

Analysis of Permeation and Diffusion Coefficients to Infer Aging Attributes in Polymers Subjected to Supercritical CO2 and H2 Gas at High Pressures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Background Theory
3.1. Classical Permeation—Solution-Diffusion Model
3.2. Diffusion Coefficient by Time-Lag Method
3.3. Diffusion Coefficient by Slope-Concentration Method
3.4. Fractional Free Volume
3.5. Fugacity Coefficient
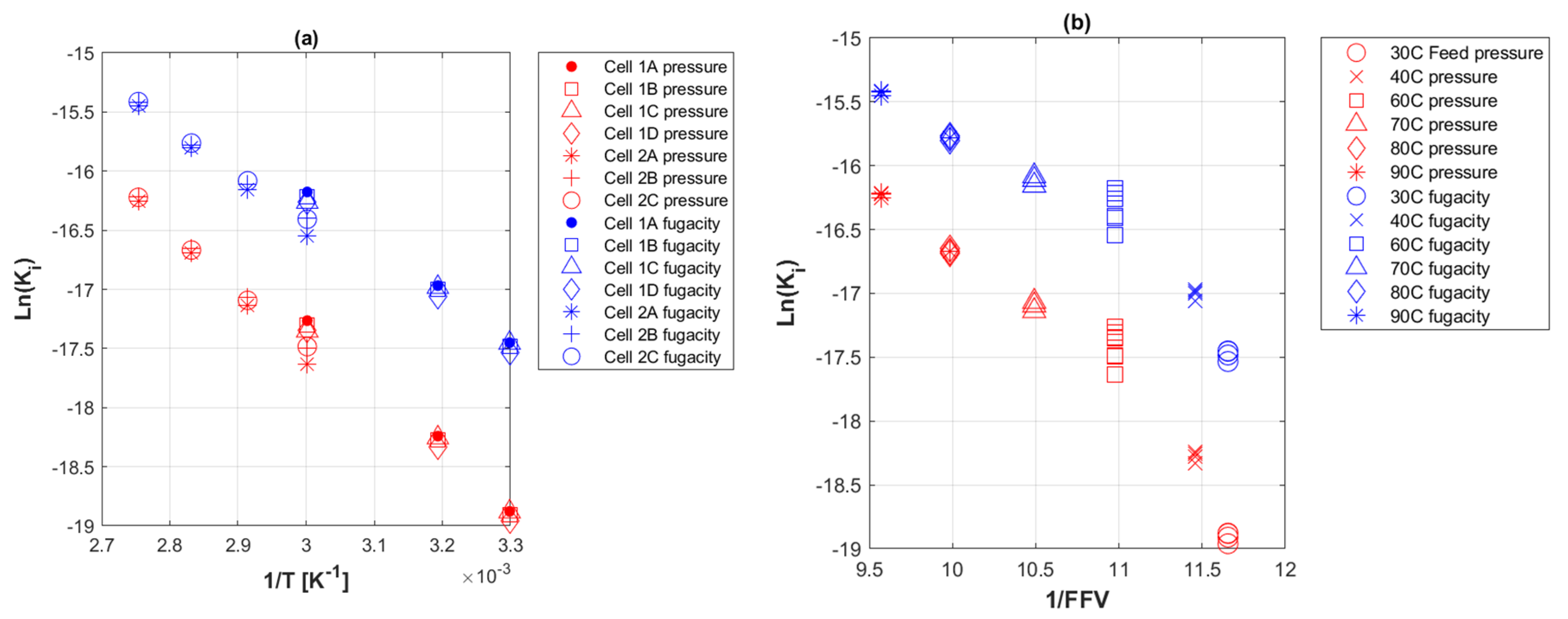
3.6. Activation Energies of Transport Coefficients
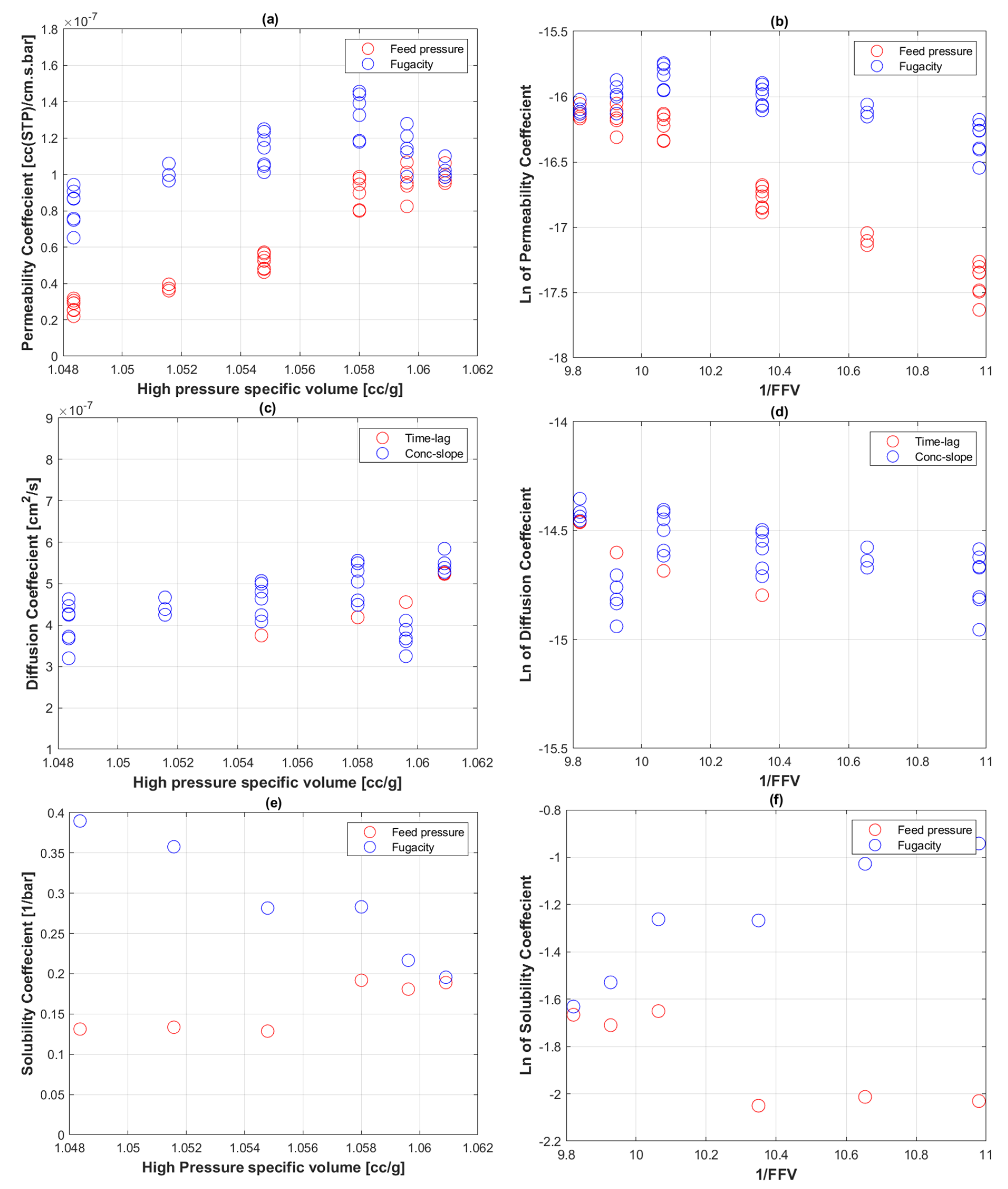
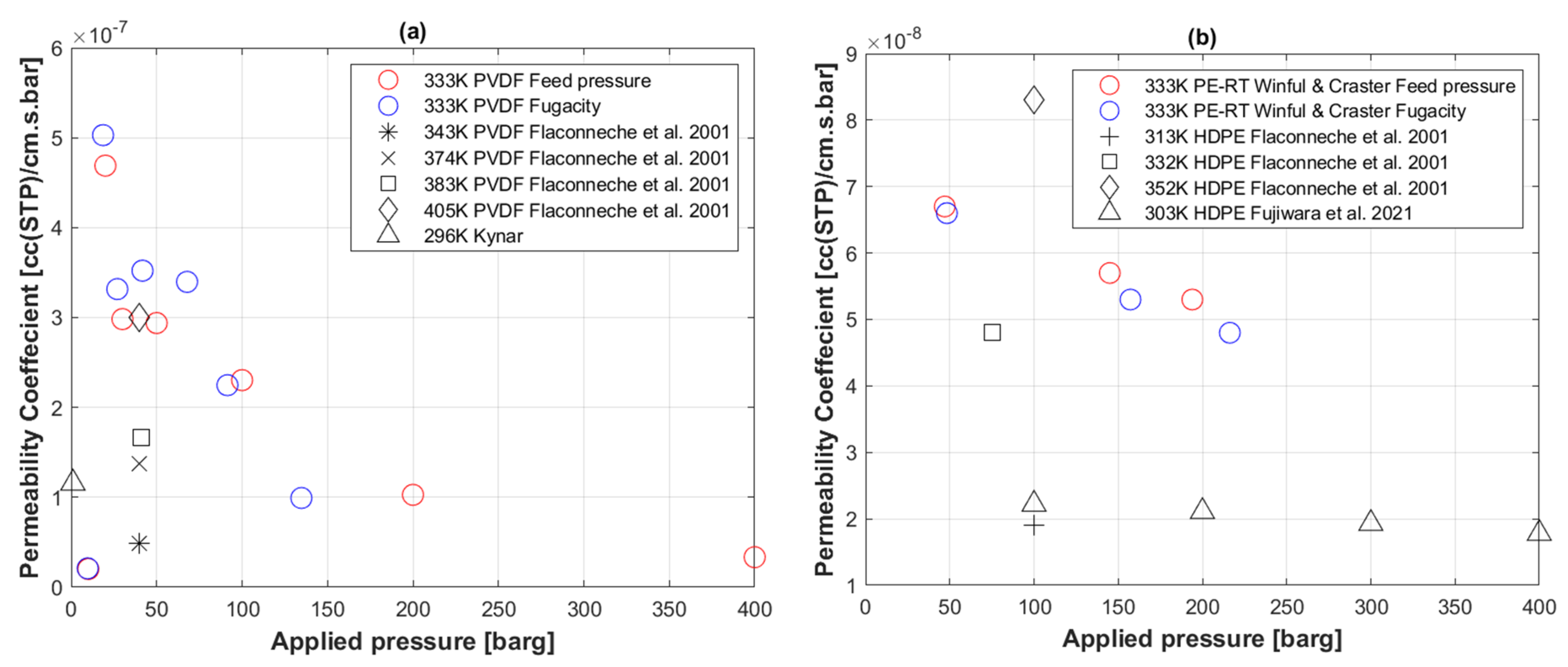
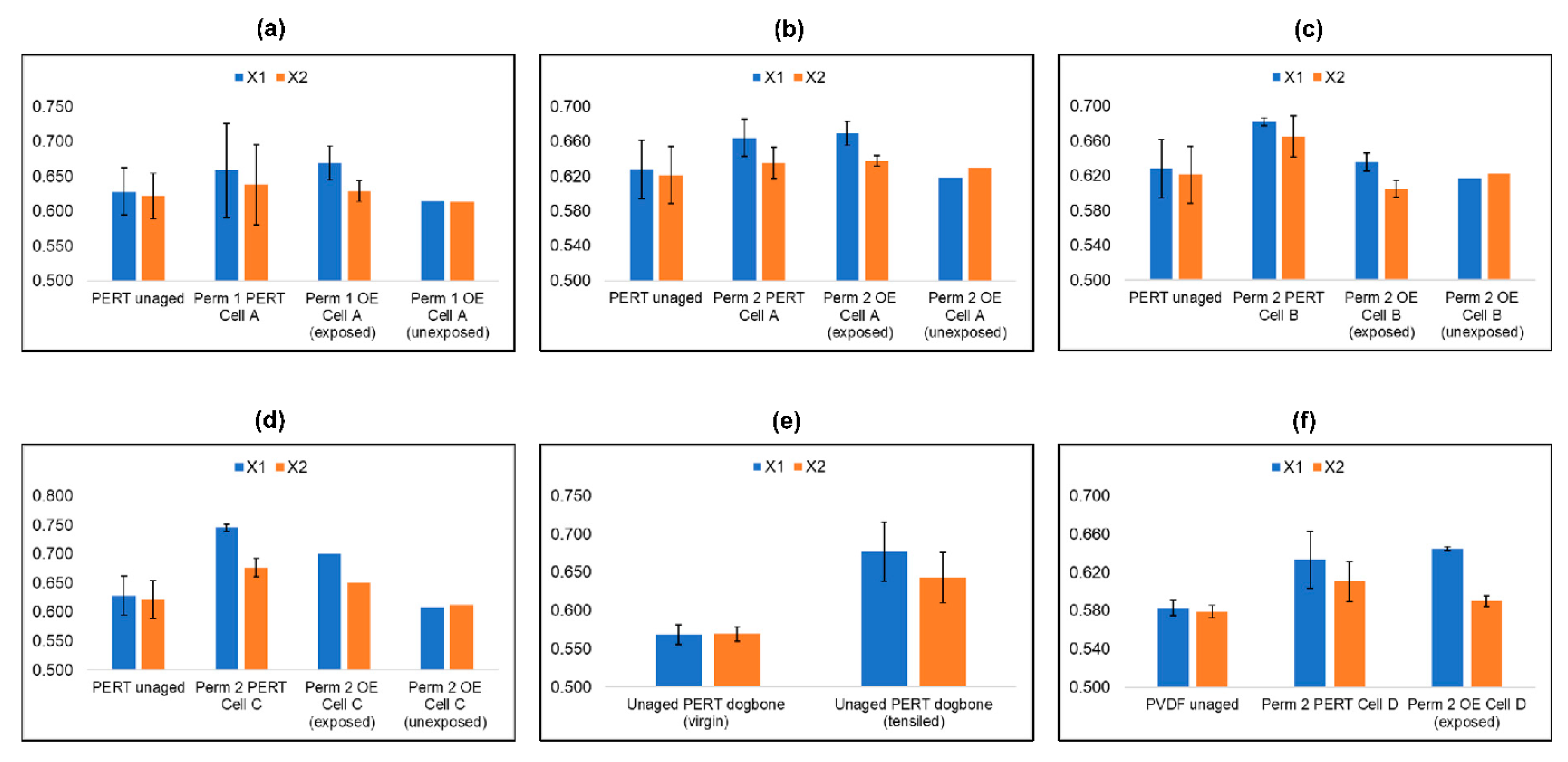
4. Results
5. Discussion
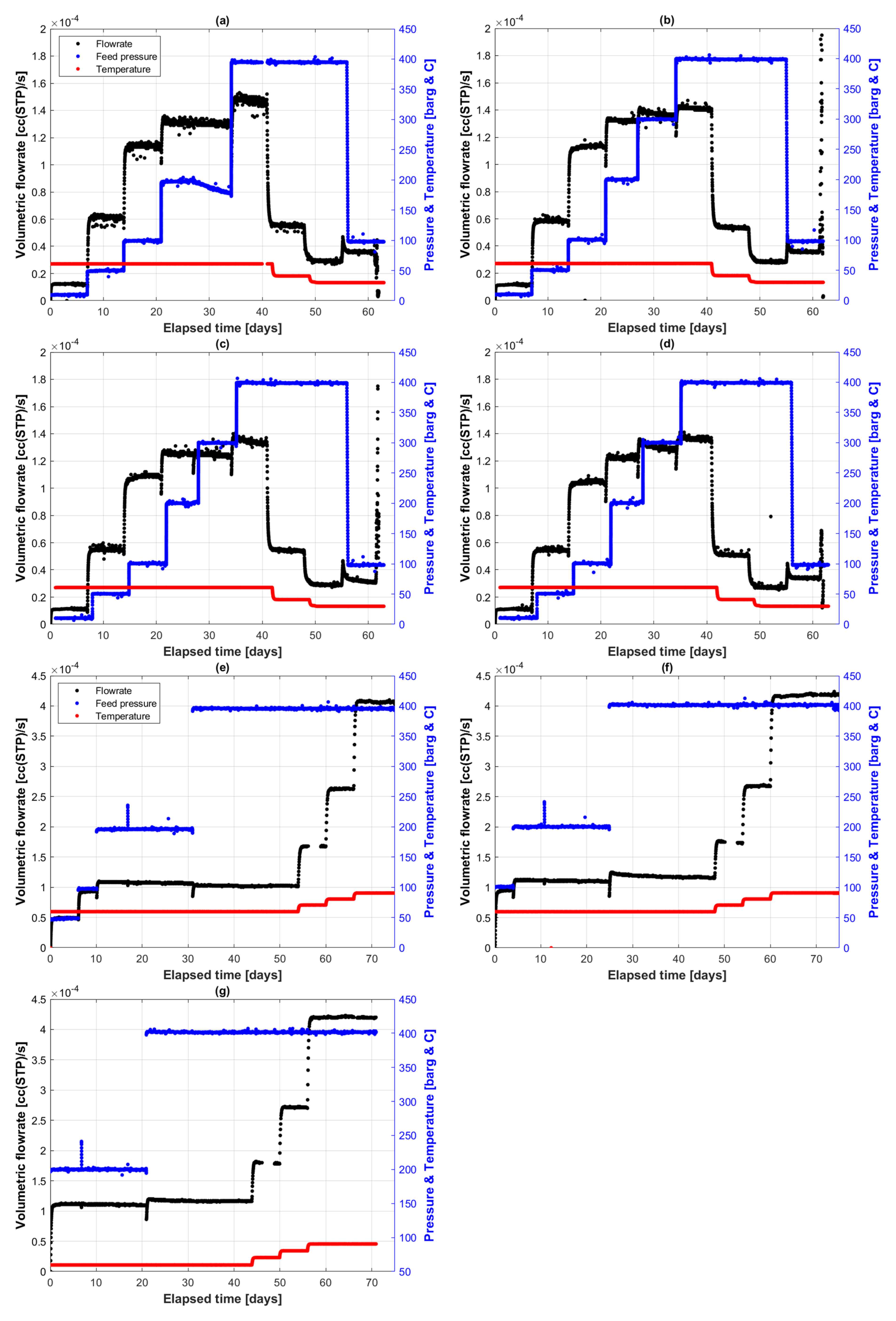
5.1. Continuous Flow Permeation Volumetric Fluxes
5.1.1. First Permeation Round
5.1.2. Second Permeation Round
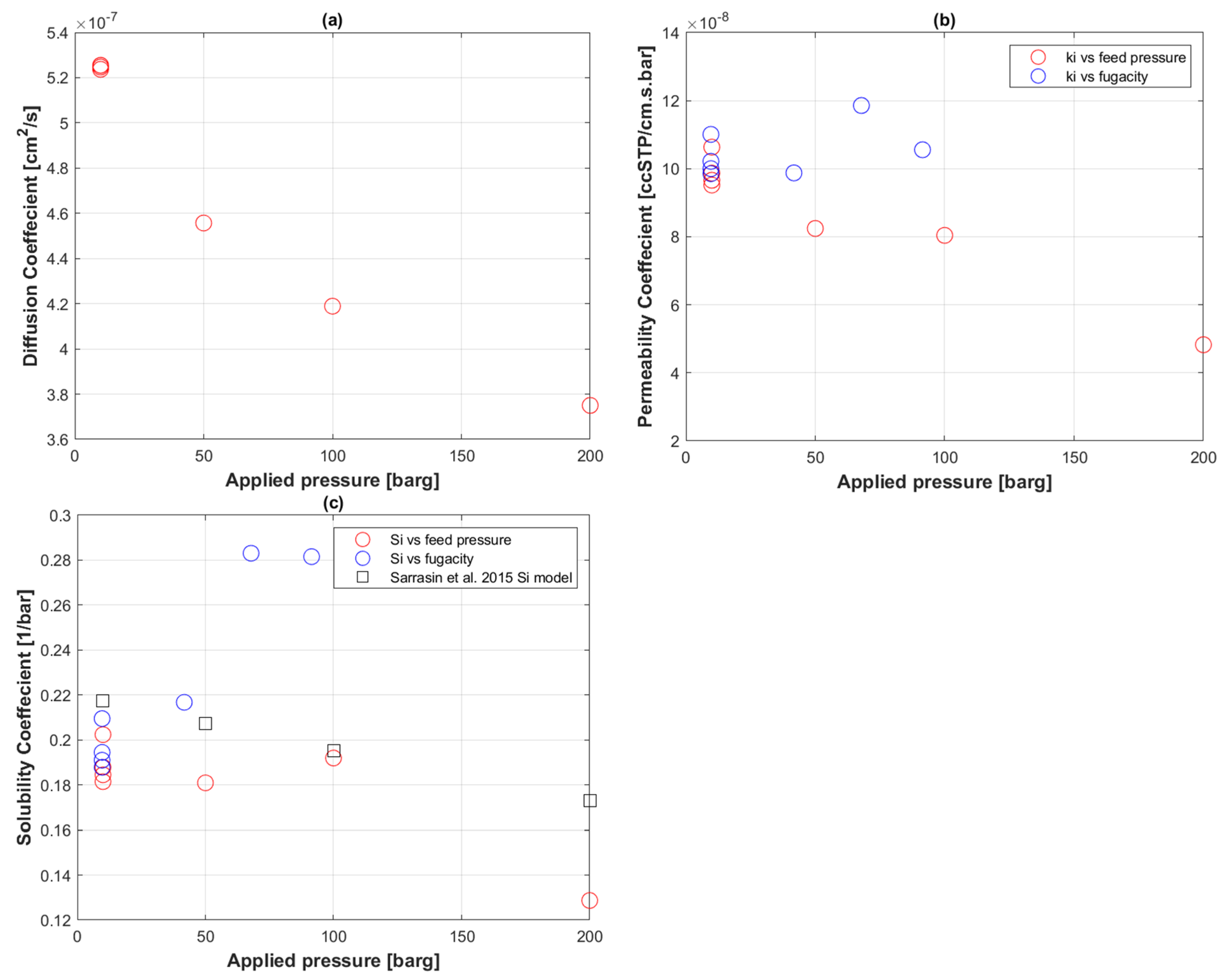
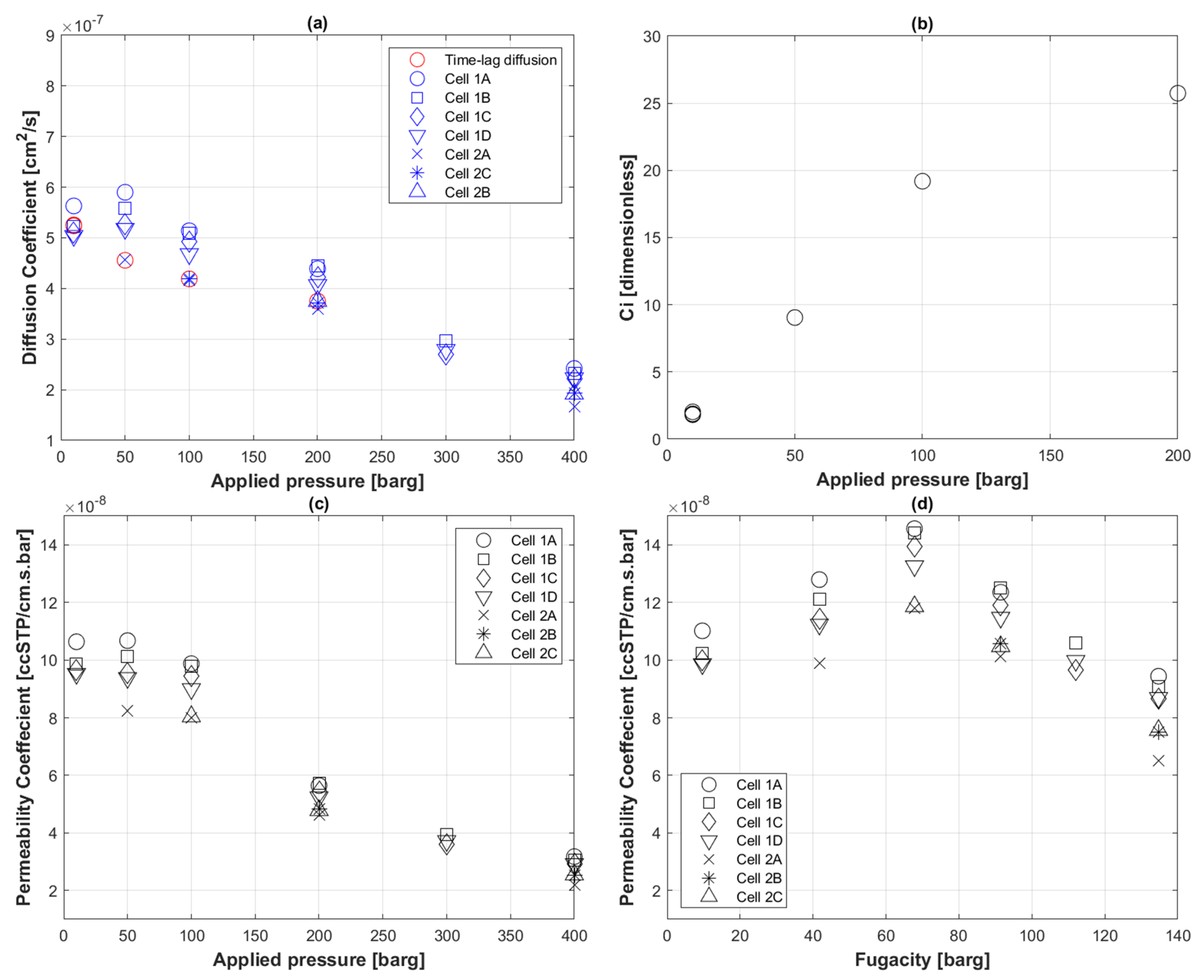
5.2. Transport Coefficients of CO2 to PE-RT
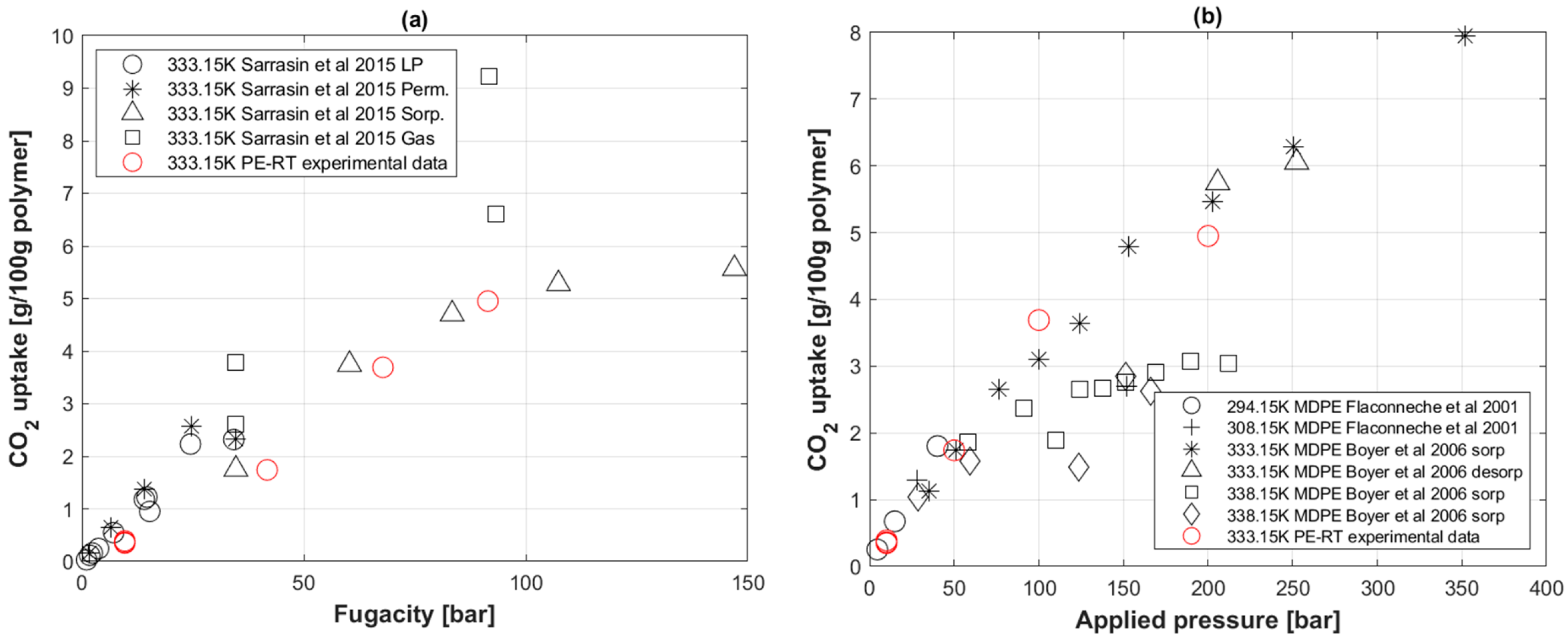
5.3. CO2 Uptake in PE-RT
5.4. Fractional free Volume of PE-RT
5.5. Transport Coefficients of CO2 to PVDF
5.6. Transport Coefficients of H2 to PE-RT
5.7. Differential Scanning Calometry of Aged Specimen
5.8. Permeation Coefficient with Temperature
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wissinger, R.G.; Paulaitis, M.E. Swelling and sorption in polymer—CO2 mixtures at elevated pressures. J. Polym. Sci. Part B Polym. Phys. 1987, 25, 2497–2510. [Google Scholar] [CrossRef]
- Wissinger, R.G.; Paulaitis, M.E. Glass transitions in polymer/CO2 mixtures at elevated pressures. J. Polym. Sci. Part B Polym. Phys. 1991, 29, 631–633. [Google Scholar] [CrossRef]
- Sanders, E.S.; Jordan, S.M.; Subramanian, R. Penetrant-plasticized permeation in polymethylmethacrylate. J. Memb. Sci. 1992, 74, 29–36. [Google Scholar] [CrossRef]
