
Control of Crystallization of PBT-PC Blends by Anisotropic SiO2 and GeO2 Glass Flakes

 ,
,  , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Wet Comminution
2.2. Polymers
2.3. Glasses
2.4. Composite Formation by Co-Comminution in a Stirred Media Mill
2.5. Characterization Methods
2.5.1. Scanning Electron Microscopy
2.5.2. Thermal Analysis
2.5.3. Helium Pycnometry
2.5.4. Volumetric Nitrogen Sorption Analyzer
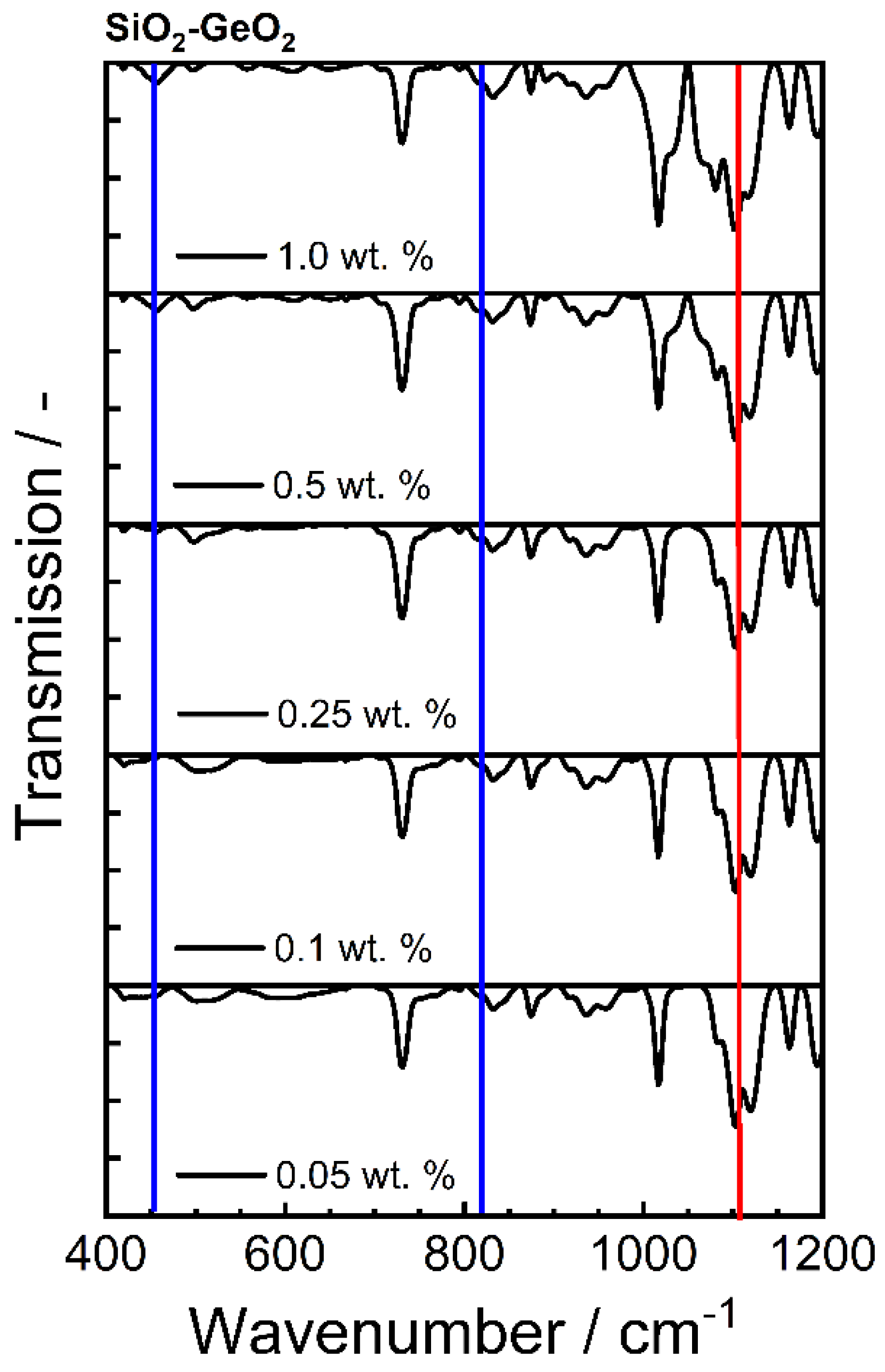
2.5.5. Fourier Transform Infrared Spectroscopy
2.5.6. Particle Size Analysis
3. Results and Discussion
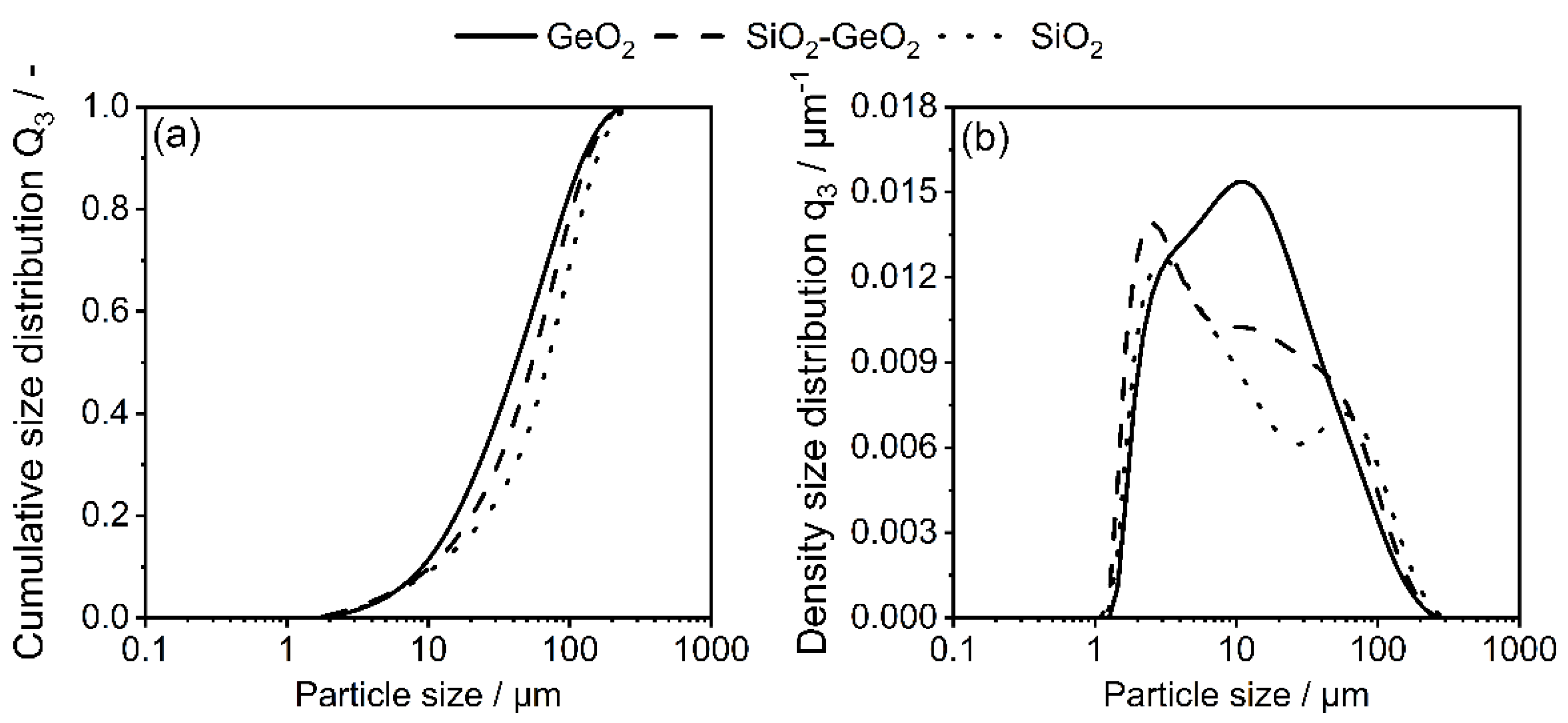
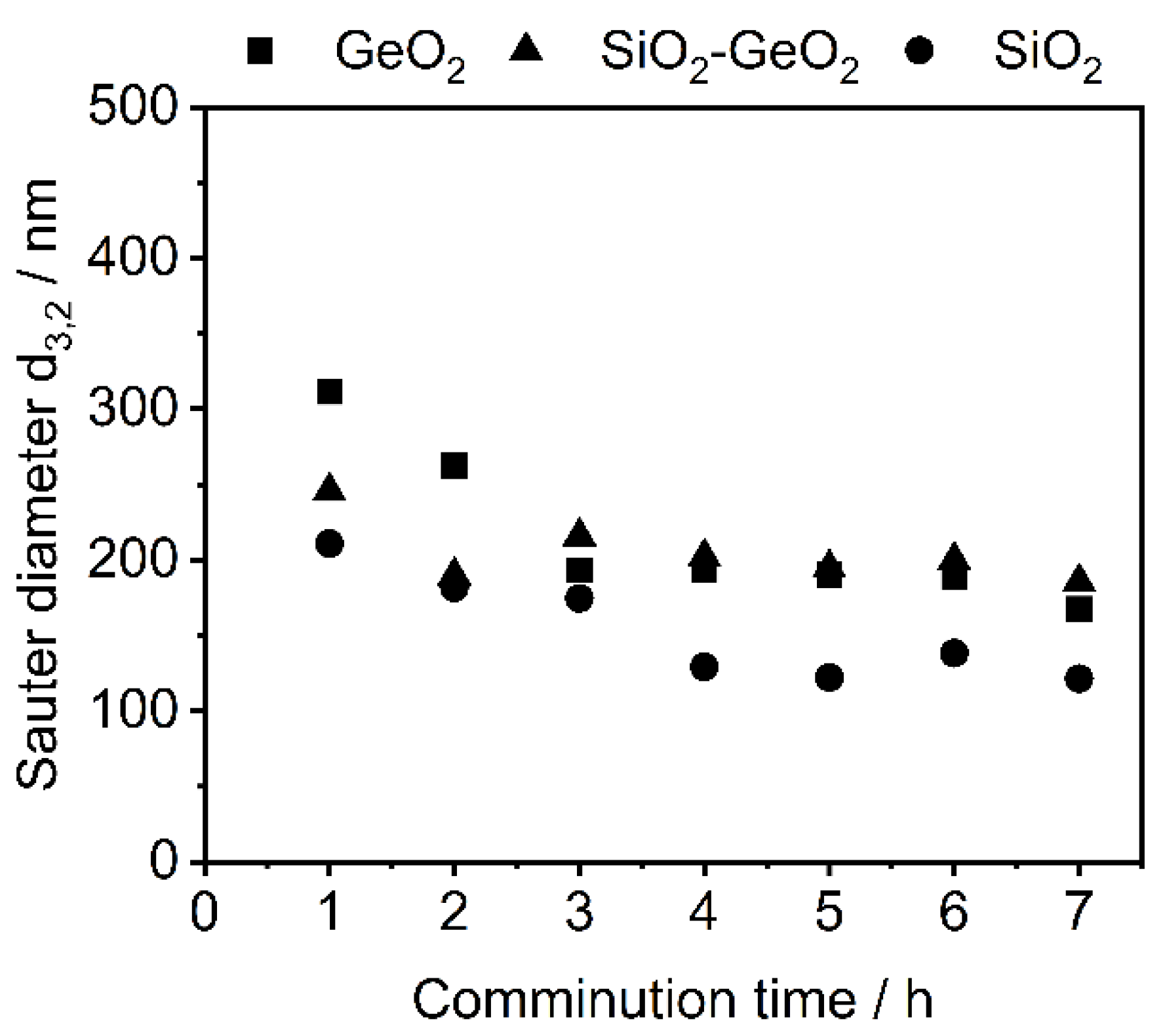
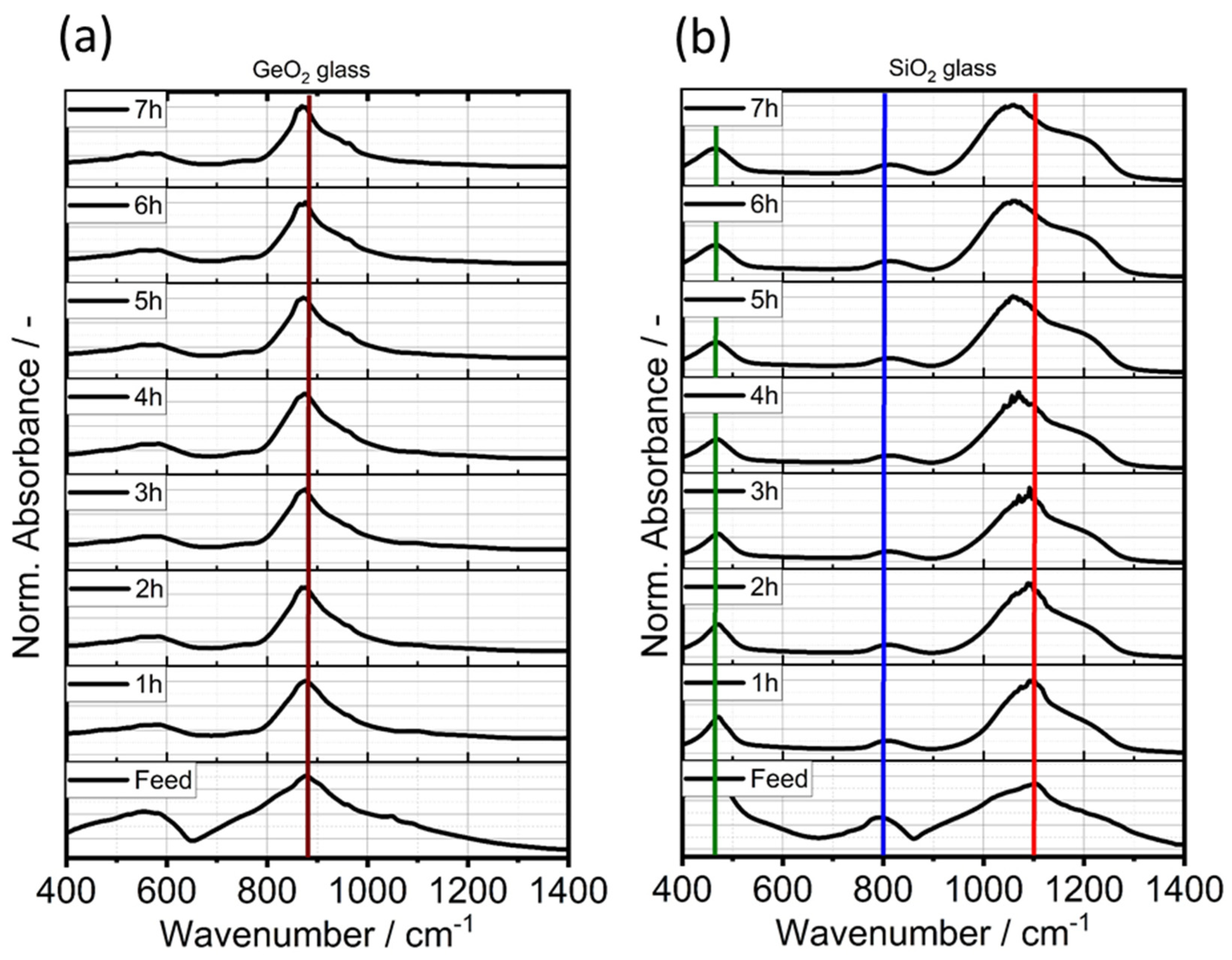
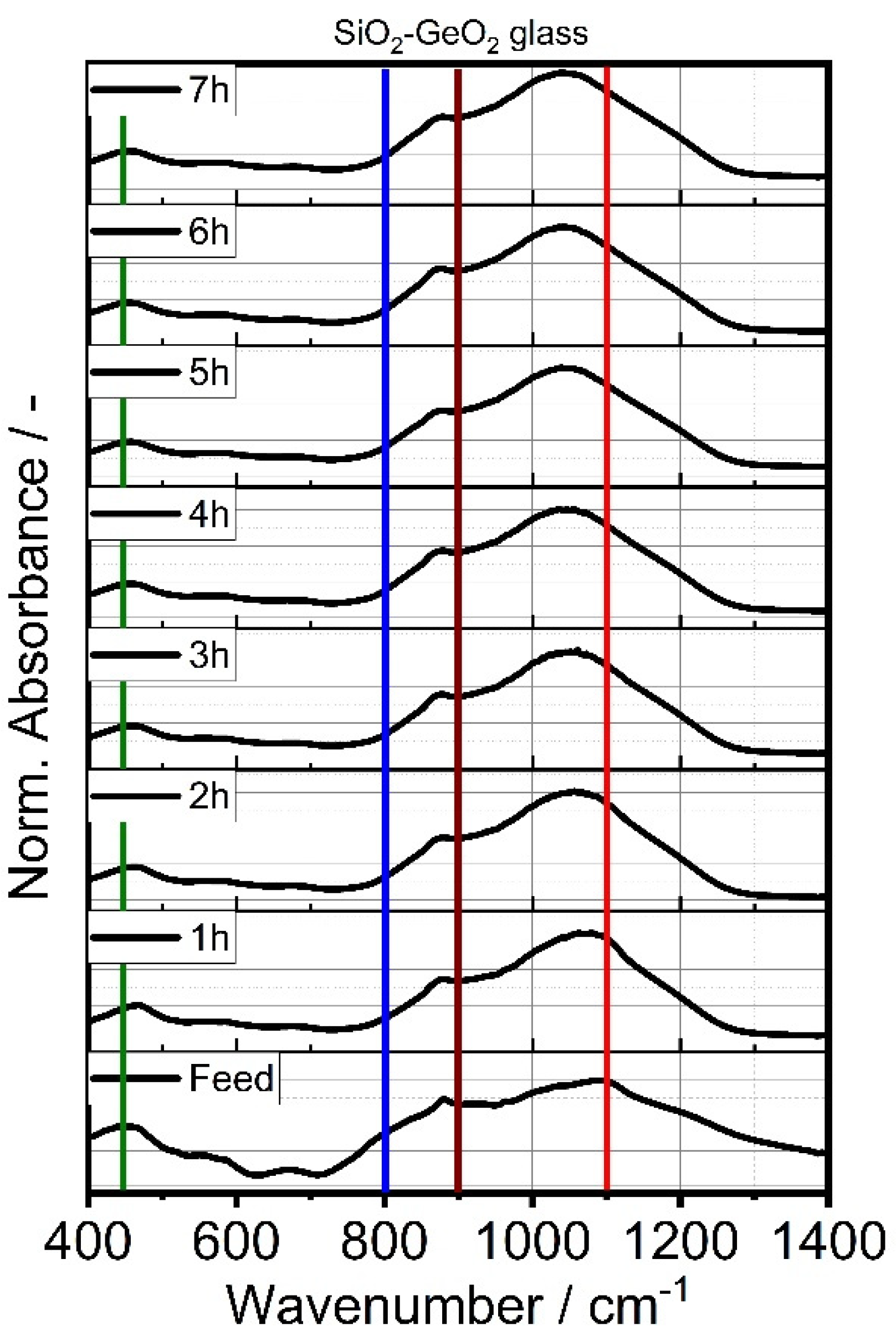
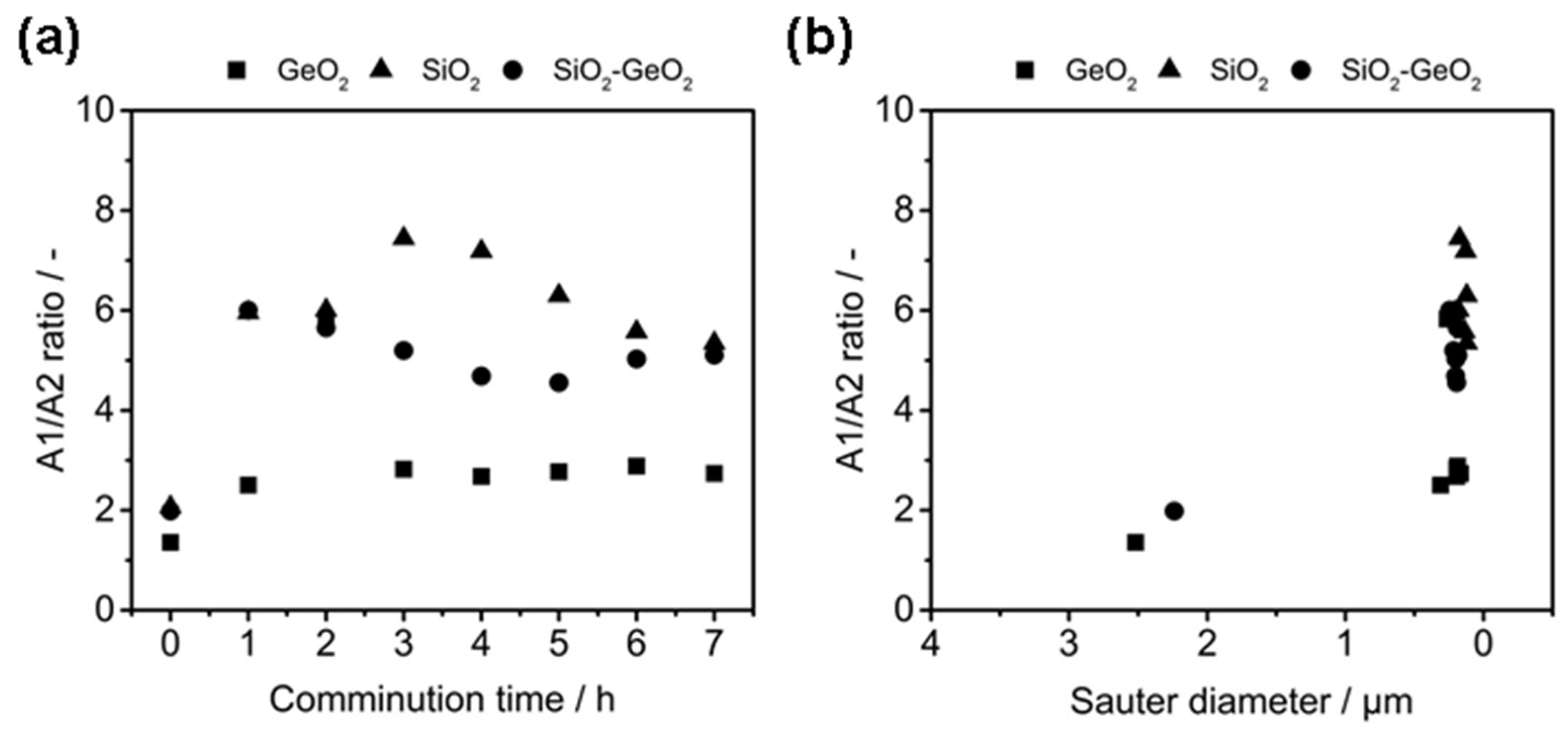
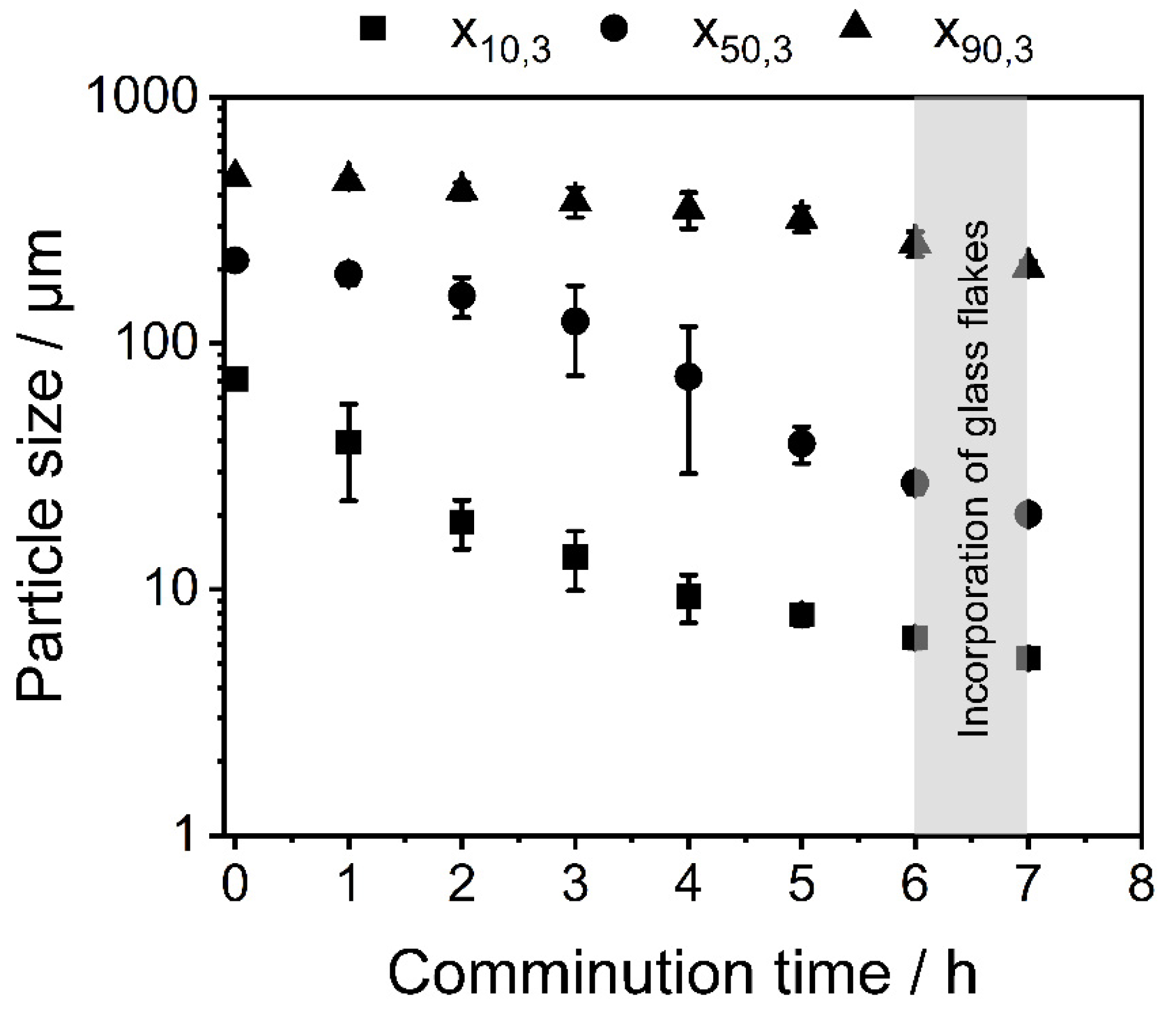
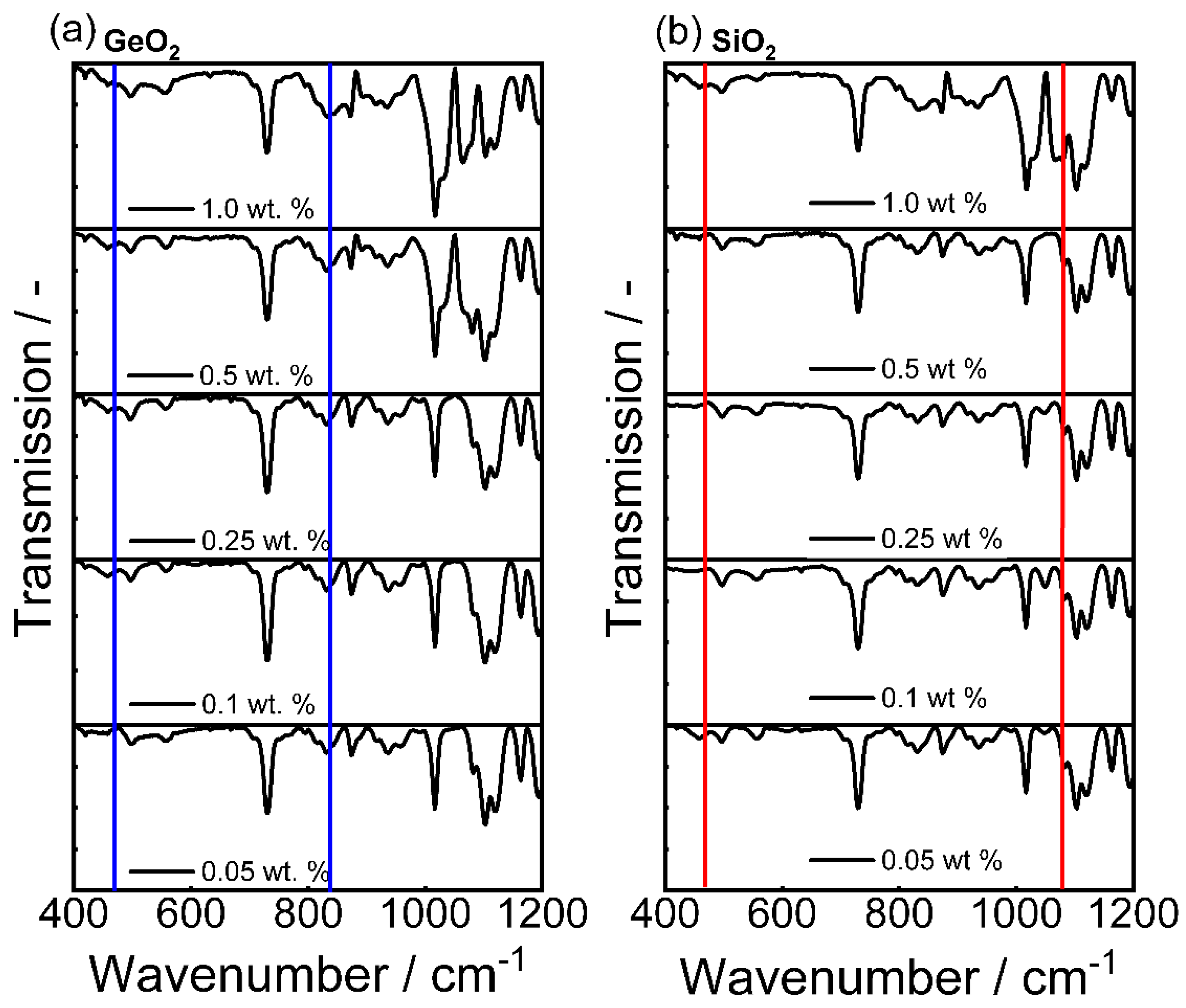
3.1. Shape Formation of Glasses Inside a Stirred Media Mill
3.2. Co-Comminution and Blend Formation
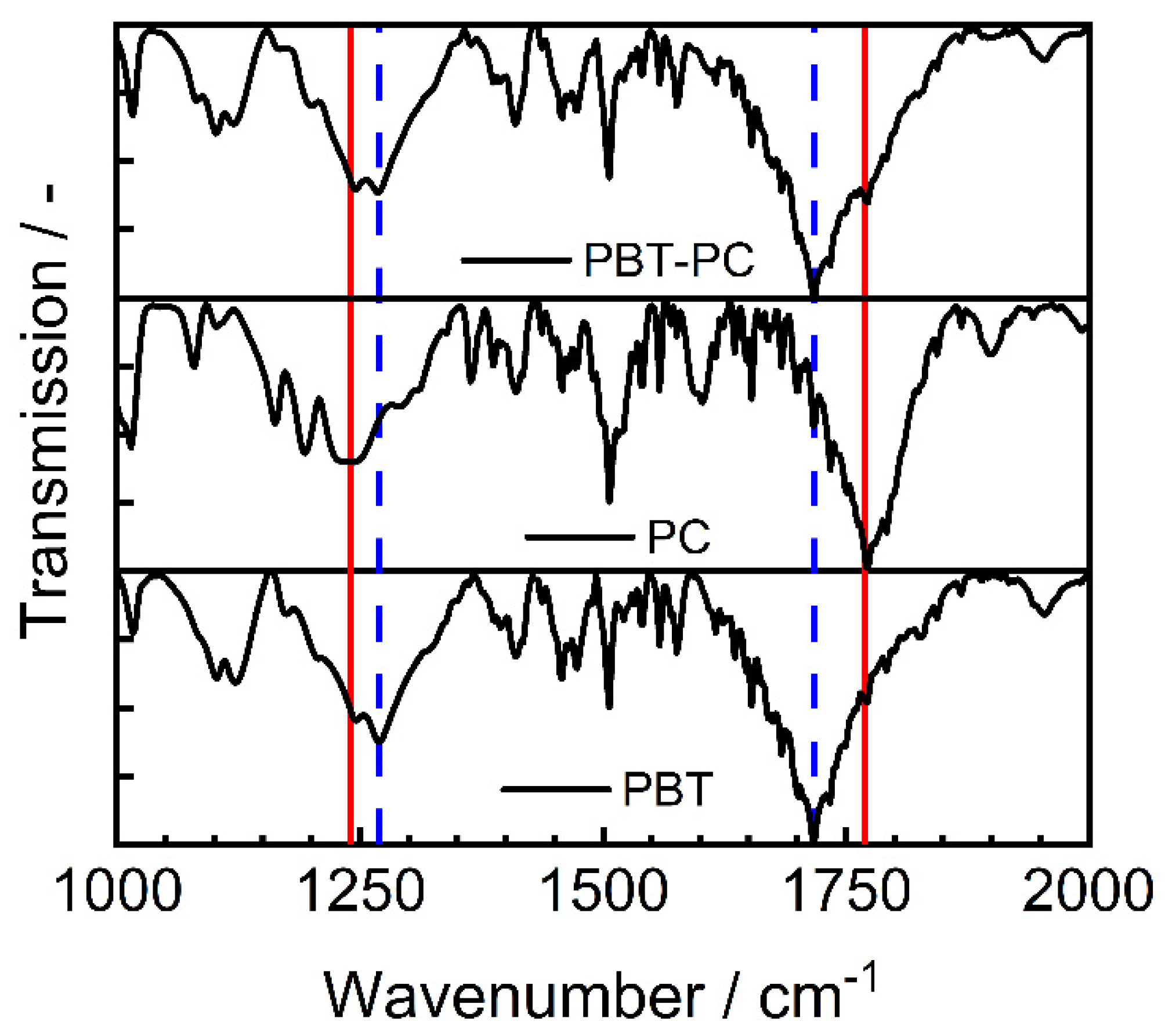
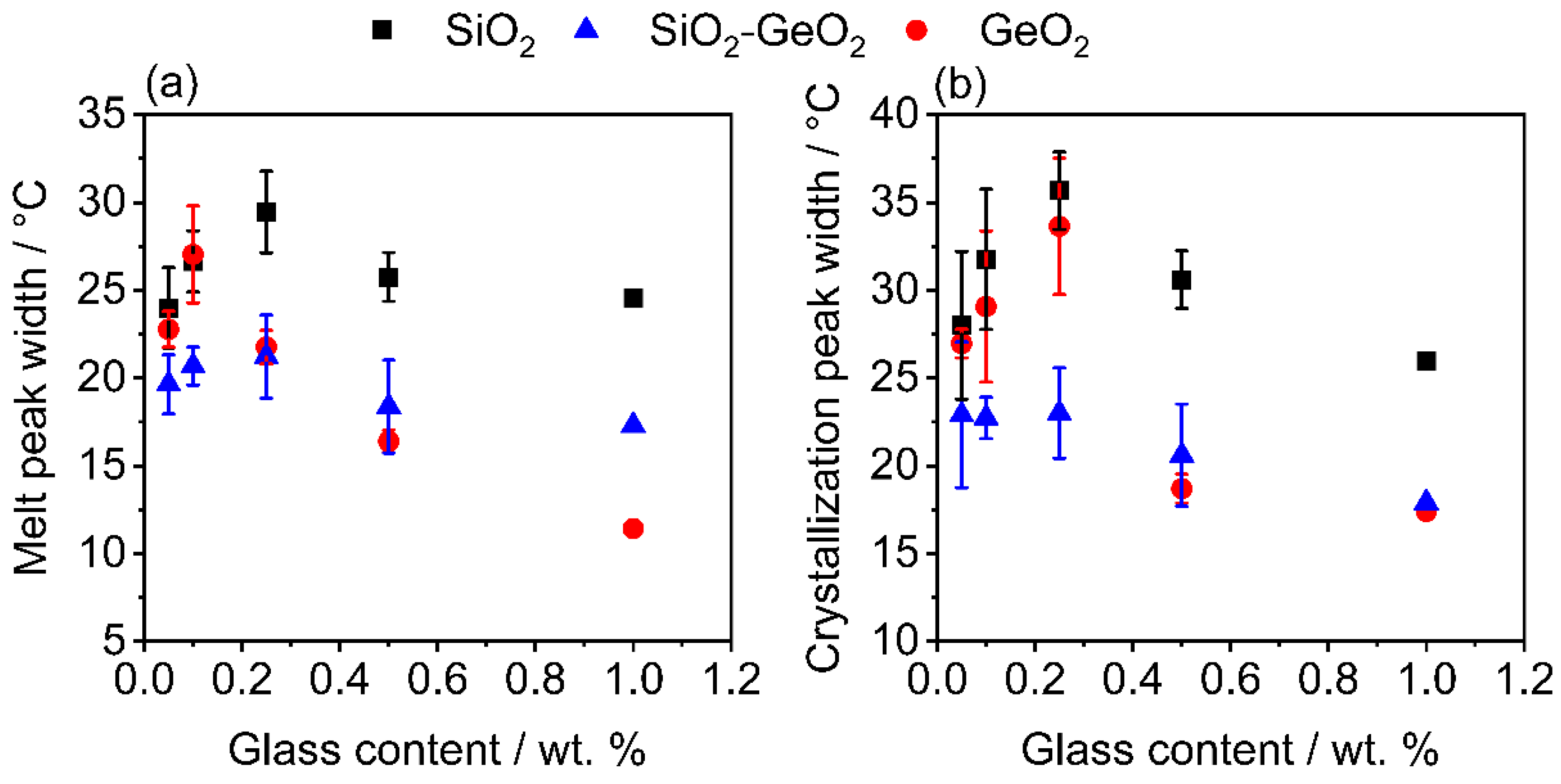
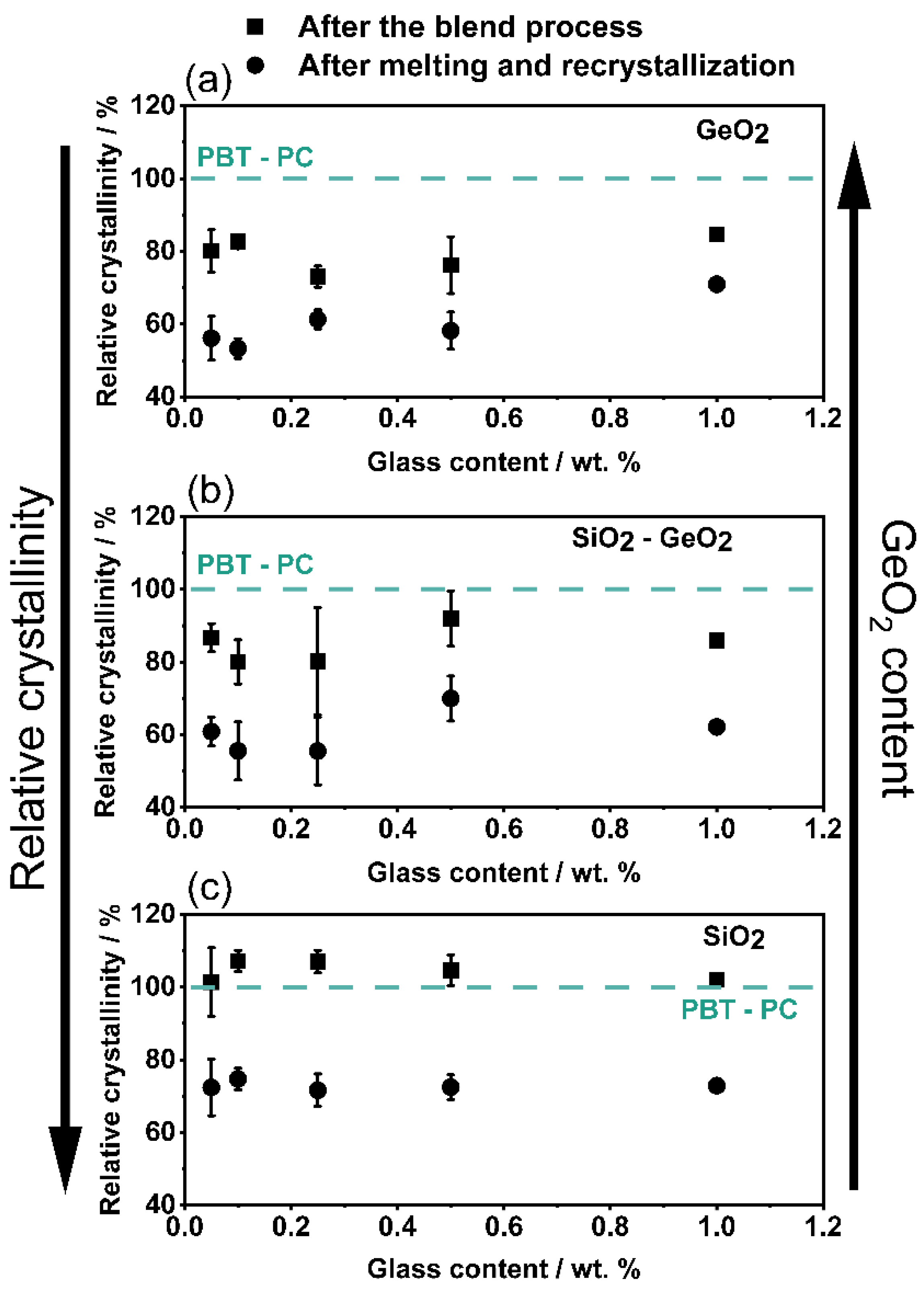
3.3. Influence on the Crystallization and Crystallinity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Park, S.J.; Seo, M.K. Intermolecular force. In Interface Science and Technology; Elsevier: Amsterdam, The Netherlands, 2011; Volume 18, pp. 1–57. [Google Scholar]
- Ma, P.-C.; Siddiqui, N.A.; Marom, G.; Kim, J.-K. Dispersion and functionalization of carbon nanotubes for polymer-based nanocomposites: A review. Compos. Part A Appl. Sci. Manuf. 2010, 41, 1345–1367. [Google Scholar] [CrossRef]
- Kalsoom, U.; Nesterenko, P.N.; Paull, B. Recent developments in 3D printable composite materials. RSC Adv. 2016, 6, 60355–60371. [Google Scholar] [CrossRef]
- Fischer, J.E.; Zhou, W.; Vavro, J.; Llaguno, M.C.; Guthy, C.; Haggenmueller, R.; Casavant, M.J.; de Walters; Smalley, R.E. Magnetically aligned single wall carbon nanotube films: Preferred orientation and anisotropic transport properties. J. Appl. Phys. 2003, 93, 2157–2163. [Google Scholar] [CrossRef]
- Khan, S.U.; Pothnis, J.R.; Kim, J.-K. Effects of carbon nanotube alignment on electrical and mechanical properties of epoxy nanocomposites. Compos. Part A Appl. Sci. Manuf. 2013, 49, 26–34. [Google Scholar] [CrossRef]
- Malhotra, S.K.; Goda, K.; Sreekala, M.S. Part one introduction to polymer composites. Polym. Compos. 2012, 1, 1–16. [Google Scholar]
- Wickramasinghe, S.; Do, T.; Tran, P. FDM-based 3D printing of polymer and associated composite: A review on mechanical properties, defects and treatments. Polymers 2020, 12, 1529. [Google Scholar] [CrossRef]
