
Coordinative Chain Transfer Polymerization of Sustainable Terpene Monomers Using a Neodymium-Based Catalyst System

 , , , , and
, , , , and 
Abstract
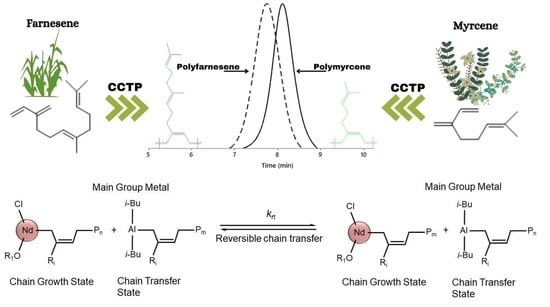
:
1. Introduction
2. Materials and Methods
2.1. Laboratory Homopolymerization Procedure
2.2. Reactor Homopolymerization Procedure
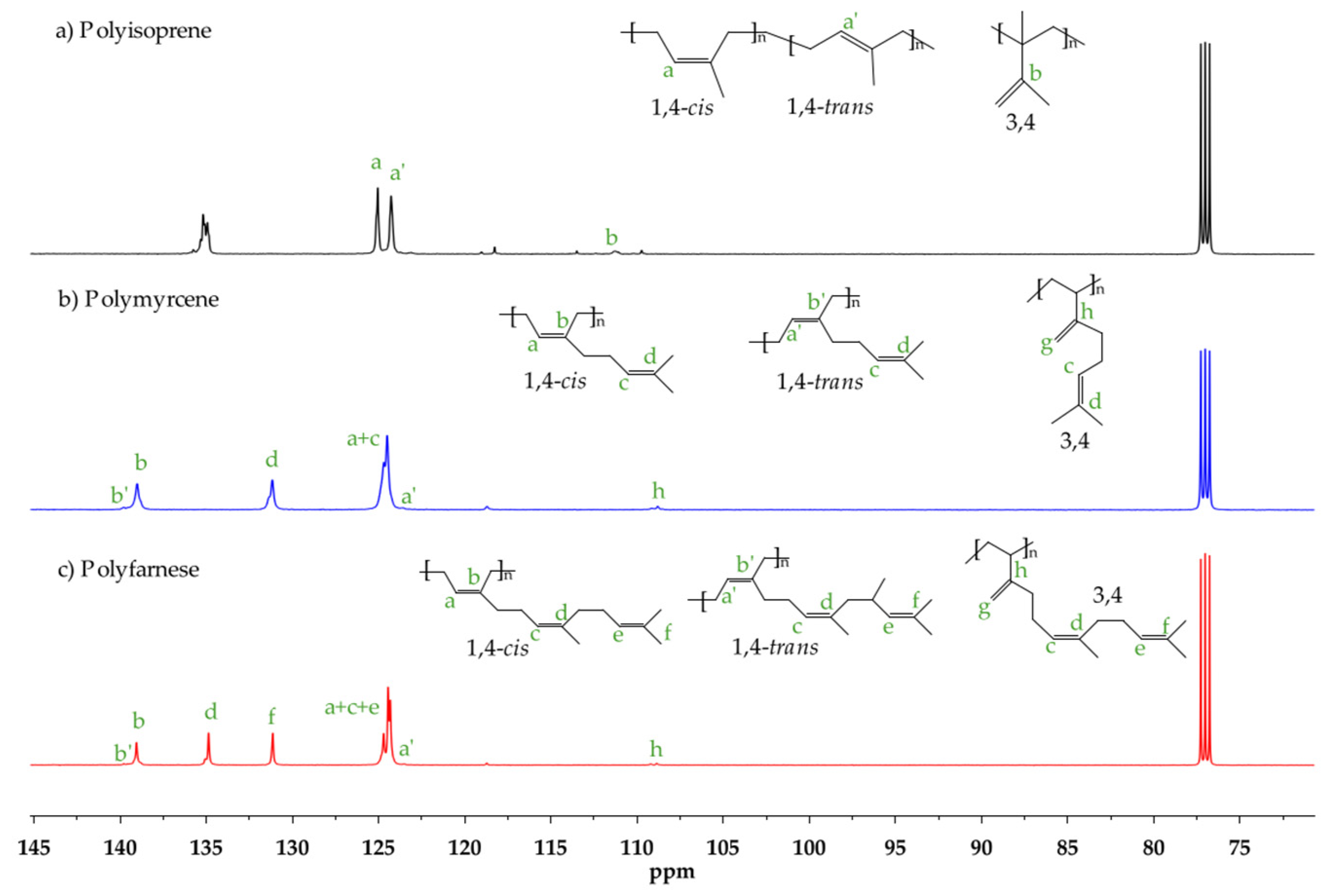
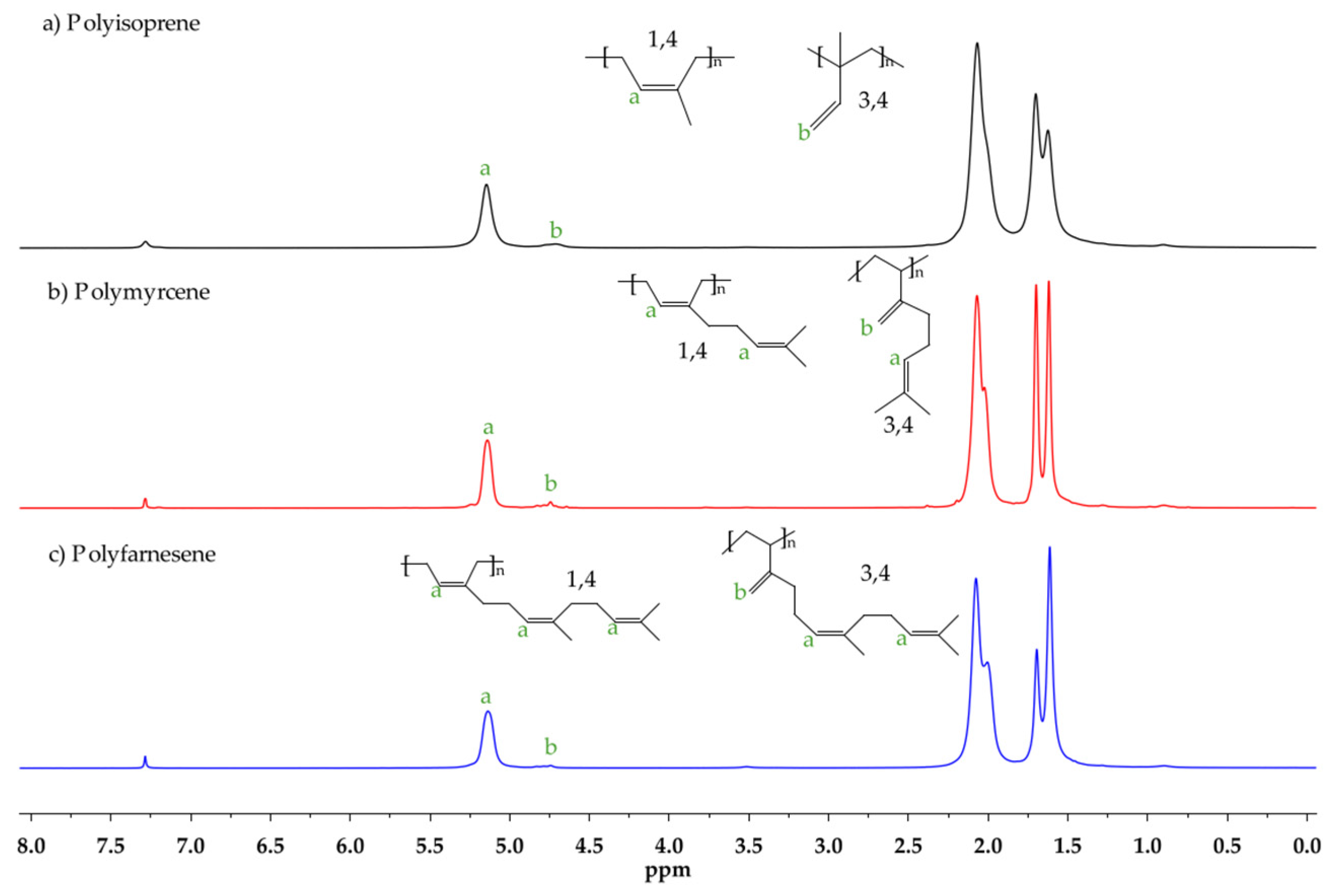
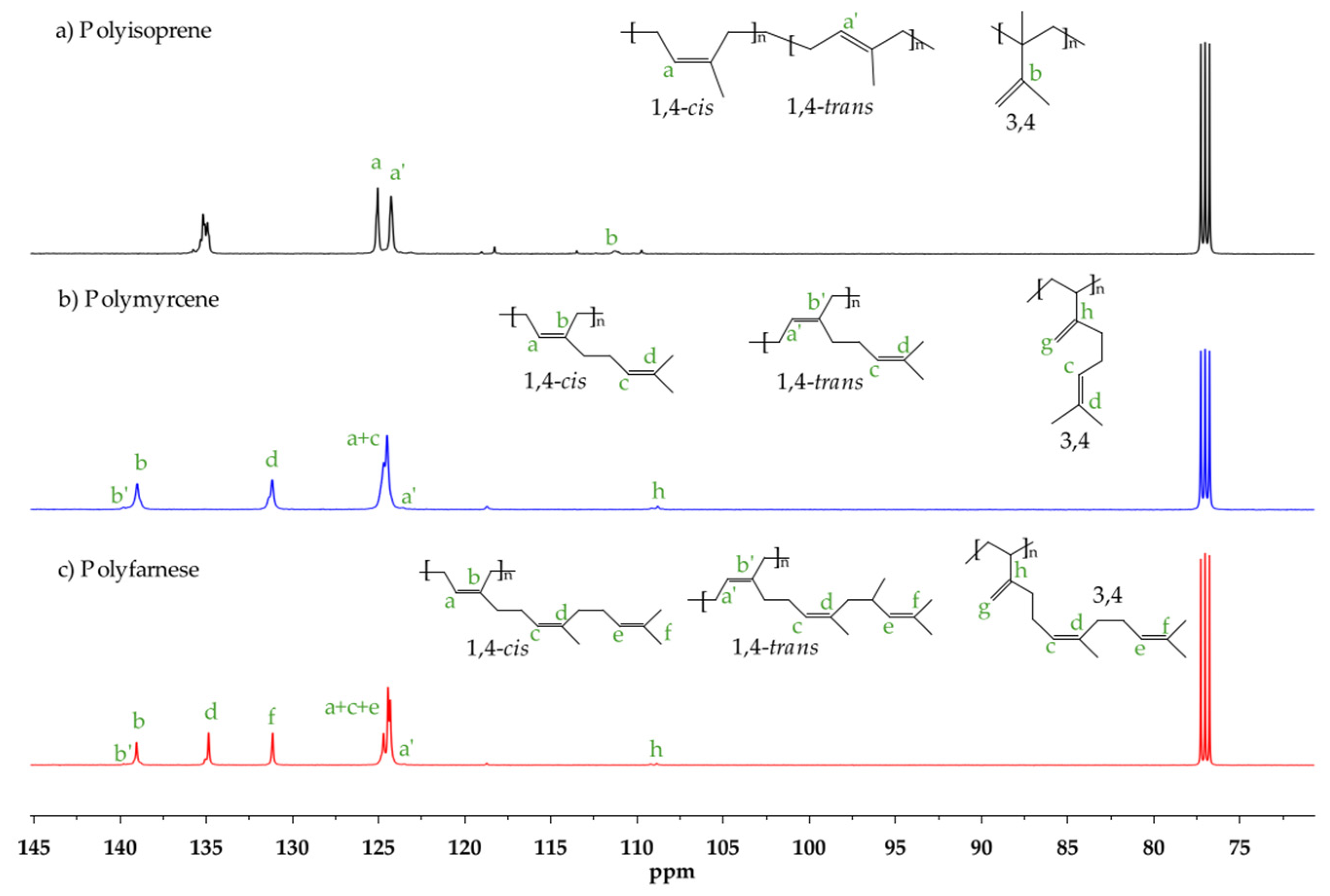
2.3. Characterization
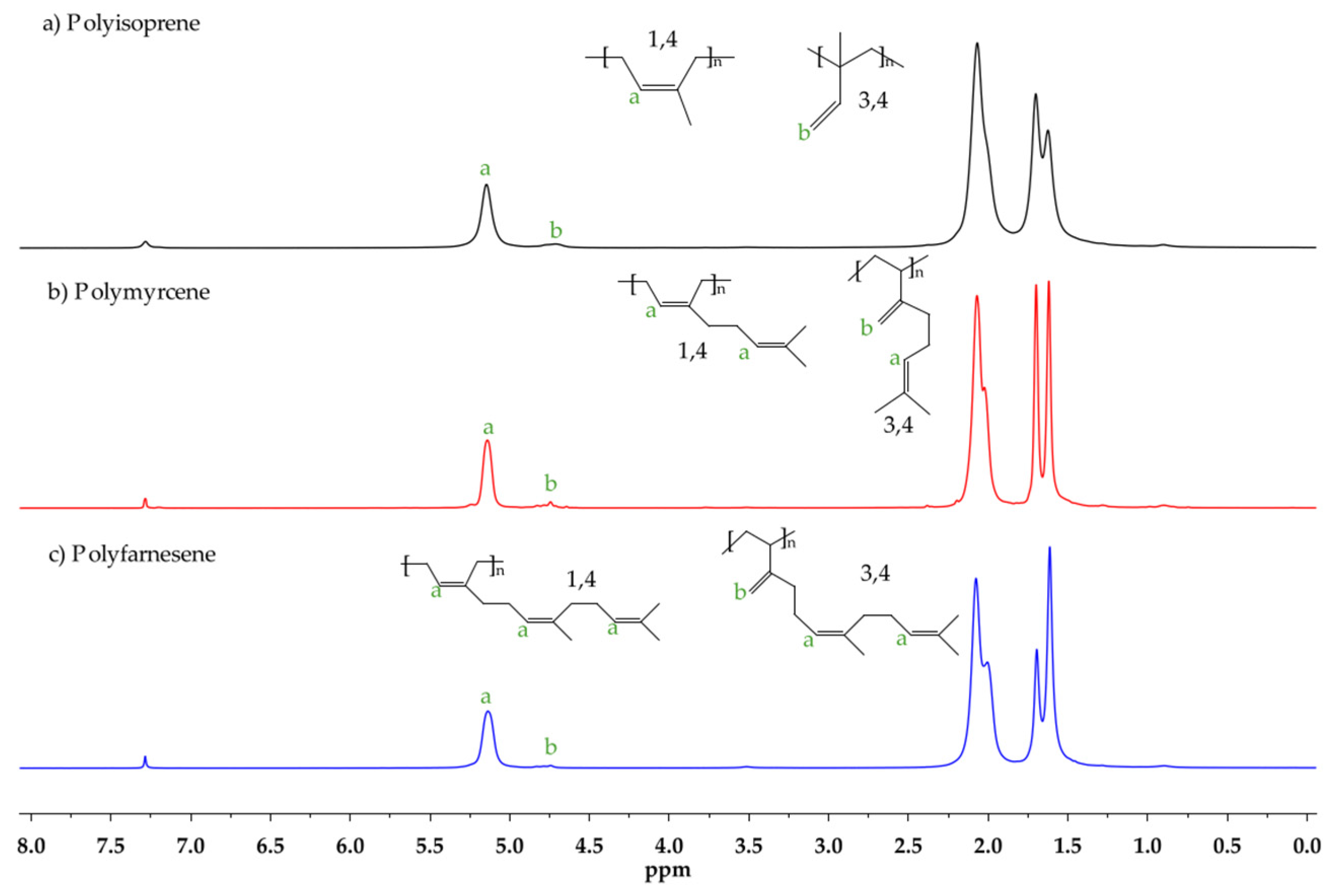
3. Results and Discussion
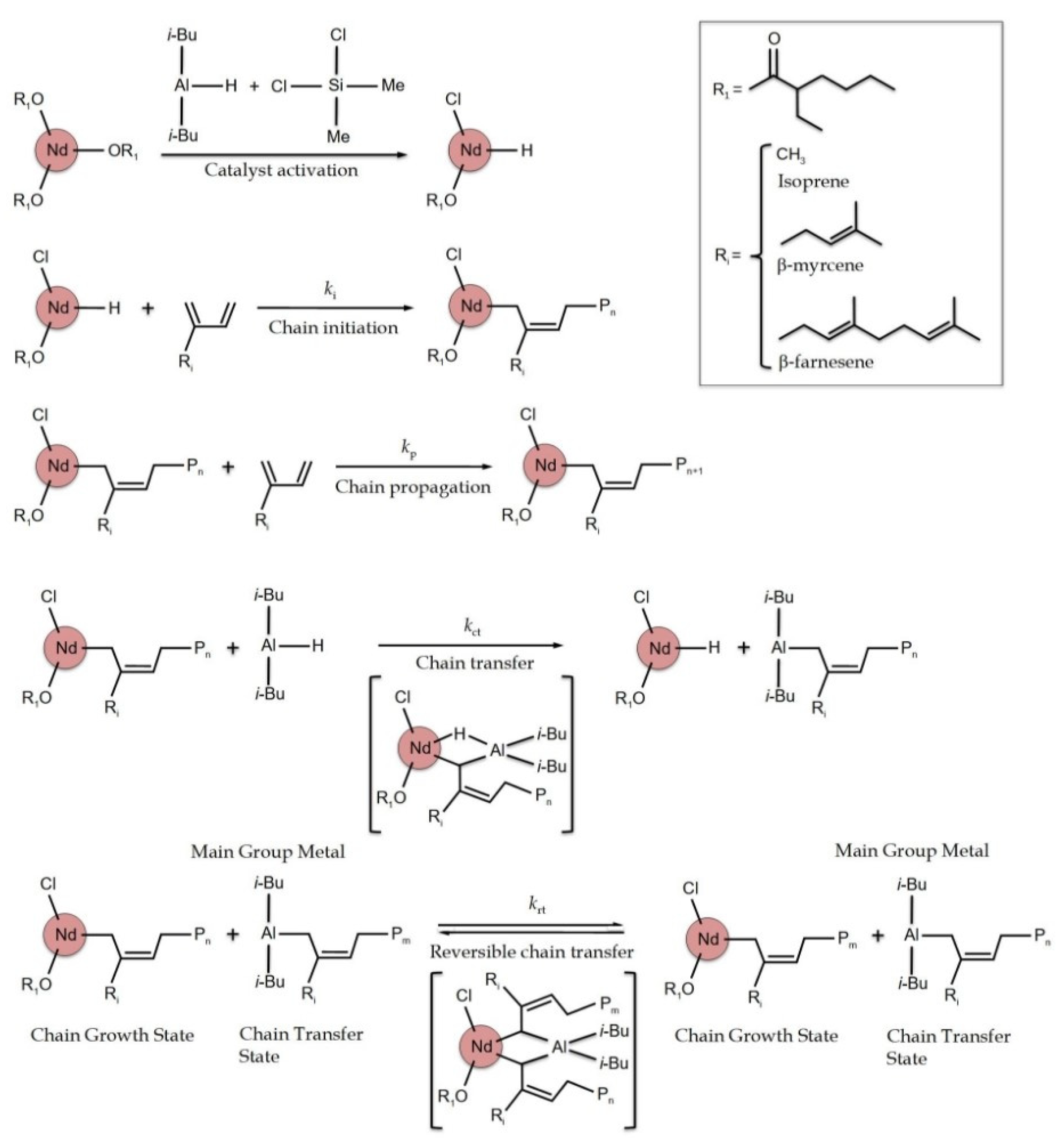
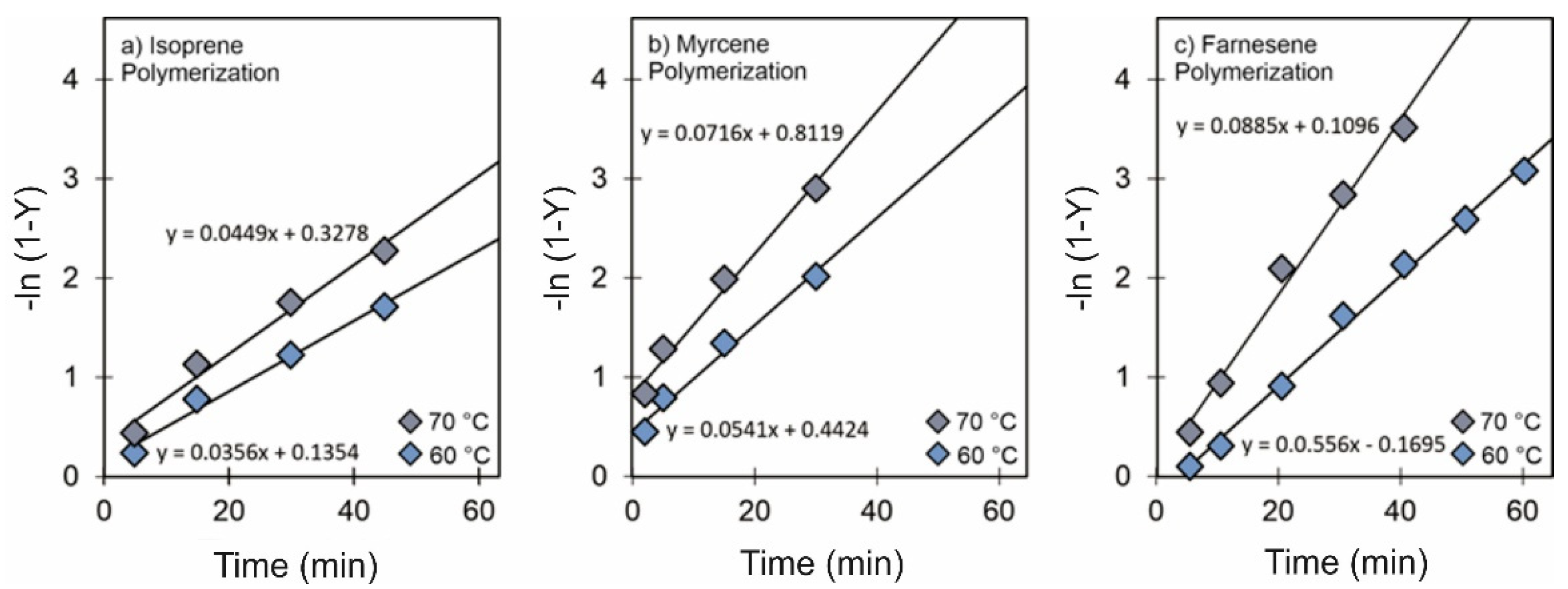
3.1. Effect of [Cl]/[Nd] Ratio
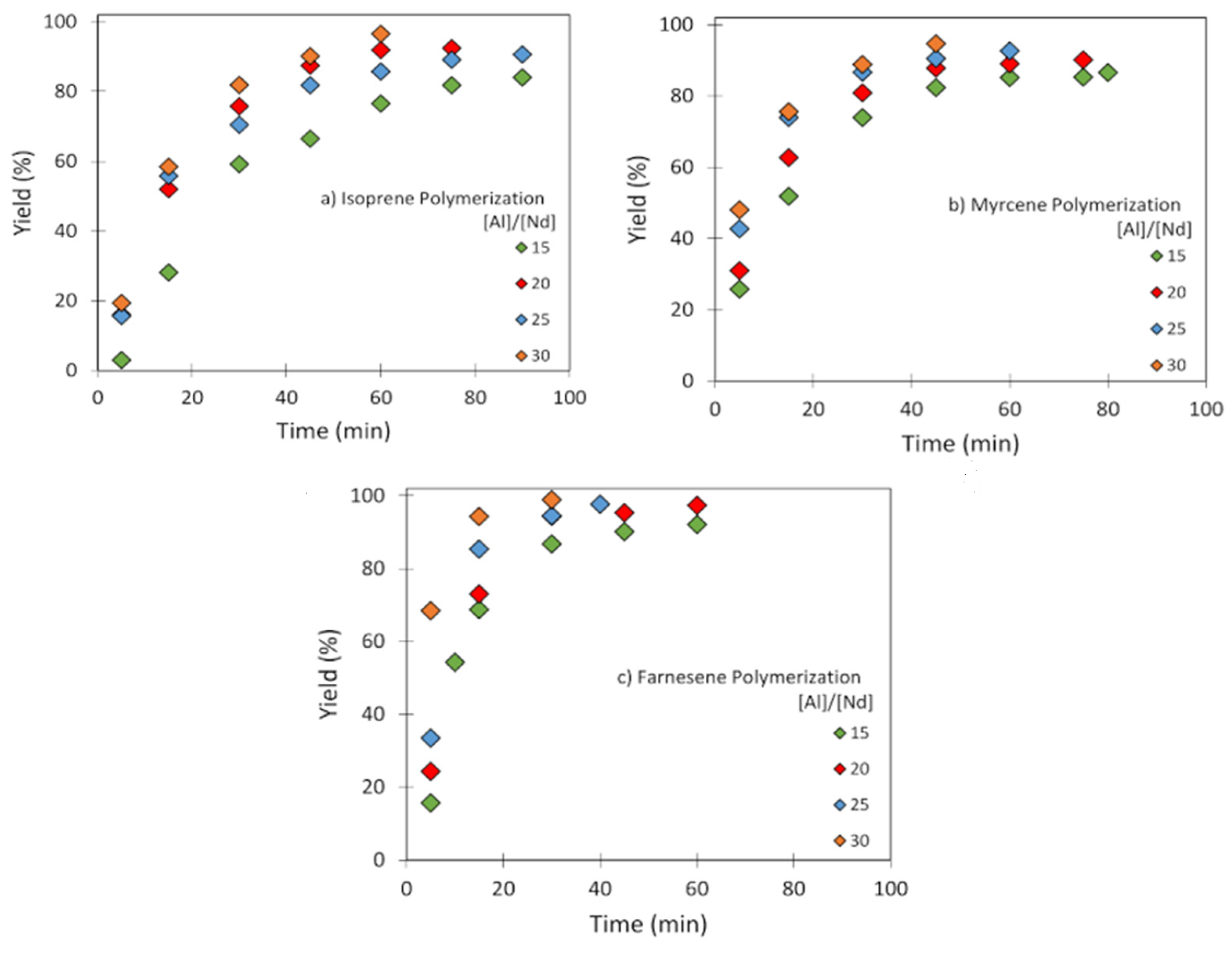
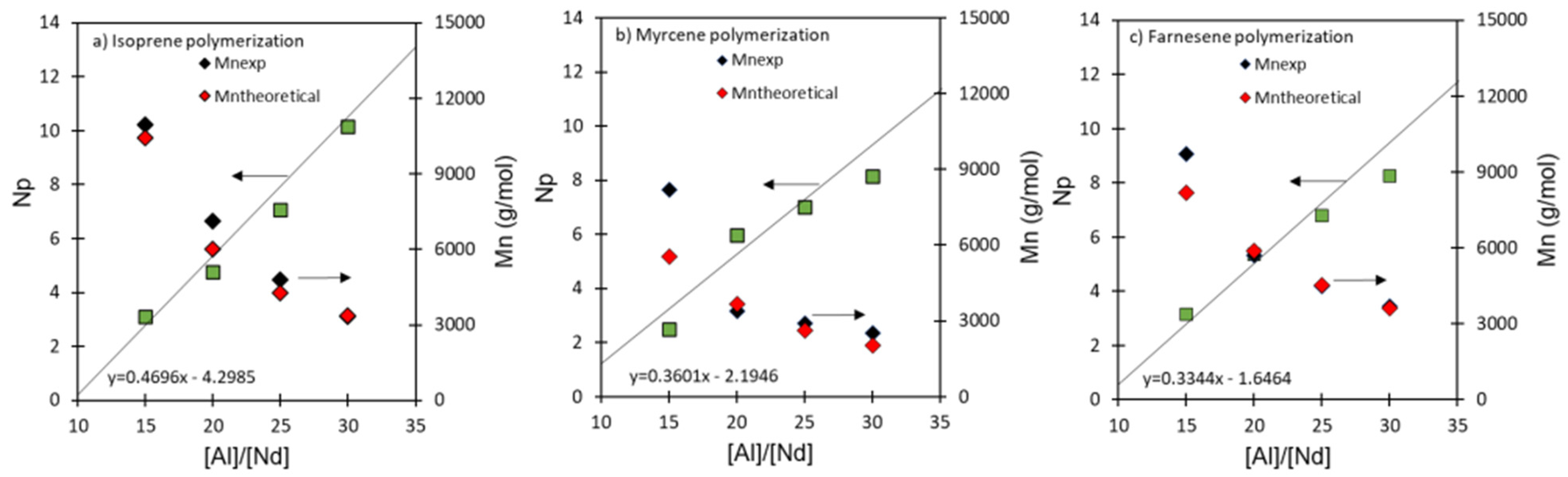
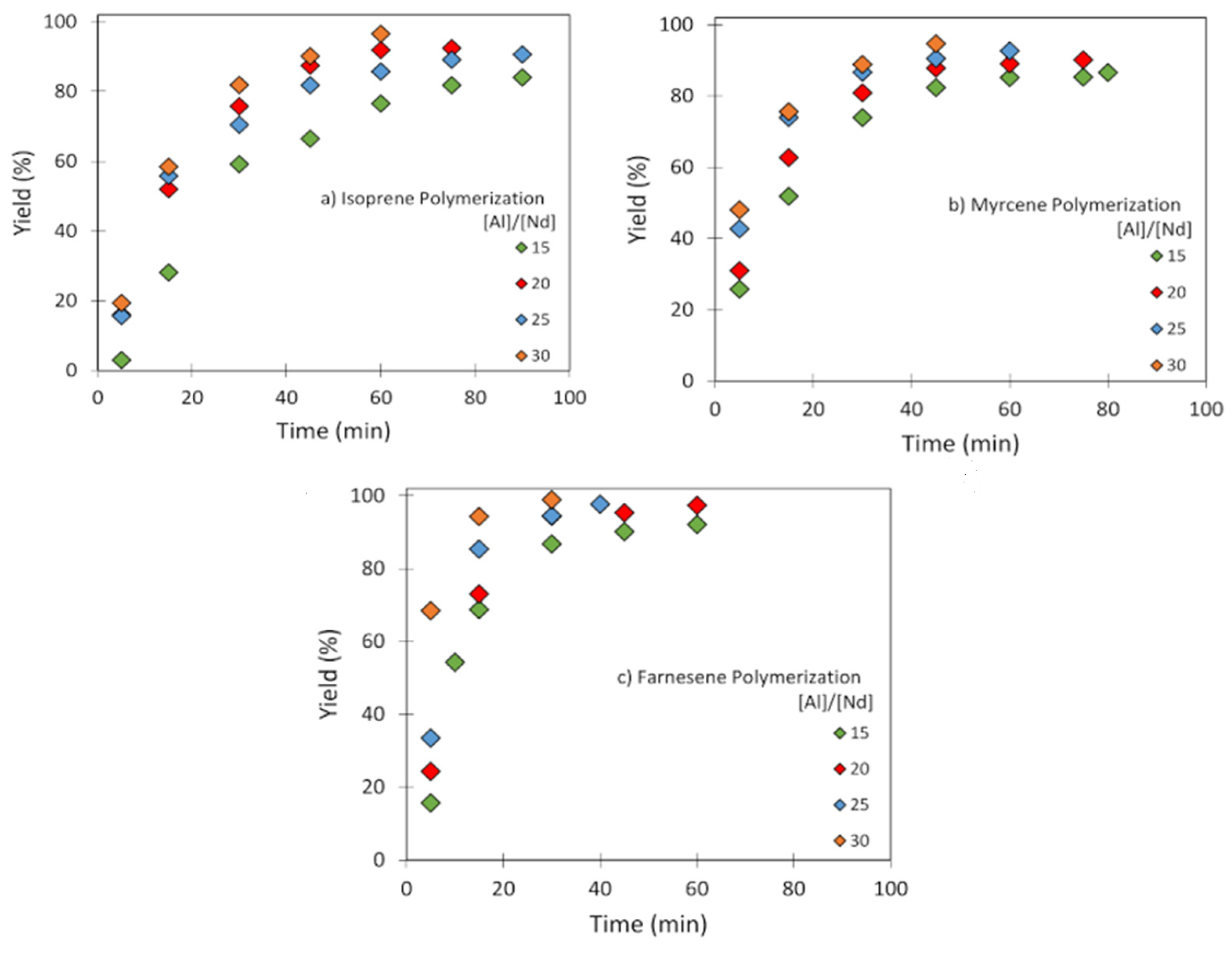
3.2. Effect of [Al]/[Nd] Ratio
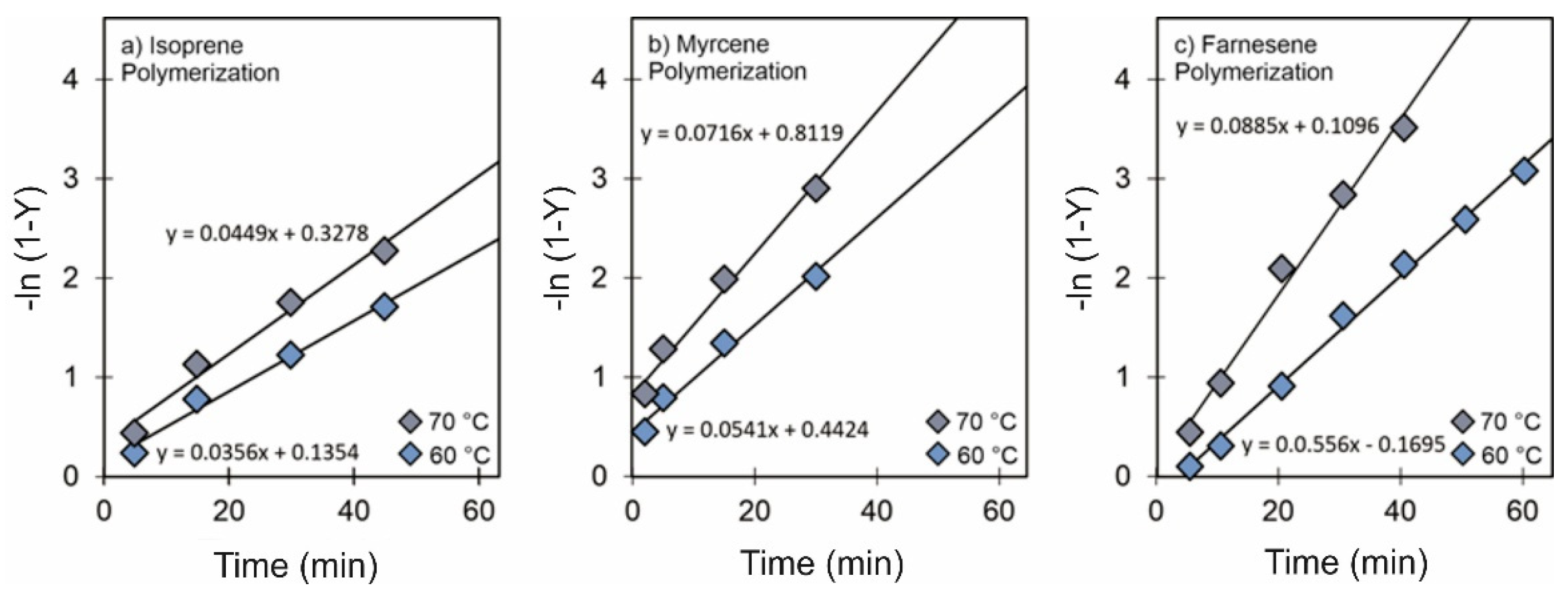
3.3. Effect of Temperature
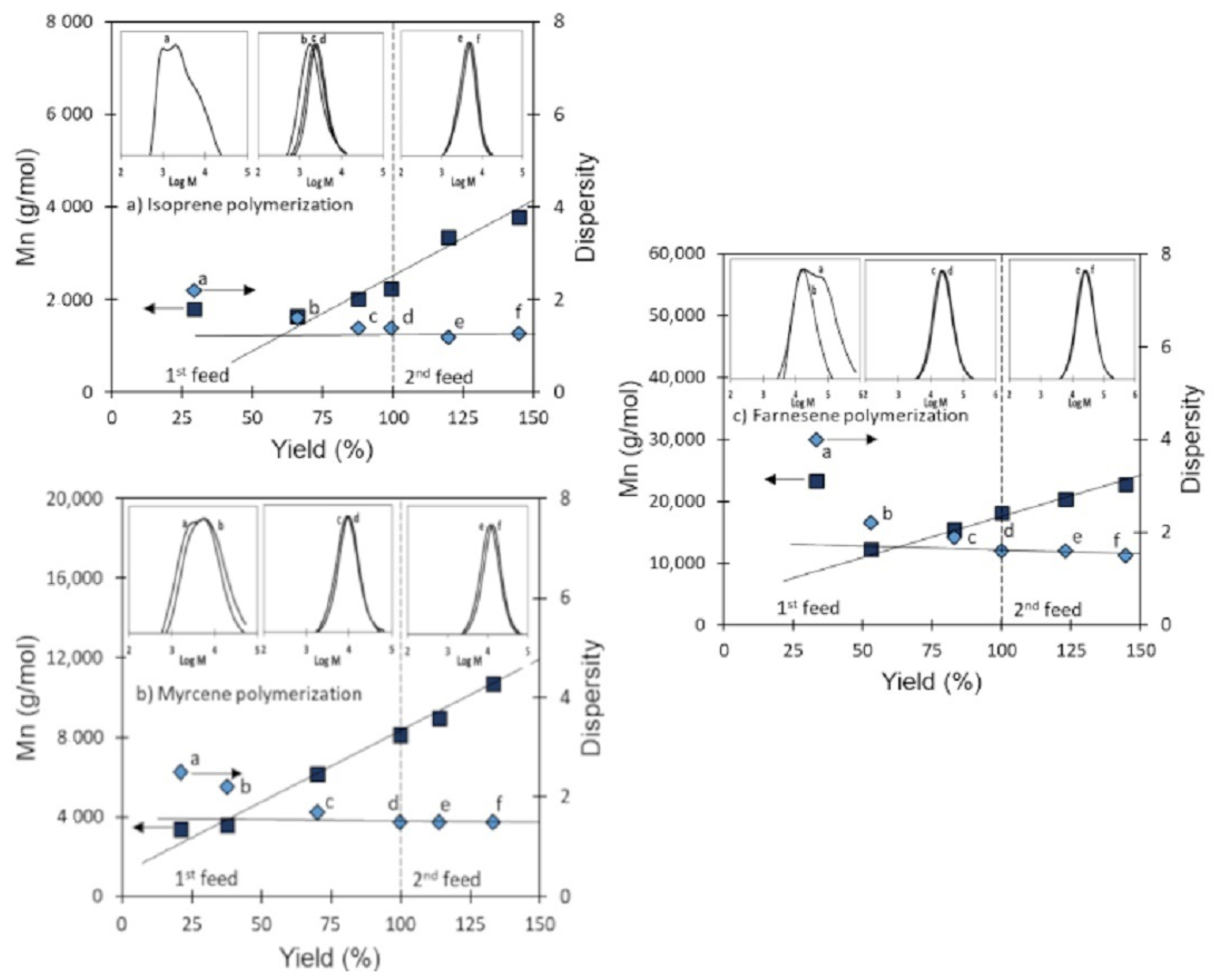
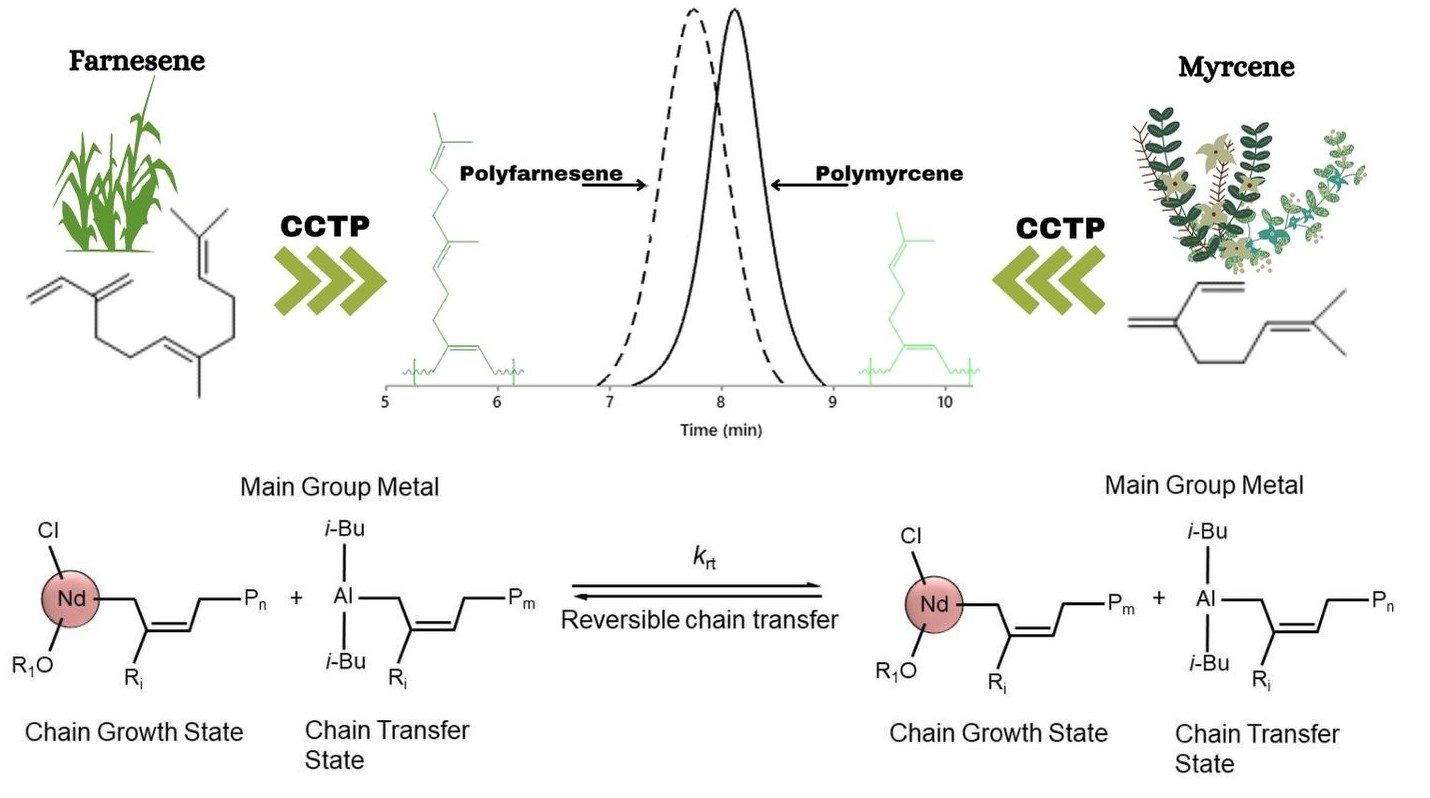
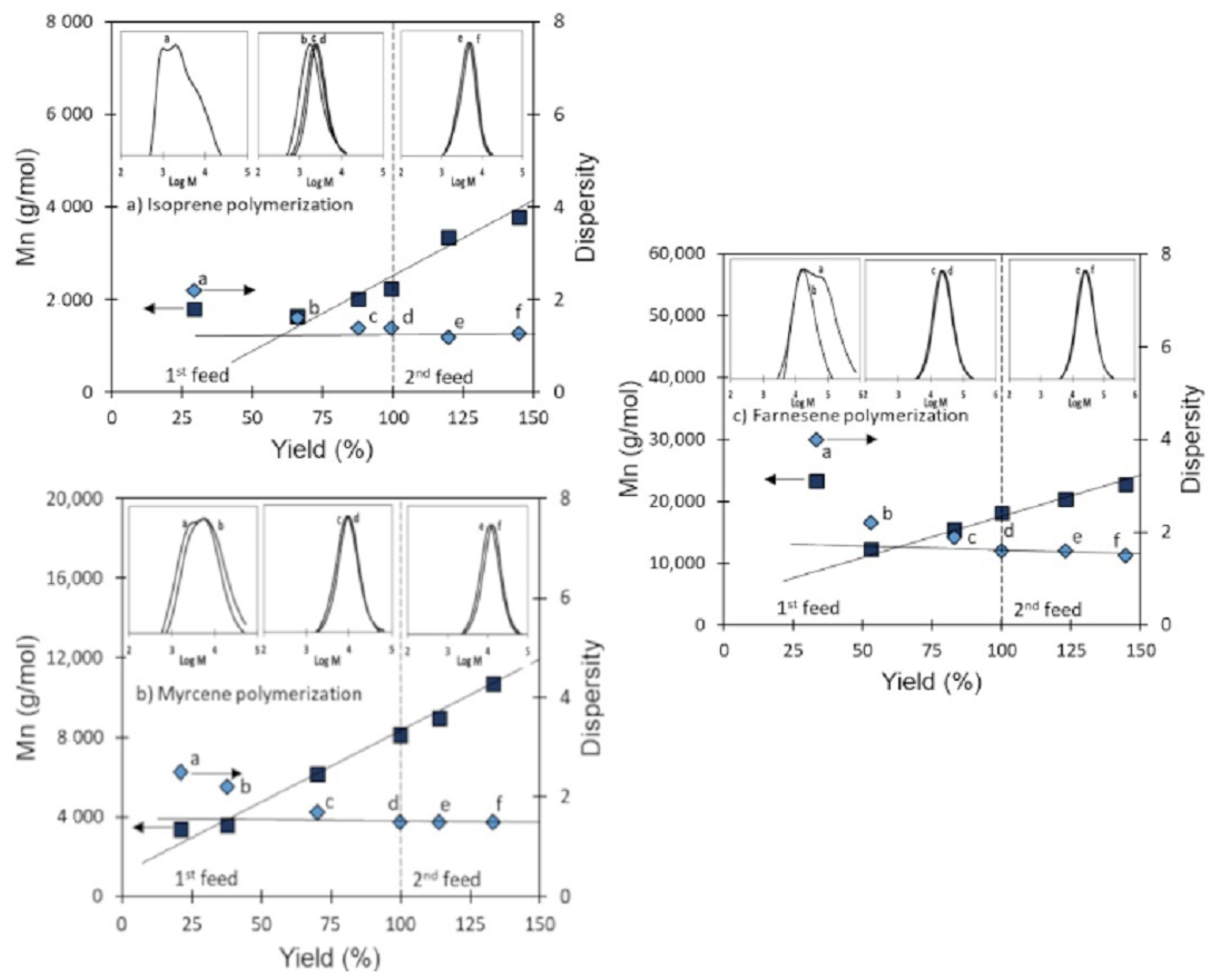
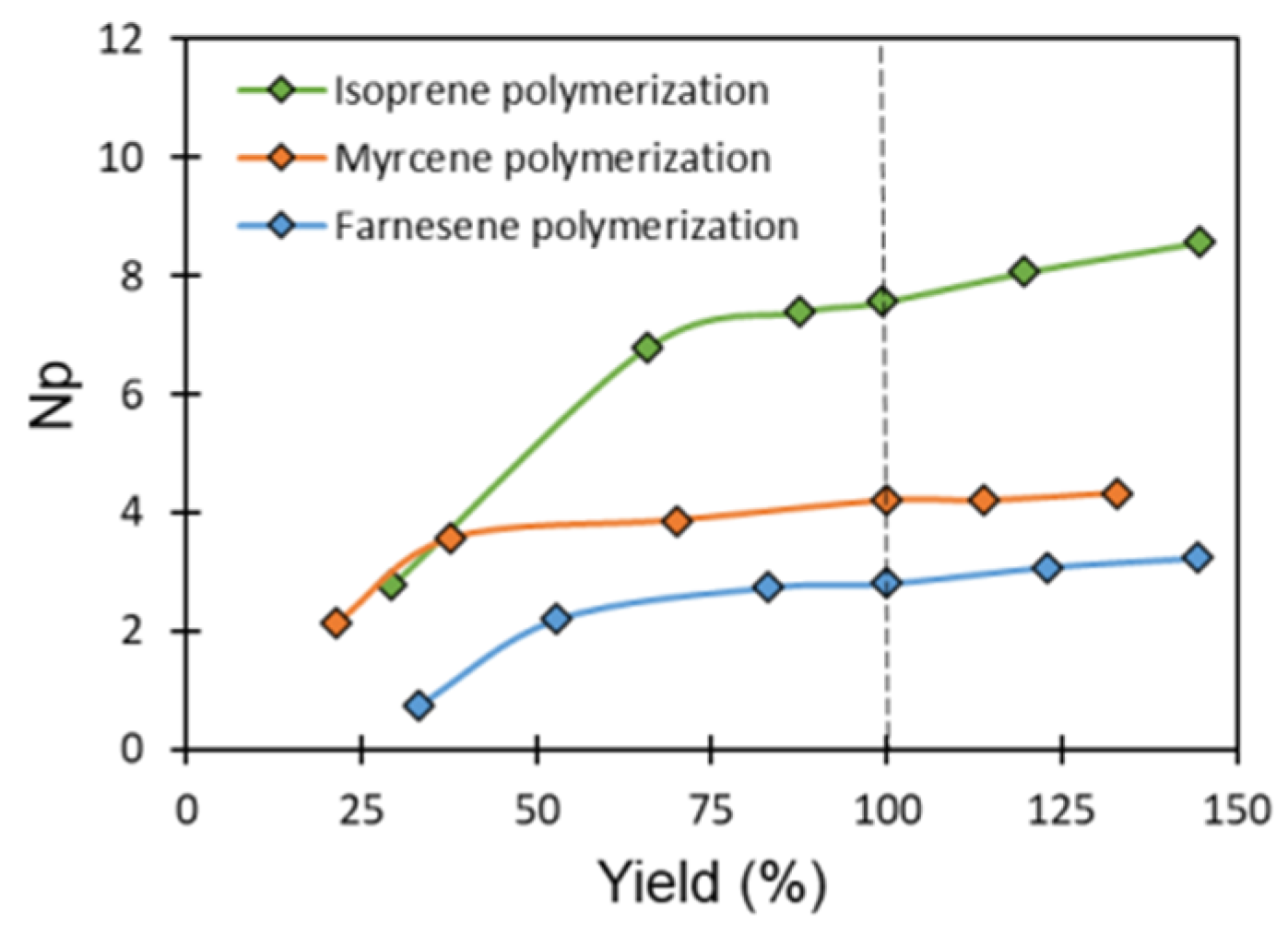
3.4. Validation of CCTP Regime through Living Polymerizations (Sequential Polymerizations)
3.5. Nd-Catalyst: Efficiency and Economy in CCTP
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lei, W.; Zhou, X.; Russell, T.P.; Hua, K.; Yang, X.; Qiao, H.; Wang, W.; Li, F.; Wang, R.; Zhang, L. High performance bio-based elastomers: Energy efficient and sustainable materials for tires. J. Mater. Chem. A 2016, 4, 13058–13062. [Google Scholar] [CrossRef]
- Parcheta, P.S.; Głowińska, E.; Datta, J. Effect of bio-based components on the chemical structure, thermal stability and mechanical properties of green thermoplastic polyurethane elastomers. Eur. Polym. J. 2020, 123, 109422. [Google Scholar] [CrossRef]
