.J.; Jo, M.J.; Nam, S.Y. A review of polymer–nanocomposite electrolyte membranes for fuel cell application. J. Ind. Eng. Chem. 2015, 21, 36–52. [Google Scholar] [CrossRef]
- Tellez-Cruz, M.M.; Escorihuela, J.; Solorza-Feria, O.; Compan, V. Proton Exchange Membrane Fuel Cells (PEMFCs): Advances and Challenges. Polymers 2021, 13, 3064. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, K.S.; Mishler, J.; Cho, S.C.; Adroher, X.C. A review of polymer electrolyte membrane fuel cells: Technology, applications, and needs on fundamental research. Appl. Energy 2011, 88, 981–1007. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.; Lv, Z.; Zhao, T.; Li, P.; Sun, Y.; Wang, J. Sulfonated Ti3C2Tx to construct proton transfer pathways in polymer electrolyte membrane for enhanced conduction. Solid State Ion. 2017, 310, 100–111. [Google Scholar] [CrossRef]
- Yu, L.; Yue, B.; Yan, L.; Zhao, H.; Zhang, J. Proton conducting composite membranes based on sulfonated polysulfone and polysulfone-g-(phosphonated polystyrene) via controlled atom-transfer radical polymerization for fuel cell applications. Solid State Ion. 2019, 338, 103–112. [Google Scholar] [CrossRef]
- Ciftcioglu, G.A.; Frank, C.W. Effect of Increased Ionic Liquid Uptake via Thermal Annealing on Mechanical Properties of Polyimide-Poly(ethylene glycol) Segmented Block Copolymer Membranes. Molecules 2021, 26, 2143. [Google Scholar] [CrossRef]
- Watanabe, M.; Thomas, M.L.; Zhang, S.G.; Ueno, K.; Yasuda, T.; Dokko, K. Application of Ionic Liquids to Energy Storage and Conversion Materials and Devices. Chem. Rev. 2017, 117, 7190–7239. [Google Scholar] [CrossRef]
- Elwan, H.A.; Mamlouk, M.; Scott, K. A review of proton exchange membranes based on protic ionic liquid/polymer blends for polymer electrolyte membrane fuel cells. J. Power Sources 2021, 484, 229197. [Google Scholar] [CrossRef]
- Schulze, M.W.; McIntosh, L.D.; Hillmyer, M.A.; Lodge, T.P. High-Modulus, High-Conductivity Nanostructured Polymer Electrolyte Membranes via Polymerization-Induced Phase Separation. Nano Lett. 2014, 14, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Y.; Cui, T.T.; Cao, X.; Zhao, C.J.; Chen, Q.; Wu, L.X.; Li, H.L. Inorganic-Macroion-Induced Formation of Bicontinuous Block Co-polymer Nanocomposites with Enhanced Conductivity and Modulus. Angew. Chem. Int. Ed. 2017, 56, 9013–9017. [Google Scholar] [CrossRef] [PubMed]
- Zhai, L.; Chai, S.C.; Wang, G.; Zhang, W.; He, H.B.; Li, H.L. Triblock Copolymer/Polyoxometalate Nanocomposite Electrolytes with Inverse Hexagonal Cylindrical Nanostructures. Macromol. Rapid Commun. 2020, 41, 2000438. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Wang, Y.C.; Wu, Z.P.; Chen, G.Y.; Yang, F.; Zhu, S.Q.; Siddharth, K.; Kong, Z.J.; Lu, A.L.; Li, J.C.; et al. Recent Advances in Electrocatalysts for Proton Exchange Membrane Fuel Cells and Alkaline Membrane Fuel Cells. Adv. Mater. 2021, 33, 2006292. [Google Scholar] [CrossRef]