- Wang, J.-S.; Naito, Y.; Kamiya, Y. Effect of penetrant-induced isothermal glass transition on sorption, dilation, and diffusion behavior of polybenzylmethacrylate/CO2. J. Polym. Sci. Part B Polym. Phys. 1996, 34, 2027–2033. [Google Scholar] [CrossRef]
- Chang, C.; Venkatesan, M.; Cho, C.; Chung, P. Thermoplastic Starch with Poly(butylene adipate-co-terephthalate) Blends Foamed by Supercritical Carbon Dioxide. Polymers 2022, 14, 1952. [Google Scholar] [CrossRef] [PubMed]
- Shirzad, M.; Karimi, M. Statistical analysis and optimal design of polymer inclusion membrane for water treatment by Co(II) removal. Desalin. Water Treat. 2020, 182, 194–207. [Google Scholar] [CrossRef]
- Crank, J. The Mathematics of Diffusion, 2nd ed.; Clarendon Press: Oxford, UK, 1975. [Google Scholar]
- Eric, C.; Rideal, K. Permeation, diffusion and solution of gases in organic polymers. Trans. Faraday Soc. 1939, 35, 628–643. [Google Scholar]
- Craster, B.; Jones, T.G.J. Permeation of a range of species through polymer layers under varying conditions of temperature and pressure: In situ measurement methods. Polymers 2019, 11, 1056. [Google Scholar] [CrossRef]
- Nguyen, X.Q.; Brož, Z.; Vašák, F.; Nguyen, Q.T. Manometric techniques for determination of gas transport parameters in membranes. Application to the study of dense and asymmetric poly(vinyltrimethylsilane) membranes. J. Memb. Sci. 1994, 91, 65–76. [Google Scholar] [CrossRef]
- Stern, S.A.; Mullhaupt, J.T.; Gareis, P.J. The effect of pressure on the permeation of gases and vapors through polyethylene. Usefulness of the corresponding states principle. AIChE J. 1969, 15, 64–73. [Google Scholar] [CrossRef]