- Sarkar, P.; Bhowmick, A.K. Synthesis, characterization and properties of a bio-based elastomer: Polymyrcene. RSC Adv. 2014, 4, 61343–61354. [Google Scholar] [CrossRef]
- Sahu, P.; Bhowmick, A.K.; Kali, G. Terpene Based Elastomers: Synthesis, Properties, and Applications. Processes 2020, 8, 553. [Google Scholar] [CrossRef]
- Sahu, P.; Sarkar, P.; Bhowmick, A.K. Synthesis and Characterization of a Terpene-Based Sustainable Polymer: Poly-alloocimene. ACS Sustain. Chem. Eng. 2017, 5, 7659–7669. [Google Scholar] [CrossRef]
- Behr, A.; Johnen, L. Myrcene as a Natural Base Chemical in Sustainable Chemistry: A Critical Review. ChemSusChem 2009, 2, 1072–1095. [Google Scholar] [CrossRef]
- Lamparelli, D.H.; Paradiso, V.; Monica, F.D.; Proto, A.; Guerra, S.; Giannini, L.; Capacchione, C. Toward More Sustainable Elastomers: Stereoselective Copolymerization of Linear Terpenes with Butadiene. Macromolecules 2020, 53, 1665–1673. [Google Scholar] [CrossRef]
- Della, M.F.; Kleij, A.W. From terpenes to sustainable and functional polymers. Polym. Chem. 2020, 11, 5109–5127. [Google Scholar] [CrossRef]
- Zhu, Y.; Romain, C.; Williams, C.K. Sustainable polymers from renewable resources. Nature 2016, 540, 354–362. [Google Scholar] [CrossRef]
- Yoo, T.; Henning, S.K. Synthesis and Characterization of Farnesene-Based Polymers. Rubber Chem. Technol. 2017, 90, 308–324. [Google Scholar] [CrossRef]
- Singh, A.; Kamal, M. Synthesis and characterization of polylimonene: Polymer of an optically active terpene. J. Appl. Polym. Sci. 2012, 125, 1456–1459. [Google Scholar] [CrossRef]