- Guerrero Moreno, N.; Cisneros Molina, M.; Gervasio, D.; Pérez Robles, J.F. Approaches to polymer electrolyte membrane fuel cells (PEMFCs) and their cost. Renew. Sust. Energ. Rev. 2015, 52, 897–906. [Google Scholar] [CrossRef]
- Postole, G.; Auroux, A. The poisoning level of Pt/C catalysts used in PEM fuel cells by the hydrogen feed gas impurities: The bonding strength. Int. J. Hydrog. Energy 2011, 36, 6817–6825. [Google Scholar] [CrossRef]
- Wang, J. System integration, durability and reliability of fuel cells: Challenges and solutions. Appl. Energy 2017, 189, 460–479. [Google Scholar] [CrossRef]
- Garzon, F.; Uribe, F.A.; Rockward, T.; Urdampilleta, I.G.; Brosha, E.L. The Impact of Hydrogen Fuel Contaminates on Long-Term PMFC Performance. ECS Trans. 2006, 3, 695–703. [Google Scholar] [CrossRef]
- Ramaswamy, N.; Mukerjee, S. Alkaline Anion-Exchange Membrane Fuel Cells: Challenges in Electrocatalysis and Interfacial Charge Transfer. Chem. Rev. 2019, 119, 11945–11979. [Google Scholar] [CrossRef]
- Varcoe, J.R.; Atanassov, P.; Dekel, D.R.; Herring, A.M.; Hickner, M.A.; Kohl, P.A.; Kucernak, A.R.; Mustain, W.E.; Nijmeijer, K.; Scott, K.; et al. Anion-exchange membranes in electrochemical energy systems. Energy Environ. Sci. 2014, 7, 3135–3191. [Google Scholar] [CrossRef]
- Chen, N.J.; Wang, H.H.; Kim, S.P.; Kim, H.M.; Lee, W.H.; Hu, C.; Bae, J.Y.; Sim, E.S.; Chung, Y.C.; Jang, J.H.; et al. Poly(fluorenyl aryl piperidinium) membranes and ionomers for anion exchange membrane fuel cells. Nat. Commun. 2021, 12, 2367. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Noonan, K.J.T.; Coates, G.W. Alkaline-stable anion exchange membranes: A review of synthetic approaches. Prog. Polym. Sci. 2020, 100, 101177. [Google Scholar] [CrossRef]
- Gao, J.F.; Wang, L.; Guo, Z.; Li, B.; Wang, H.; Luo, J.C.; Huang, X.W.; Xue, H.G. Flexible, superhydrophobic, and electrically conductive polymer nanofiber composite for multifunctional sensing applications. Chem. Eng. J. 2020, 381, 122778. [Google Scholar] [CrossRef]
- Zhu, J.Q.; Birgisson, B.; Kringos, N. Polymer modification of bitumen: Advances and challenges. Eur. Polym. J. 2014, 54, 18–38. [Google Scholar] [CrossRef]
- Zhu, S.; So, J.H.; Mays, R.; Desai, S.; Barnes, W.R.; Pourdeyhimi, B.; Dickey, M.D. Ultrastretchable Fibers with Metallic Conductivity Using a Liquid Metal Alloy Core. Adv. Funct. Mater. 2013, 23, 2308–2314. [Google Scholar] [CrossRef]
- Park, H.S.; Hong, C.K. Anion Exchange Membrane Based on Sulfonated Poly (Styrene-Ethylene-Butylene-Styrene) Copolymers. Polymers 2021, 13, 1669. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Su, Y.X.; Wu, Y.B.; Zhang, M.; Wang, Y.G.; Chen, Q.; Li, N.W. Synthesis and properties of quaternized polyolefins with bulky poly(4-phenyl-1-butene) moieties as anion exchange membranes. J. Membr. Sci. 2017, 541, 244–252. [Google Scholar] [CrossRef]
- Zhang, X.J.; Chu, X.M.; Zhang, M.; Zhu, M.; Huang, Y.D.; Wang, Y.G.; Liu, L.; Li, N.W. Molecularly designed, solvent processable tetraalkylammonium-functionalized fluoropolyolefin for durable anion exchange membrane fuel cells. J. Membr. Sci. 2019, 574, 212–221. [Google Scholar] [CrossRef]