- Dechet, M.A.; Gómez Bonilla, J.S.; Lanzl, L.; Drummer, D.; Bück, A.; Schmidt, J.; Peukert, W. Spherical Polybutylene Terephthalate (PBT)-Polycarbonate (PC) Blend Particles by Mechanical Alloying and Thermal Rounding. Polymers 2018, 10, 1373. [Google Scholar] [CrossRef]
- Schmidt, J.; Sachs, M.; Blümel, C.; Winzer, B.; Toni, F.; Wirth, K.-E.; Peukert, W. A novel process route for the production of spherical LBM polymer powders with small size and good flowability. Powder Technol. 2014, 261, 78–86. [Google Scholar] [CrossRef]
- Riedel, F.; Düsenberg, B.; Schmidt, J.; Bück, A.; Peukert, W. Production of PBT/PC multi-material particles via a combination of co-grinding and spray-agglomeration for powder bed fusion. Procedia CIRP 2020, 94, 100–104. [Google Scholar] [CrossRef]
- Canziani, H.; Chiera, S.; Schuffenhauer, T.; Kopp, S.-P.; Metzger, F.; Bück, A.; Schmidt, M.; Vogel, N. Bottom-Up Design of Composite Supraparticles for Powder-Based Additive Manufacturing. Small 2020, 16, 2002076. [Google Scholar] [CrossRef]
- Zhai, Z.; Groeschel, C.; Drummer, D. Tensile behavior of quasi-unidirectional glass fiber/polypropylene composites at room and elevated temperatures. Polym. Test. 2016, 54, 126–133. [Google Scholar] [CrossRef]
- Niendorf, K.; Raeymaekers, B. Additive Manufacturing of Polymer Matrix Composite Materials with Aligned or Organized Filler Material: A Review. Adv. Eng. Mater. 2021, 23, 2001002. [Google Scholar] [CrossRef]
- Gómez Bonilla, J.S.; Düsenberg, B.; Lanyi, F.; Schmuki, P.; Schubert, D.W.; Schmidt, J.; Peukert, W.; Bück, A. Improvement of polymer properties for powder bed fusion by combining in situ PECVD nanoparticle synthesis and dry coating. Plasma Process. Polym. 2021, 18, e2000247. [Google Scholar] [CrossRef]
- Dashtizadeh, A.; Abdouss, M.; Mahdavi, H.; Khorassani, M. Acrylic coatings exhibiting improved hardness, solvent resistance and glossiness by using silica nano-composites. Appl. Surf. Sci. 2011, 257, 2118–2125. [Google Scholar] [CrossRef]
- Liang, J.-Z. Tensile and Impact Properties of Hollow Glass Bead-Filled PVC Composites. Macromol. Mater. Eng. 2002, 287, 588–591. [Google Scholar] [CrossRef]
- Beck, H.N. Effect of molecular weight upon the heterogeneous nucleation of crystallization in polypropylene. J. Polym. Sci. Part A-2 Polym. Phys. 1966, 4, 631–638. [Google Scholar] [CrossRef]
- Beck, H.N.; Ledbetter, H.D. DTA study of heterogeneous nucleation of crystallization in polypropylene. J. Appl. Polym. Sci. 1965, 9, 2131–2142. [Google Scholar] [CrossRef]