- Sharma, S.; Srivastava, A.K. Synthesis and characterization of copolymers of limonene with styrene initiated by azobisisobutyronitrile. Eur. Polym. J. 2004, 40, 2235–2240. [Google Scholar] [CrossRef]
- Sharma, S.; Srivastava, A.K. Azobisisobutyronitrile-initiated free-radical copolymerization of limonene with vinyl acetate: Synthesis and characterization. J. Appl. Polym. Sci. 2007, 106, 2689–2695. [Google Scholar] [CrossRef]
- Sharma, S.; Srivastava, A.K. Radical Copolymerization of Limonene with Acrylonitrile: Kinetics and Mechanism. Polym. Plast. Technol. Eng. 2003, 42, 485–502. [Google Scholar] [CrossRef]
- Guiné, R.P.F.; Castro, J.A.A.M. Polymerization of β-pinene with ethylaluminum dichloride (C2H5AlCl2). J. Appl. Polym. Sci. 2001, 82, 2558–2565. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F. Cationic polymerization of β-pinene. J. Polym. Sci. Polym. Chem. Ed. 1984, 22, 673–677. [Google Scholar] [CrossRef]
- Lu, J.; Kamigaito, M.; Sawamoto, M.; Higashimura, T.; Deng, Y.X. Cationic polymerization of β-pinene with the AlCl3/SbCl3 binary catalyst: Comparison with α-pinene polymerization. J. Appl. Polym. Sci. 1996, 61, 1011–1016. [Google Scholar] [CrossRef]
- Lu, J.; Kamigaito, M.; Sawamoto, M.; Higashimura, T.; Deng, Y.X. Living Cationic Isomerization Polymerization of β-Pinene. 1. Initiation with HCl−2-Chloroethyl Vinyl Ether Adduct/TiCl3(OiPr) in Conjunction with nBu4NCl. Macromolecules 1997, 30, 22–26. [Google Scholar] [CrossRef]