- Trivedi, V.; Ajiboye, A.L.; Coleman, N.J.; Bhomia, R.; Bascougnano, M. Melting point depression of poly (ethylene oxide)-poly (propylene oxide)-poly (ethylene oxide) triblock polymers in supercritical carbon dioxide in the presence of menthol as a solid co-plasticiser. Polymers 2022, 14, 2825. [Google Scholar] [CrossRef]
- Yasuda, H.; Lamaze, C.; Ikenberry, L.D. Permeability of solutes through hydrated polymer membranes. Part 1 Diffusion of sodium chloride. Makromol. Chem. Phys. 1968, 118, 19–35. [Google Scholar] [CrossRef]
- Harogoppad, S.B.; Aminabhavi, T.M.; Balundgi, R.H. Sorption and transport of aqueous salt solution in polyurethane membrane at 25, 44, and 60 °C. J. Appl. Polym. Sci. 1991, 42, 1297–1306. [Google Scholar] [CrossRef]
- Schultze, J.D.; Böhning, M.; Springer, J. Sorption and permeation properties of poly(p-phenylene sulfide) crystallized in the presence of sorbed gas molecules. Die Makromol. Chem. 1993, 194, 431–444. [Google Scholar] [CrossRef]
- Matteucci, S.; Yampolskii, Y.; Freeman, B.D.; Pinnau, I. Transport of gases and vapors in glassy and rubbery polymers. In Material Science of Membranes for Gas and Vapor Separation; John Wiley & Sons: Chichester, UK, 2006; pp. 1–47. [Google Scholar]
- Minelli, M.; Sarti, G.C. Permeability and diffusivity of CO2 in glassy polymers with and without plasticization. J. Memb. Sci. 2013, 435, 176–185. [Google Scholar] [CrossRef]
- McKeen, L.W. Permeability Properties of Plastics and Elastomers, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Cowling, R.; Park, G.S. Permeability, solubility and diffusion of gases in amorphous and crystalline 1,4-polybutadiene membranes. J. Memb. Sci. 1979, 5, 199–207. [Google Scholar] [CrossRef]
- Heilman, W.; Tammela, V.; Meyer, J.A.; Stannett, V.; Szwarc, M. Permeability of Polymer Films to Hydrogen Sulfide Gas. Ind. Eng. Chem. 1956, 48, 821–824. [Google Scholar] [CrossRef]