- Godovsky, Y.K.; Slonimsky, G.L. Kinetics of polymer crystallization from the melt (calorimetric approach). J. Polym. Sci. Polym. Phys. Ed. 1974, 12, 1053–1080. [Google Scholar] [CrossRef]
- Song, R.-Q.; Cölfen, H. Additive controlled crystallization. CrystEngComm 2011, 13, 1249–1276. [Google Scholar] [CrossRef]
- Lanzl, L.; Wudy, K.; Drummer, D. The effect of short glass fibers on the process behavior of polyamide 12 during selective laser beam melting. Polym. Test. 2020, 83, 106313. [Google Scholar] [CrossRef]
- Pompe, G.; Meyer, E.; Komber, H.; Hamann, H. Influence of PBT crystallization on miscibility degree of amorphous phase in PC/PBT melt blends. Thermochim. Acta 1991, 187, 185–200. [Google Scholar] [CrossRef]
- Sanchez, P.; Remiro, P.M.; Nazabal, J. Physical properties and structure of unreacted PC/PBT blends. J. Appl. Polym. Sci. 1993, 50, 995–1005. [Google Scholar] [CrossRef]
- Liu, S.; Qin, S.; Luo, Z.; Yu, J.; Guo, J.; He, M. Reactive Compatibilization of Poly(Butylene Terephthalate)/Acrylonitrile-Styrene-Acrylate Blends by Epoxy Resin: Morphology and Mechanical Properties. J. Macromol. Sci. Part B 2011, 50, 1780–1790. [Google Scholar] [CrossRef]
- Kuram, E.; Ozcelik, B.; Yilmaz, F. The influence of recycling number on the mechanical, chemical, thermal and rheological properties of poly(butylene terephthalate)/polycarbonate binary blend and glass-fibre-reinforced composite. J. Thermoplast. Compos. Mater. 2016, 29, 1443–1457. [Google Scholar] [CrossRef]
- Zhang, F.; Sun, S.; Liu, X.; Zhang, L.; Zhang, H. Toughening of PBT/PC blends with epoxy functionalized core-shell modifiers. e-Polymers 2009, 9. [Google Scholar] [CrossRef]
- Schnell, H. Chemistry and Physics of Polycarbonates; Interscience: New York, NY, USA, 1964. [Google Scholar]
- Freitag, D.; Fengler, G.; Morbitzer, L. Routes to New Aromatic Polycarbonates with Special Material Properties. Angew. Chem. Int. Ed. Engl. 1991, 30, 1598–1610. [Google Scholar] [CrossRef]
- Shah, N.; Sandhu, H.; Choi, D.S.; Chokshi, H.; Malick, A.W. Amorphous solid dispersions. In Theory and Practice; Springer: Berlin, Germany, 2014. [Google Scholar]
- Ivanisevic, I. Physical stability studies of miscible amorphous solid dispersions. J. Pharm. Sci. 2010, 99, 4005–4012. [Google Scholar] [CrossRef]
- DePolo, W.S.; Baird, D.G. Particulate reinforced PC/PBT composites. I. Effect of particle size (nanotalc versus fine talc particles) on dimensional stability and properties. Polym. Compos. 2009, 30, 188–199. [Google Scholar] [CrossRef]
- DePolo, W.S.; Baird, D.G. Particulate reinforced PC/PBT composites. II. Effect of nano-clay particles on dimensional stability and structure-property relationships. Polym. Compos. 2009, 30, 200–213. [Google Scholar] [CrossRef]