- Pan, Y.; Jiang, K.; Sun, X.R.; Ma, S.Y.; So, Y.M.; Ma, H.W.; Yan, X.M.; Zhang, N.; He, G.H. Facilitating ionic conduction for anion exchange membrane via employing star-shaped block copolymer. J. Membr. Sci. 2021, 630, 119290. [Google Scholar] [CrossRef]
- Tarascon, J.M.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef]
- Sun, Y.M.; Liu, N.A.; Cui, Y. Promises and challenges of nanomaterials for lithium-based rechargeable batteries. Nat. Energy 2016, 1, 16071. [Google Scholar] [CrossRef]
- Ye, Y.S.; Chou, L.Y.; Liu, Y.Y.; Wang, H.S.; Lee, H.K.; Huang, W.X.; Wan, J.Y.; Liu, K.; Zhou, G.M.; Yang, Y.F.; et al. Ultralight and fire-extinguishing current collectors for high-energy and high-safety lithium-ion batteries. Nat. Energy 2020, 5, 786–793. [Google Scholar] [CrossRef]
- Liu, K.; Liu, Y.; Lin, D.; Pei, A.; Cui, Y. Materials for lithium-ion battery safety. Sci. Adv. 2018, 4, eaas9820. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Yanilmaz, M.; Toprakci, O.; Fu, K.; Zhang, X.W. A review of recent developments in membrane separators for rechargeable lithium-ion batteries. Energy Environ. Sci. 2014, 7, 3857–3886. [Google Scholar] [CrossRef]
- Zhang, L.P.; Li, X.L.; Yang, M.R.; Chen, W.H. High-safety separators for lithium-ion batteries and sodium-ion batteries: Advances and perspective. Energy Storage Mater. 2021, 41, 522–545. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, J.J.; Song, Y.Z.; Yin, X.; Sun, C.C.; Zhu, L.P.; Zhu, B.K. Tannic acid/polyethyleneimine-decorated polypropylene separators for Li-Ion batteries and the role of the interfaces between separator and electrolyte. Electrochim. Acta 2018, 275, 25–31. [Google Scholar] [CrossRef]
- Wang, X.; Peng, L.Q.; Hua, H.M.; Liu, Y.Z.; Zhang, P.; Zhao, J.B. Magnesium Borate Fiber Coating Separators with High Lithium-Ion Transference Number for Lithium-Ion Batteries. Chemelectrochem 2020, 7, 1187–1192. [Google Scholar] [CrossRef]
- Zahn, R.; Lagadec, M.F.; Hess, M.; Wood, V. Improving Ionic Conductivity and Lithium-Ion Transference Number in Lithium-Ion Battery Separators. ACS Appl. Mater. Interfaces 2016, 8, 32637–32642. [Google Scholar] [CrossRef]
- Ren, D.S.; Feng, X.N.; Liu, L.S.; Hsu, H.J.; Lu, L.G.; Wang, L.; He, X.M.; Ouyang, M.G. Investigating the relationship between internal short circuit and thermal runaway of lithium-ion batteries under thermal abuse condition. Energy Storage Mater. 2021, 34, 563–573. [Google Scholar] [CrossRef]
- Zheng, Q.F.; Yamada, Y.; Shang, R.; Ko, S.; Lee, Y.Y.; Kim, K.; Nakamura, E.; Yamada, A. A cyclic phosphate-based battery electrolyte for high voltage and safe operation. Nat. Energy 2020, 5, 291–298. [Google Scholar] [CrossRef]
- Zhang, X.Q.; Chen, X.; Cheng, X.B.; Li, B.Q.; Shen, X.; Yan, C.; Huang, J.Q.; Zhang, Q. Highly Stable Lithium Metal Batteries Enabled by Regulating the Solvation of Lithium Ions in Nonaqueous Electrolytes. Angew. Chem. Int. Ed. 2018, 57, 5301–5305. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Shi, X.S.; Chu, S.Y.; Shao, Z.P.; Wang, Y. Design of Block-Copolymer Nanoporous Membranes for Robust and Safer Lithium-Ion Battery Separators. Adv. Sci. 2021, 8, 2003096. [Google Scholar] [CrossRef]