- Derdar, H.; Belbachir, M.; Hennaoui, F.; Akeb, M.; Harrane, A. Green Copolymerization of Limonene with β-Pinene Catalyzed by an Eco-Catalyst Maghnite-H+. Polym. Sci. Ser. B 2018, 60, 555–562. [Google Scholar] [CrossRef]
- Brum, F.J.B.; Laux, F.N.; Forte, M.M.C. Synthesis of hydrocarbon polymers by cationic polymerization and their thermal properties. Des. Monomers Polym. 2013, 16, 291–301. [Google Scholar] [CrossRef]
- Jenkins, D.K. Butadiene polymerization with a rare earth compound using a magnesium alkyl cocatalyst: 1. Polymer 1985, 26, 147–151. [Google Scholar] [CrossRef]
- Liu, B.; Sun, G.; Li, S.; Liu, D.; Cui, D. Isoprene Polymerization with Iminophosphonamide Rare-Earth-Metal Alkyl Complexes: Influence of Metal Size on the Regio- and Stereoselectivity. Organometallics 2015, 34, 4063–4068. [Google Scholar] [CrossRef]
- Liu, L.; Song, Y.; Li, H. Carbene polymerization: Characterization of poly(carballyloxycarbene). Polym. Int. 2002, 51, 1047–1049. [Google Scholar] [CrossRef]
- Hollfelder, C.O.; Jende, L.N.; Diether, D. 1,3-Diene Polymerization Mediated by Homoleptic Tetramethylaluminates of the Rare-Earth Metals. Catalysts 2018, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Martins, N.; Bonnet, F.; Visseaux, M. Highly efficient cis-1,4 polymerisation of isoprene using simple homoleptic amido rare earth-based catalysts. Polymer 2014, 55, 5013–5016. [Google Scholar] [CrossRef]
- Göttker, S.I.; Kenyon, P.; Mecking, S. Coordinative Chain Transfer Polymerization of Butadiene with Functionalized Aluminum Reagents. Angew. Chemie. Int. Ed. 2019, 58, 17777–17781. [Google Scholar] [CrossRef]