- Flaconnèche, B.; Klopffer, M.H.; Taravel-Condat, C. Transport properties of gas mixtures in polymers: Measurement method, experimental data obtained on thermoplastics. In Oilfield Engineering with Polymers; Rapra Technology: London, UK, 2006; pp. 1–16. [Google Scholar]
- Yasuda, H.; Stannett, V.; Frisch, H.L.; Peterlin, A. The permeability of strained polymer films. Die Makromol. Chem. Macromol. Chem. Phys. 1964, 73, 188–202. [Google Scholar] [CrossRef]
- Yasuda, H.; Peterlin, A. Gas permeability of deformed polyethylene films. J. Appl. Polym. Sci. 1974, 18, 531–546. [Google Scholar] [CrossRef]
- Sha, H.; Harrison, I.R. CO2 permeability and amorphous fractional free-volume in uniaxially drawn HDPE. J. Polym. Sci. Part B Polym. Phys. 1992, 30, 915–922. [Google Scholar] [CrossRef]
- Boyer, S.A.E.; Klopffer, M.-H.; Martin, J.; Grolier, J.-P.E. Supercritical Gas–Polymer Interactions with Applications in the Petroleum Industry. Determination of Thermophysical Properties. J. Appl. Polym. Sci. 2006, 103, 1706–1722. [Google Scholar] [CrossRef]
- Sarrasin, F.; Memari, P.; Klopffer, M.H.; Lachet, V.; Condat, C.T.; Rousseau, B.; Espuche, E. Influence of high spressures on CH4, CO2 and H2S solubility in polyethylene: Experimental and molecular simulation approaches for pure gas and gas mixtures. Modelling of the sorption isotherms. J. Memb. Sci. 2015, 490, 380–388. [Google Scholar] [CrossRef]
- Naito, Y.; Kamiya, Y.; Terada, K.; Mizoguchi, K.; Wang, J.-S. Pressure dependence of gas permeability in a rubbery polymer. J. Appl. Polym. Sci. 1996, 61, 945–950. [Google Scholar] [CrossRef]
- Kumazawa, H.; Wang, J.-S.; Naito, K.; Messaoudi, B.; Sada, E. Gas transport in polymer membrane at temperatures above and below glass transition point. J. Appl. Polym. Sci. 1994, 51, 1015–1020. [Google Scholar] [CrossRef]