- Khamsehnezhad, A.; Hashemi, S. Mechanical properties of single- and double-gated injection moulded short glass fibre reinforced PBT/PC composites. J. Mater. Sci. 2008, 43, 6344–6352. [Google Scholar] [CrossRef]
- Wang, K.; Wu, J.S. Mechanical Properties and Fracture Mechanisms of Fiber Reinforced PBT/PC/Elastomer Blends. KEM 2000, 177–180, 363–368. [Google Scholar] [CrossRef]
- Spoerk, M.; Arbeiter, F.; Raguž, I.; Weingrill, G.; Fischinger, T.; Traxler, G.; Schuschnigg, S.; Cardon, L.; Holzer, C. Polypropylene Filled With Glass Spheres in Extrusion-Based Additive Manufacturing: Effect of Filler Size and Printing Chamber Temperature. Macromol. Mater. Eng. 2018, 303, 1800179. [Google Scholar] [CrossRef]
- Lanzl, L.; Wudy, K.; Greiner, S.; Drummer, D. Selective laser sintering of copper filled polyamide 12: Characterization of powder properties and process behavior. Polym. Compos. 2019, 40, 1801–1809. [Google Scholar] [CrossRef]
- Franklin, P.; Wood, D.J.; Bubb, N.L. Reinforcement of poly (methyl methacrylate) denture base with glass flake. Dent. Mater. 2005, 21, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Esper, J.D.; Liu, L.; Willnauer, J.; Strobel, A.; Schwenger, J.; Romeis, S.; Peukert, W. Scalable production of glass flakes via compression in the liquid phase. Adv. Powder Technol. 2020, 31, 4145–4156. [Google Scholar] [CrossRef]
- Esper, J.D.; Zhuo, Y.; Barr, M.K.; Yokosawa, T.; Spiecker, E.; de Ligny, D.; Bachmann, J.; Peukert, W.; Romeis, S. Shape-anisotropic cobalt-germanium-borate glass flakes as novel Li-ion battery anodes. Powder Technol. 2020, 363, 218–231. [Google Scholar] [CrossRef]
- Devaux, J.; Godard, P.; Mercier, J.P. Bisphenol-A polycarbonate–poly(butylene terephthalate) transesterification. IV. Kinetics and mechanism of the exchange reaction. J. Polym. Sci. Polym. Phys. Ed. 1982, 20, 1901–1907. [Google Scholar] [CrossRef]
- Wang, K.; Wu, J.; Zeng, H. Microstructures and fracture behavior of glass-fiber reinforced PBT/PC/E-GMA elastomer blends—1: Microstructures. Compos. Sci. Technol. 2001, 61, 1529–1538. [Google Scholar] [CrossRef]
- Wilkinson, A.N.; Tattum, S.B.; Ryan, A.J. Melting, reaction and recrystallization in a reactive PC-PBT blend. Polymer 1997, 38, 1923–1928. [Google Scholar] [CrossRef]
- Gulati, S.T.; Helfinstine, J.D. Fatigue behavior of GeO2-SiO2 glasses. MRS Online Proc. Libr. 1998, 531, 133–141. [Google Scholar] [CrossRef]
- Butterman, W.C.; Jorgenson, J.D. Mineral commodity profiles: Germanium. US Geol. Surv. Open-File Rep. 2004, 1218, 19. [Google Scholar]