- Zheng, W.; Yan, N.; Zhu, Y.; Zhao, W.; Zhang, C.; Zhang, X.; Bai, C.; Hu, Y.; Zhang, X. Highly trans-1,4-stereoselective coordination chain transfer polymerization of 1,3-butadiene and copolymerization with cyclic esters by a neodymium-based catalyst system. Polym. Chem. 2015, 6, 6088–6095. [Google Scholar] [CrossRef]
- Wang, F.; Dong, B.; Liu, H.; Guo, J.; Zheng, W.; Zhang, C.; Zhao, L.; Bai, C.; Hu, Y.; Zhang, X. Synthesis of Block Copolymers Containing Polybutadiene Segments by Combination of Coordinative Chain Transfer Polymerization, Ring-Opening Polymerization, and Atom Transfer Radical Polymerization. Macromol. Chem. Phys. 2015, 216, 321–328. [Google Scholar] [CrossRef]
- Tang, Z.; Liang, A.; Liang, H.; Zhao, J.; Xu, L.; Zhang, J. Reversible Coordinative Chain Transfer Polymerization of Butadiene Using a Neodymium Phosphonate Catalyst. Macromol. Res. 2019, 27, 789–794. [Google Scholar] [CrossRef]
- Wang, F.; Liu, H.; Zheng, W.; Guo, J.; Zhang, C.; Zhao, L.; Zhang, H.; Hu, Y.; Bai, C.; Zhang, X. Fully-reversible and semi-reversible coordinative chain transfer polymerizations of 1,3-butadiene with neodymium-based catalytic systems. Polymer 2013, 54, 6716–6724. [Google Scholar] [CrossRef]
- Liu, B.; Cui, D. Regioselective Chain Shuttling Polymerization of Isoprene: An Approach To Access New Materials from Single Monomer. Macromolecules 2016, 49, 6226–6231. [Google Scholar] [CrossRef]
- Fan, C.; Bai, C.; Cai, H.; Dai, Q.; Zhang, X.; Wang, F. Preparation of high cis-1,4 polyisoprene with narrow molecular weight distribution via coordinative chain transfer polymerization. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 4768–4774. [Google Scholar] [CrossRef]