- Wang, Y.; Zhong, W.H. Development of Electrolytes towards Achieving Safe and High-Performance Energy-Storage Devices: A Review. Chemelectrochem 2015, 2, 22–36. [Google Scholar] [CrossRef]
- Xiao, Y.R.; Turcheniuk, K.; Narla, A.; Song, A.Y.; Ren, X.L.; Magasinski, A.; Jain, A.; Huang, S.; Lee, H.; Yushin, G. Electrolyte melt infiltration for scalable manufacturing of inorganic all-solid-state lithium-ion batteries. Nat. Mater. 2021, 20, 984–990. [Google Scholar] [CrossRef]
- Schnell, J.; Gunther, T.; Knoche, T.; Vieider, C.; Kohler, L.; Just, A.; Keller, M.; Passerini, S.; Reinhart, G. All-solid-state lithium-ion and lithium metal batteries-paving the way to large-scale production. J. Power Sources 2018, 382, 160–175. [Google Scholar] [CrossRef]
- Wang, Q.S.; Jiang, L.H.; Yu, Y.; Sun, J.H. Progress of enhancing the safety of lithium ion battery from the electrolyte aspect. Nano Energy 2019, 55, 93–114. [Google Scholar] [CrossRef]
- Manthiram, A.; Yu, X.W.; Wang, S.F. Lithium battery chemistries enabled by solid-state electrolytes. Nat. Rev. Mater. 2017, 2, 16103. [Google Scholar] [CrossRef]
- Lin, Z.Y.; Guo, X.W.; Yang, Y.B.; Tang, M.X.; Wei, Q.; Yu, H.J. Block copolymer electrolyte with adjustable functional units for solid polymer lithium metal battery. J. Energy Chem. 2021, 52, 67–74. [Google Scholar] [CrossRef]
- He, Y.B.; Liu, N.; Kohl, P.A. Difunctional block copolymer with ion solvating and crosslinking sites as solid polymer electrolyte for lithium batteries. J. Power Sources 2021, 481, 228832. [Google Scholar] [CrossRef]
- Liu, D.; Yang, W.L.; Liu, Y.; Yang, S.C.; Shen, Z.H.; Fan, X.H.; Yang, H.; Zhou, Q.F. Enhancing ionic conductivity in tablet-bottlebrush block copolymer electrolytes with well-aligned nanostructures via solvent vapor annealing. J. Mater. Chem. C 2022, 10, 4247–4256. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, S.; Hou, Y.; Hao, J.; Lin, C.; Zhao, J.; Sui, X. Well-Defined Nanostructures by Block Copolymers and Mass Transport Applications in Energy Conversion. Polymers 2022, 14, 4568. https://doi.org/10.3390/polym14214568
Ma S, Hou Y, Hao J, Lin C, Zhao J, Sui X. Well-Defined Nanostructures by Block Copolymers and Mass Transport Applications in Energy Conversion. Polymers. 2022; 14(21):4568. https://doi.org/10.3390/polym14214568
Chicago/Turabian StyleMa, Shuhui, Yushuang Hou, Jinlin Hao, Cuncai Lin, Jiawei Zhao, and Xin Sui. 2022. "Well-Defined Nanostructures by Block Copolymers and Mass Transport Applications in Energy Conversion" Polymers 14, no. 21: 4568. https://doi.org/10.3390/polym14214568
APA StyleMa, S., Hou, Y., Hao, J., Lin, C., Zhao, J., & Sui, X. (2022). Well-Defined Nanostructures by Block Copolymers and Mass Transport Applications in Energy Conversion. Polymers, 14(21), 4568. https://doi.org/10.3390/polym14214568