- Choi, S.H.; Lee, S.J.; Kim, H.J.; Park, S.B.; Choi, J.W. Li2O–B2O3–GeO2 glass as a high performance anode material for rechargeable lithium-ion batteries. J. Mater. Chem. A 2018, 6, 6860–6866. [Google Scholar] [CrossRef]
- Production of PBT/PC Particle Systems by Wet Grinding; Schmidt, J., Romeis, S., Peukert, W., Eds.; AIP Publishing LLC: Melville, NY, USA, 2017; AIP Conf. Proc. 1914, 050003. [Google Scholar]
- Yang, C.; Xu, X.; Wang, J.; An, Z. Use of the co-grinding method to enhance the dissolution behavior of a poorly water-soluble drug: Generation of solvent-free drug–polymer solid dispersions. Chem. Pharm. Bull. 2012, 60, 837–845. [Google Scholar] [CrossRef]
- Penkina, A.; Semjonov, K.; Hakola, M.; Vuorinen, S.; Repo, T.; Yliruusi, J.; Aruväli, J.; Kogermann, K.; Veski, P.; Heinämäki, J. Towards improved solubility of poorly water-soluble drugs: Cryogenic co-grinding of piroxicam with carrier polymers. Drug Dev. Ind. Pharm. 2016, 42, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Kwade, A. Wet comminution in stirred media mills—Research and its practical application. Powder Technol. 1999, 105, 14–20. [Google Scholar] [CrossRef]
- Becker, M.; Kwade, A.; Schwedes, J. Stress intensity in stirred media mills and its effect on specific energy requirement. Int. J. Miner. Process. 2001, 61, 189–208. [Google Scholar] [CrossRef]
- Kwade, A. Determination of the most important grinding mechanism in stirred media mills by calculating stress intensity and stress number. Powder Technol. 1999, 105, 382–388. [Google Scholar] [CrossRef]
- Schmidt, J.; Plata, M.; Tröger, S.; Peukert, W. Production of polymer particles below 5 μm by wet grinding. Powder Technol. 2012, 228, 84–90. [Google Scholar] [CrossRef]
- Romeis, S.; Schmidt, J.; Peukert, W. Mechanochemical aspects in wet stirred media milling. Int. J. Miner. Process. 2016, 156, 24–31. [Google Scholar] [CrossRef]
- Zhang, H.; Wu, J.; Wan, Y.; Romeis, S.; Esper, J.D.; Peukert, W.; Zheng, K.; Boccaccini, A.R. Bioactive Glass Flakes as Innovative Fillers in Chitosan Membranes for Guided Bone Regeneration. Adv. Eng. Mater. 2021, 24, 2101042. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Zhang, L.Y.; Li, H.; Hu, L.L. Statistical structure analysis of GeO2 modified Yb3+: Phosphate glasses based on Raman and FTIR study. J. Alloys Compd. 2017, 698, 103–113. [Google Scholar] [CrossRef]
- Majérus, O.; Cormier, L.; Neuville, D.R.; Galoisy, L.; Calas, G. The structure of SiO2–GeO2 glasses: A spectroscopic study. J. Non-Cryst. Solids 2008, 354, 2004–2009. [Google Scholar] [CrossRef]