- Loughmari, S.; Hafid, A.; Bouazza, A.; Bouadili, E.A.; Zinck, P.; Visseaux, M. Highly stereoselective coordination polymerization of β-myrcene from a lanthanide-based catalyst: Access to bio-sourced elastomers. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 2898–2905. [Google Scholar] [CrossRef]
- Valente, A.; Zinck, P.; Vitorino, M.J.; Mortreux, A.; Visseaux, M. Rare earths/main group metal alkyls catalytic systems for the 1,4-trans stereoselective coordinative chain transfer polymerization of isoprene. J. Polym. Sci. Part A Polym. Chem. 2010, 48, 4640–4647. [Google Scholar] [CrossRef]
- Liu, B.; Han, B.; Zhang, C.; Li, S.; Sun, G.; Cui, D. Renewable β-myrcene polymerization initiated by lutetium alkyl complexes ligated by imidophosphonamido ligand. Chin. J. Polym. Sci. 2015, 33, 792–796. [Google Scholar] [CrossRef]
- Liu, B.; Li, L.; Sun, G.; Liu, D.; Li, S.; Cui, D. Isoselective 3,4-(co)polymerization of bio-renewable myrcene using NSN-ligated rare-earth metal precursor: An approach to a new elastomer. Chem. Commun. 2015, 51, 1039–1041. [Google Scholar] [CrossRef]
- Liu, B.; Liu, D.; Li, S.; Sun, G.; Cui, D. High trans-1,4 (co)polymerization of β-myrcene and isoprene with an iminophosphonamide lanthanum catalyst. Chin. J. Polym. Sci. 2016, 34, 104–110. [Google Scholar] [CrossRef]
- Díaz de León, R.; López, R.; Valencia, L.; Mendoza, R.; Cabello, J.; Enríquez, J. Towards Bioelastomers via Coordination Polymerization of Renewable Terpenes Using Neodymium-Based Catalyst Systems. Key Eng. Mater. 2018, 779, 115–121. [Google Scholar] [CrossRef]