- Yampolskii, Y.P.; Kamiya, Y.; Alentiev, A.Y. Transport parameters and solubility coefficients of polymers at their glass transition temperatures. J. Appl. Polym. Sci. 2000, 76, 1691–1705. [Google Scholar] [CrossRef]
- Toi, K.; Maeda, Y.; Tokuda, T. Mechanism of diffusion and sorption of carbon dioxide in poly(vinyl acetate) above and below the glass transition temperature. J. Memb. Sci. 1983, 13, 15–27. [Google Scholar] [CrossRef]
- Rowe, B.W.; Freeman, B.D.; Paul, D.R. Physical aging of ultrathin glassy polymer films tracked by gas permeability. Polymer 2009, 50, 5565–5575. [Google Scholar] [CrossRef]
- Bernardo, P.; Bazzarelli, F.; Tasselli, F.; Clarizia, G.; Mason, C.; Maynard-Atem, L.; Budd, P.; Lanč, M.; Pilnáček, K.; Vopička, O.; et al. Effect of physical aging on the gas transport and sorption in PIM-1 membranes. Polymer 2017, 113, 283–294. [Google Scholar] [CrossRef]
- Schramm, D. PE-RT, a new class of polyethylene for industrial pipes. In Proceedings of the International Conference on Offshore Mechanics and Arctic Engineering, Hamburg, Germany, 4–9 June 2006. [Google Scholar] [CrossRef]
- Allara, D.L. Aging of polymers. Environ. Health Perspect. 1975, 11, 29–33. [Google Scholar] [CrossRef]
- Tremblay, P.; Savard, M.M.; Vermette, J.; Paquin, R. Gas permeability, diffusivity and solubility of nitrogen, helium, methane, carbon dioxide and formaldehyde in dense polymeric membranes using a new on-line permeation apparatus. J. Membr. Sci. 2006, 282, 245–256. [Google Scholar] [CrossRef]