- Mansour, E.; El-Damrawi, G.; Fetoh, R.E.; Doweidar, H. Structure-properties changes in ZnO-PbO-GeO2 glasses. Eur. Phys. J. B 2011, 83, 133–141. [Google Scholar] [CrossRef]
- Jiao, K.; Zhang, Z.; Xu, X.; Lv, Z.; Song, J.; Lin, C.; Sun, J.; He, M.; Gies, H. Synthesis and characterization of germanosilicate molecular sieves: GeO2/SiO2 ratio, H2O/TO2 ratio and temperature. Dalton Trans. 2017, 46, 2270–2280. [Google Scholar] [CrossRef] [PubMed]
- Mark, H.F.; Kroschwitz, J.I. Encyclopedia of Polymer Science and Engineering; Wiley-Interscience: New York, NY, USA, 1985; ISBN 0-471-80948-9. [Google Scholar]
- Lin, G.-P.; Lin, L.; Wang, X.-L.; Chen, L.; Wang, Y.-Z. PBT/PC Blends Compatibilized and Toughened via Copolymers in Situ Formed by MgO-Catalyzed Transesterification. Ind. Eng. Chem. Res. 2015, 54, 1282–1291. [Google Scholar] [CrossRef]
- Günzler, H.; Gremlich, H.-U. IR-Spektroskopie: Eine Einführung; John Wiley & Sons: New York, NY, USA, 2012; ISBN 9783527662876. [Google Scholar]
- Delimoy, D.; Bailly, C.; Devaux, J.; Legras, R. Morphological studies of polycarbonate-poly(butylene terephthalate) blends by transmission electron microscopy. Polym. Eng. Sci. 1988, 28, 104–112. [Google Scholar] [CrossRef]
- Seal, M.; Mukherjee, S. Approach for selection of a synthesis procedure of GeO2 ultra-small nanoparticles and its characterization. Int. J. Phys 2015, 3, 133–138. [Google Scholar]
- Uchino, T.; Aboshi, A.; Kohara, S.; Ohishi, Y.; Sakashita, M.; Aoki, K. Microscopic structure of nanometer-sized silica particles. Phys. Rev. B 2004, 69, 155409. [Google Scholar] [CrossRef]
- Ngo, D.T.; Kalubarme, R.S.; Le, H.T.T.; Park, C.-N.; Park, C.-J. Conducting additive-free amorphous GeO 2/C composite as a high capacity and long-term stability anode for lithium ion batteries. Nanoscale 2015, 7, 2552–2560. [Google Scholar] [CrossRef]
- Iwao, M.; Okuno, M.; Koyano, M.; Katayama, S. Structural changes of SiO2 glass by mechanical milling. J. Mineral. Petrol. Sci. 2010, 13, 1003120131. [Google Scholar] [CrossRef]
- Romeis, S.; Paul, J.; Hanisch, M.; Marthala, V.R.; Hartmann, M.; Taylor, R.N.K.; Schmidt, J.; Peukert, W. Correlation of enhanced strength and internal structure for heat-treated submicron Stöber silica particles. Part. Part. Syst. Charact. 2014, 31, 664–674. [Google Scholar] [CrossRef]
- Kamitsos, E.I.; Patsis, A.P.; Kordas, G. Infrared-reflectance spectra of heat-treated sol-gel-derived silica. Phys. Rev. B 1993, 48, 12499. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.J.; Hibbins-Butler, D.C. Infrared activity of normal modes in vitreous silica, germania and beryllium fluoride. J. Phys. C Solid State Phys. 1976, 9, 1171. [Google Scholar] [CrossRef]