- Georges, S.; Touré, A.O.; Visseaux, M.; Zinck, P. Coordinative Chain Transfer Copolymerization and Terpolymerization of Conjugated Dienes. Macromolecules 2014, 47, 4538–4547. [Google Scholar] [CrossRef]
- Cavalcante de Sá, M.C.; Córdova, A.M.T.; Díaz de León Gómez, R.E.; Pinto, J.C. Modeling of Isoprene Solution Coordinative Chain Transfer Polymerization. Macromol. React. Eng. 2021, 15, 2100005. [Google Scholar] [CrossRef]
- González-Zapata, J.L.; Enríquez-Medrano, F.J.; López González, H.R.; Revilla-Vázquez, J.; Carrizales, R.M.; Georgouvelas, D.; Valencia, L.; de León Gómez, R.E.D. Introducing random bio-terpene segments to high cis-polybutadiene: Making elastomeric materials more sustainable. RSC Adv. 2020, 10, 44096–44102. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, C.; Hu, Y.; Jia, X.; Bai, C.; Zhang, X. Reversible coordinative chain transfer polymerization of isoprene and copolymerization with ε-caprolactone by neodymium-based catalyst. Polymer 2012, 53, 6027–6032. [Google Scholar] [CrossRef]
- Fischbach, A.; Perdih, F.; Herdtweck, E.; Anwander, R. Structure−Reactivity Relationships in Rare-Earth Metal Carboxylate-Based Binary Ziegler-Type Catalysts. Organometallics 2006, 25, 1626–1642. [Google Scholar] [CrossRef]
- Ubaldo-Alarcón, A.; Soriano-Corral, F.; Córdova, T.; Zapata-González, I.; and Díaz-de-León, R. Terpene Coordinative Chain Transfer Polymerization: Understanding the Process through Kinetic Modeling. Polymers 2022, 14, 2352. [Google Scholar] [CrossRef]
- Shehu, H.; Norazilawati, M.S.; Azizah, M. Synthesis and characterisation of highly branched polyisoprene: Exploiting the “Strathclyde route” in anionic polymerization. RSC Adv. 2018, 8, 11684–11692. [Google Scholar] [CrossRef] [Green Version]