- Taravel-Condat, C.; Epsztein, T. The Use of Flexible Pipe for CO2 Enhanced Oil Recovery Applications. In Proceedings of the ASME 2012 31st International Conference on Ocean, Offshore and Arctic Engineering, Volume 3: Pipeline and Riser Technology, Rio de Janeiro, Brazil, 1–6 July 2012; pp. 251–259. [Google Scholar] [CrossRef]
- Smith, Z.P.; Hernández, G.; Gleason, K.L.; Anand, A.; Doherty, C.; Konstas, K.; Alvarez, C.; Hill, A.J.; E Lozano, A.; Paul, D.R.; et al. Effect of polymer structure on gas transport properties of selected aromatic polyimides, polyamides and TR polymers. J. Memb. Sci. 2015, 493, 766–781. [Google Scholar] [CrossRef]
- Minelli, M.; Doghieri, F. Predictive model for gas and vapor solubility and swelling in glassy polymers I: Application to different polymer/penetrant systems. Fluid Phase Equilib. 2014, 381, 1–11. [Google Scholar] [CrossRef]
- Cussler, E. Diffusion: Mass Transfer in Fluid Systems, 3rd ed.; Cambridge University Press: Cambridge, UK, 2009. [Google Scholar]
- Rutherford, S.W.; Do, D.D. Review of time lag permeation technique as a method for characterisation of porous media and membranes. Adsorption 1997, 3, 283–312. [Google Scholar] [CrossRef]
- Memari, P.; Lachet, V.; Klopffer, M.-H.; Flaconnèche, B.; Rousseau, B. Gas mixture solubilities in polyethylene below its melting temperature: Experimental and molecular simulation studies. J. Memb. Sci. 2012, 390, 194–200. [Google Scholar] [CrossRef]
- Tsujita, Y. Gas sorption and permeation of glassy polymers with microvoids. Prog. Polym. Sci. 2003, 28, 1377–1401. [Google Scholar] [CrossRef]
- Wadhawan, J.D.; Craster, B.; Lawrence, N.S.; Kelly, S.M. Regular Solution Theory for Polymer Permeation Transients: A Toolkit for Understanding Experimental Waveshapes. Langmuir 2020, 36, 5003–5020. [Google Scholar] [CrossRef] [PubMed]