- Flizikowski, J.; Kruszelnicka, W.; Macko, M. The Development of Efficient Contaminated Polymer Materials Shredding in Recycling Processes. Polymers 2021, 13, 713. [Google Scholar] [CrossRef]
- Danch, A. Effect of Supermolecular Structure Changes on the Glass Transition of Polymer. J. Therm. Anal. Calorim. 2001, 65, 525–535. [Google Scholar] [CrossRef]
- Zhu, D.-M.; Kosugi, T. Thermal conductivity of GeO2□ SiO2 and TiO2□ SiO2 mixed glasses. J. Non-Cryst. Solids 1996, 202, 88–92. [Google Scholar] [CrossRef]
- Richet, P.; de Ligny, D.; Westrum, E.F., Jr. Low-temperature heat capacity of GeO2 and B2O3 glasses: Thermophysical and structural implications. J. Non-Cryst. Solids 2003, 315, 20–30. [Google Scholar] [CrossRef]
- Richet, P. GeO2 vs SiO2: Glass transitions and thermodynamic properties of polymorphs. Phys. Chem. Miner. 1990, 17, 79–88. [Google Scholar] [CrossRef]
- Andersson, S.; Dzhavadov, L. Thermal conductivity and heat capacity of amorphous SiO2: Pressure and volume dependence. J. Phys. Condens. Matter 1992, 4, 6209. [Google Scholar] [CrossRef]
- Siódmiak, J.; Gadomski, A. Spherulites: How Do They Emerge at an Onset of Nonequilibrium Kinetic-Thermodynamic and Structural Singularity Addressing Conditions? Entropy 2022, 24, 663. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.M.; Sikka, S.K. Pressure induced amorphization of materials. Prog. Mater. Sci. 1996, 40, 1–77. [Google Scholar] [CrossRef]
- Liimatainen, H.; Sirviö, J.; Haapala, A.; Hormi, O.; Niinimäki, J. Characterization of highly accessible cellulose microfibers generated by wet stirred media milling. Carbohydr. Polym. 2011, 83, 2005–2010. [Google Scholar] [CrossRef]
- Fillon, B.; Thierry, A.; Lotz, B.; Wittmann, J.C. Efficiency scale for polymer nucleating agents. J. Therm. Anal. 1994, 42, 721–731. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | PBT | PC |
|---|---|---|
| Particle size x10,3/µm | 70 | 77 |
| Particle size x50,3/µm | 213 | 230 |
| Particle size x90,3/µm | 465 | 495 |
| Density ρbulk/g cm−3 | 1.33 | 1.22 |
| Material | Particle Size µm | Specific Surface Area m2 g−1 | Density g cm−3 | ||
|---|---|---|---|---|---|
| x10,3 | x50,3 | x90,3 | |||
| SiO2 | 8 | 47 | 129 | 0.51 | 2.2717 +/− 0.0009 |
| SiO2-GeO2 | 8 | 57 | 143 | 0.68 | 2.7771 +/− 0.0002 |
| GeO2 | 6 | 36 | 99 | 0.49 | 3.7935 +/− 0.0003 |
| Parameter | Experimental Condition |
|---|---|
| Rotor revolution/min−1 | 2000 |
| Rotational speed/m s−1 | 6.50 |
| Grinding bead media | Yttria stabilized zirconia (5 wt. % Y2O3) |
| Grinding bead density/g cm−3 | 6.5 |
| Grinding bead diameter dGM/mm | 1.0 |
| Solids concentration cw/wt. % | 3.0 |
| Grinding time/h | 5 |
| Temperature grinding chamber/°C | 20 °C |
| Parameter | Experimental Condition |
|---|---|
| Rotor revolution/min−1 | 2000 |
| Rotational speed/m s−1 | 6.50 |
| Grinding bead media | Yttria stabilized zirconia (5 wt. % Y2O3) |
| Grinding bead density/g cm−3 | 6.5 |
| Grinding bead diameter dGM/mm | 2.0 |
| Solids concentration cw/wt. % | 20.0 |
| Grinding time/h | 7 |
| Temperature grinding chamber/°C | 15 °C |
| No | PBT-PC | SiO2 | SiO2-GeO2 | GeO2 |
|---|---|---|---|---|
| wt. % | wt. % | wt. % | ||
| 1 | 15 g PBT 5 g PC | 0.05 | ||
| 2 | 0.1 | |||
| 3 | 0.25 | |||
| 4 | 0.5 | |||
| 5 | 1.0 | |||
| 6 | 15 g PBT 5 g PC | 0.05 | ||
| 7 | 0.1 | |||
| 8 | 0.25 | |||
| 9 | 0.5 | |||
| 10 | 1.0 | |||
| 11 | 15 g PBT 5 g PC | 0.05 | ||
| 12 | 0.1 | |||
| 13 | 0.25 | |||
| 14 | 0.5 | |||
| 15 | 1.0 |
| Bond | Type | Wavenumber/cm−1 | Literature |
|---|---|---|---|
| Ge-O-Ge | Bending vibration | 300–470 | [59] |
| Si-O-Si | Asymmetric vibration | 450 | [59] |
| Ge-O-Ge | Symmetric vibration | 515–565 | [56,58] |
| Si-O-Ge | Stretching vibration | 660 | [57,59] |
| Si-O-Si | Symmetric vibration | 800 | [65] |
| Ge-O-Ge | Asymmetric vibration | 820–890 | [58,66] |
| Si-O-Ge | Stretching vibration | 990 | [57,59] |
| Si-O-Si | Stretching vibration | 1050–1200 | [59,67] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Düsenberg, B.; Esper, J.D.; Maußner, F.; Mayerhofer, M.; Schmidt, J.; Peukert, W.; Bück, A. Control of Crystallization of PBT-PC Blends by Anisotropic SiO2 and GeO2 Glass Flakes. Polymers 2022, 14, 4555. https://doi.org/10.3390/polym14214555
Düsenberg B, Esper JD, Maußner F, Mayerhofer M, Schmidt J, Peukert W, Bück A. Control of Crystallization of PBT-PC Blends by Anisotropic SiO2 and GeO2 Glass Flakes. Polymers. 2022; 14(21):4555. https://doi.org/10.3390/polym14214555
Chicago/Turabian StyleDüsenberg, Björn, Julian D. Esper, Felix Maußner, Magdalena Mayerhofer, Jochen Schmidt, Wolfgang Peukert, and Andreas Bück. 2022. "Control of Crystallization of PBT-PC Blends by Anisotropic SiO2 and GeO2 Glass Flakes" Polymers 14, no. 21: 4555. https://doi.org/10.3390/polym14214555
APA StyleDüsenberg, B., Esper, J. D., Maußner, F., Mayerhofer, M., Schmidt, J., Peukert, W., & Bück, A. (2022). Control of Crystallization of PBT-PC Blends by Anisotropic SiO2 and GeO2 Glass Flakes. Polymers, 14(21), 4555. https://doi.org/10.3390/polym14214555