- Yen, S.K.; Shih, H.C. Determination of the Critical Hydrogen Concentration for Brittle Fracture of a High Chromium Ferritic Steel. J. Electrochem. Soc. 1990, 137, 2028–2031. [Google Scholar] [CrossRef]
- Beck, W.; Bockris, J.O.M.; McBreen, J.; Nanis, L. Hydrogen permeation in metals as a function of stress, temperature and dissolved hydrogen concentration. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1966, 290, 220–235. [Google Scholar] [CrossRef]
- McBreen, J.; Nonis, L.; Beck, W. A Method for Determination of the Permeation Rate of Hydrogen through Metal Membranes. J. Electrochem. Soc. 1966, 113, 1218. [Google Scholar] [CrossRef]
- Kimble, M.C.; White, R.E.; Tsou, Y.; Beaver, R.N. Estimation of the Diffusion Coefficient and Solubility for a Gas Diffusing through a Membrane. J. Electrochem. Soc. 1990, 137, 2510–2514. [Google Scholar] [CrossRef]
- Wu, J.K. Electrochemical method for studying hydrogen in iron, nickel and palladium. Int. J. Hydrog. Energy 1992, 17, 917–921. [Google Scholar] [CrossRef]
- Osoba, W. Investigation of the free volume changes in thermally treated polyethylene by positron annihilation. Acta Phys. Pol. Ser. A 1999, 95, 632–636. [Google Scholar] [CrossRef]
- Dlubek, G.; Saarinen, K.; Fretwell, H.M. The temperature dependence of the local free volume in polyethylene and polytetrafluoroethylene: A positron lifetime study. J. Polym. Sci. Part B Polym. Phys. 1998, 36, 1513–1528. [Google Scholar] [CrossRef]
- Fleischer, G. Temperature dependence of self diffusion of polystyrene and polyethylene in the melt. An interpretation in terms of the free volume theory. Polym. Bull. 1980, 440, 437–440. [Google Scholar] [CrossRef]
- Polymer Database, Polyethylene, Thermo-Physical Properties: Calculated Data. p. 1. Available online: https://polymerdatabase.com/polymers/polyethylene.html (accessed on 19 May 2016).
- Campion, G.J.; Morgan, R.P. High pressure permeation and diffusion of gases in polymers of different structures. Plast. Rubber Compos. Process. Appl. 1992, 17, 51–58. [Google Scholar]
- Spyriouni, T.; Boulougouris, G.C.; Theodorou, D.N. Prediction of sorption of CO2 in glassy atactic polystyrene at elevated pressures through a new computational scheme. Macromolecules 2009, 42, 1759–1769. [Google Scholar] [CrossRef]
- Bonavoglia, B.; Storti, G.; Morbidelli, M.; Rajendran, A. Sorption and Swelling of Semicrystalline Polymers in Supercritical CO2. Polym. Phys. 2006, 44, 1531–1546. [Google Scholar] [CrossRef]
- Nilsson, F.; Hallstensson, K.; Johansson, K.; Umar, Z.; Hedenqvist, M.S. Predicting solubility and diffusivity of gases in polymers under high pressure: N2 in polycarbonate and poly(ether-ether-ketone). Ind. Eng. Chem. Res. 2013, 52, 8655–8663. [Google Scholar] [CrossRef]
- Hedenqvist, M.; Angelstok, A.; Edsberg, L.; Larssont, P.T.; Gedde, U.W. Diffusion of small-molecule penetrants in polyethylene: Free volume and morphology. Polymer 1996, 37, 2887–2902. [Google Scholar] [CrossRef]
- Fujiwara, H.; Ono, H.; Ohyama, K.; Kasai, M.; Kaneko, F.; Nishimura, S. Hydrogen permeation under high pressure conditions and the destruction of exposed polyethylene-property of polymeric materials for high-pressure hydrogen devices (2)-. Int. J. Hydrog. Energy 2021, 46, 11832–11848. [Google Scholar] [CrossRef]
- Minelli, M.; Sarti, G.C. Permeability and solubility of carbon dioxide in different glassy polymer systems with and without plasticization. J. Memb. Sci. 2013, 444, 429–439. [Google Scholar] [CrossRef]
- Redlich, O.; Kwong, J.N.S. On the Thermodynamics of Solutions. V. An Equation of State. Fugacities of Gaseous Solutions. Chem. Rev. 1949, 44, 233–244. [Google Scholar] [CrossRef]
- Celina, M.; Gillen, K.T. Oxygen Permeability Measurements on Elastomers at Temperatures up to 225 °C. Macromolecules 2005, 38, 2754–2763. [Google Scholar] [CrossRef]
- Flaconneche, B.; Martin, J.; Klopffer, M.H. Permeability, Diffusion and Solubility of Gases in Polyethylene, Polyamide 11 and Poly (Vinylidene Fluoride). Oil Gas Sci. Technol. 2001, 56, 261–278. [Google Scholar] [CrossRef]
- Fu, J.; Ma, Y. Mold modification methods to fix warpage problems for plastic molding products. Comput. Aided. Des. Appl. 2016, 13, 138–151. [Google Scholar] [CrossRef][Green Version]
- Winful, D.; Craster, B. Testing of metallic and non-metallic material in gaseous hydrogen. In Proceedings of the Steel and Hydrogen, 4th International Conference on Metals Hydrogen Steel and Hydrogen, Ghent, Belgium, 11–13 October 2022. [Google Scholar]
- Li, D.; Zhou, L.; Wang, X.; He, L.; Yang, X. Effect of crystallinity of polyethylene with different densities on breakdown strength and conductance property. Materials 2019, 12, 1746. [Google Scholar] [CrossRef]
- NETZSCH. Determination of the Degree of Crystallinity of Polymers (Pe and Pp). Polymer Manufacturing, Application Sheet. 2006. Available online: https://analyzing-testing.netzsch.com/_Resources/Persistent/5/8/5/0/58508d8bc19cd910b11627c4beee913fdaa31231/2006-111_Determination%20of%20the%20Degree%20of%20Crystallinity%20of%20Polymers%20%28PE%20and%20PP%29.pdf (accessed on 30 July 2022).
- Lin, Y.; Tang, Y.; Wang, L.; Wang, X. Non-Isothermal Crystallization Behavior of Poly(vinylidene fluoride) in Dialkyl Phthalate Diluents during Thermally Induced Phase Separation Process. Crystals 2020, 10, 782. [Google Scholar] [CrossRef]
- Zohuri, B. Properties of Pure Substances. Phys. Cryog. 2018, 53–79. [Google Scholar] [CrossRef]
- Dhingra, S.S.; Marand, E. Mixed gas transport study through polymeric membranes. J. Memb. Sci. 1998, 141, 45–63. [Google Scholar] [CrossRef]
- Li, H.; Yang, D.; Zhang, D.; Zhu, Y.; Ge, P.; Qi, D.; Zhang, Z. Permeation Characteristic and Mechanism of CO2 in High Density Polyethylene. In Proceedings of the Chinese Materials Conference, Xiamen, China, 12–16 July 2018. [Google Scholar] [CrossRef]
- Maxwell, A.S.; Roberts, S.J. Directorate, Review of Data on Gas Migration through Polymer Encapsulants. SERCO/TAS/000500/001—Issue 2. 2010, 1–50. Available online: https://www.scribd.com/document/519915797/Review-of-Data-on-Gas-Migration-Through-Polymer-Encapsulants (accessed on 30 July 2022).
- Raine, T.P. Polymer/Graphene Nanocomposites for Improved Barrier Performance; University of Manchester: Manchester, UK, 2018. [Google Scholar]
- Borek, J.; Osoba, W. Free volume changes in physically aged polyethylene by positron annihilation. Polymer 2001, 42, 2901–2905. [Google Scholar] [CrossRef]
- Monge, M.A.; Díaz, J.A.; Pareja, R. Strain-induced changes of free volume measured by positron lifetime spectroscopy in ultrahigh molecular weight polyethylene. Macromolecules 2004, 37, 7223–7230. [Google Scholar] [CrossRef]
- Raheem, H.; Seshia, A.; Craster, B. Resonant Coupling of Piezoelectric Micromachined Ultrasound Transducers with Polymer Specimens in Different Media. In Proceedings of the 2021 IEEE International Ultrasonics Symposium (IUS), Virtual (Online) Symposium, 11–16 September 2021; pp. 21–24. [Google Scholar] [CrossRef]
- Curbell Plastics, Materials—PVDF, High Purity Engineering Plastic with Excellent Chemical, Abrasion, and Flame Resistance. Website 2022. Available online: https://www.curbellplastics.com/Research-Solutions/Materials/PVDF (accessed on 30 July 2022).
- Zhang, D.; Li, H.; Qi, D.; Ding, N.; Shao, X.; Wei, B.; Cai, X. Gas permeation behaviors of high-density polyethylene as a liner material of flexible pipes. Nat. Gas Ind. 2017, 37, 104–110. [Google Scholar] [CrossRef]
- Michaels, H.J.; Bixler, A.S. Flow of Gases through Polyethylene. Polym. Sci. 1961, 50, 413–439. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raheem, H.; Craster, B.; Seshia, A. Analysis of Permeation and Diffusion Coefficients to Infer Aging Attributes in Polymers Subjected to Supercritical CO2 and H2 Gas at High Pressures. Polymers 2022, 14, 3741. https://doi.org/10.3390/polym14183741
Raheem H, Craster B, Seshia A. Analysis of Permeation and Diffusion Coefficients to Infer Aging Attributes in Polymers Subjected to Supercritical CO2 and H2 Gas at High Pressures. Polymers. 2022; 14(18):3741. https://doi.org/10.3390/polym14183741
Chicago/Turabian StyleRaheem, Hamad, Bernadette Craster, and Ashwin Seshia. 2022. "Analysis of Permeation and Diffusion Coefficients to Infer Aging Attributes in Polymers Subjected to Supercritical CO2 and H2 Gas at High Pressures" Polymers 14, no. 18: 3741. https://doi.org/10.3390/polym14183741
APA StyleRaheem, H., Craster, B., & Seshia, A. (2022). Analysis of Permeation and Diffusion Coefficients to Infer Aging Attributes in Polymers Subjected to Supercritical CO2 and H2 Gas at High Pressures. Polymers, 14(18), 3741. https://doi.org/10.3390/polym14183741