- Monakov, Y.B.; Sabirov, Z.M.; Urazbaev, V.N.; Efimov, V.P. Relationship between the Stereospecificity of Lanthanide Catalysts and the Structures of Active Sites and Dienes, the Nature of a Cocatalyst, and Preparation Conditions. Kinet. Catal. 2001, 42, 310–316. [Google Scholar] [CrossRef]
- Kempe, R. How to Polymerize Ethylene in a Highly Controlled Fashion? Chem. A Eur. J. 2007, 13, 2764–2773. [Google Scholar] [CrossRef]
- Díaz de León, G.R.E.; Enríquez-Medrano, F.J.; Maldonado, T.H.; Mendoza, C.R.; Reyes, A.K.; López, G.H.R.; Olivares, R.J.L.; Lugo, U.L.E. Synthesis and characterization of high cis-polymyrcene using neodymium-based catalysts. Can. J. Chem. Eng. 2016, 94, 823–832. [Google Scholar] [CrossRef]
- Quirk, R.P.; Kells, A.M. Polymerization of butadiene using neodymium versatate-based catalyst systems: Performed catalysts with SiCl4 as halide source. Polym. Int. 2000, 49, 751–756. [Google Scholar] [CrossRef]
- Mendoza, R.; López, R.; Rodríguez, F.; Díaz, A.; Maldonado, H.; Cabello, J.; Enrique, J.; Díaz de León, R. Polymerization of β-myrcene with Neodymium Versatate and Neodymium Isoproxide as Catalysts. In Proceedings of the XV Simposio Latinoamericano de Polímeros—VIII Congreso Iberoamericano de Polímeros, Cancún, Mexico, 23–27 October 2016; p. 433. [Google Scholar]
- Pires, N.M.T.; Coutinho, F.M.B.; Costa, M.A.S. Synthesis and characterization of high cis-polybutadiene: Influence of monomer concentration and reaction temperatura. Eur. Polym. J. 2004, 40, 2599–2603. [Google Scholar] [CrossRef]
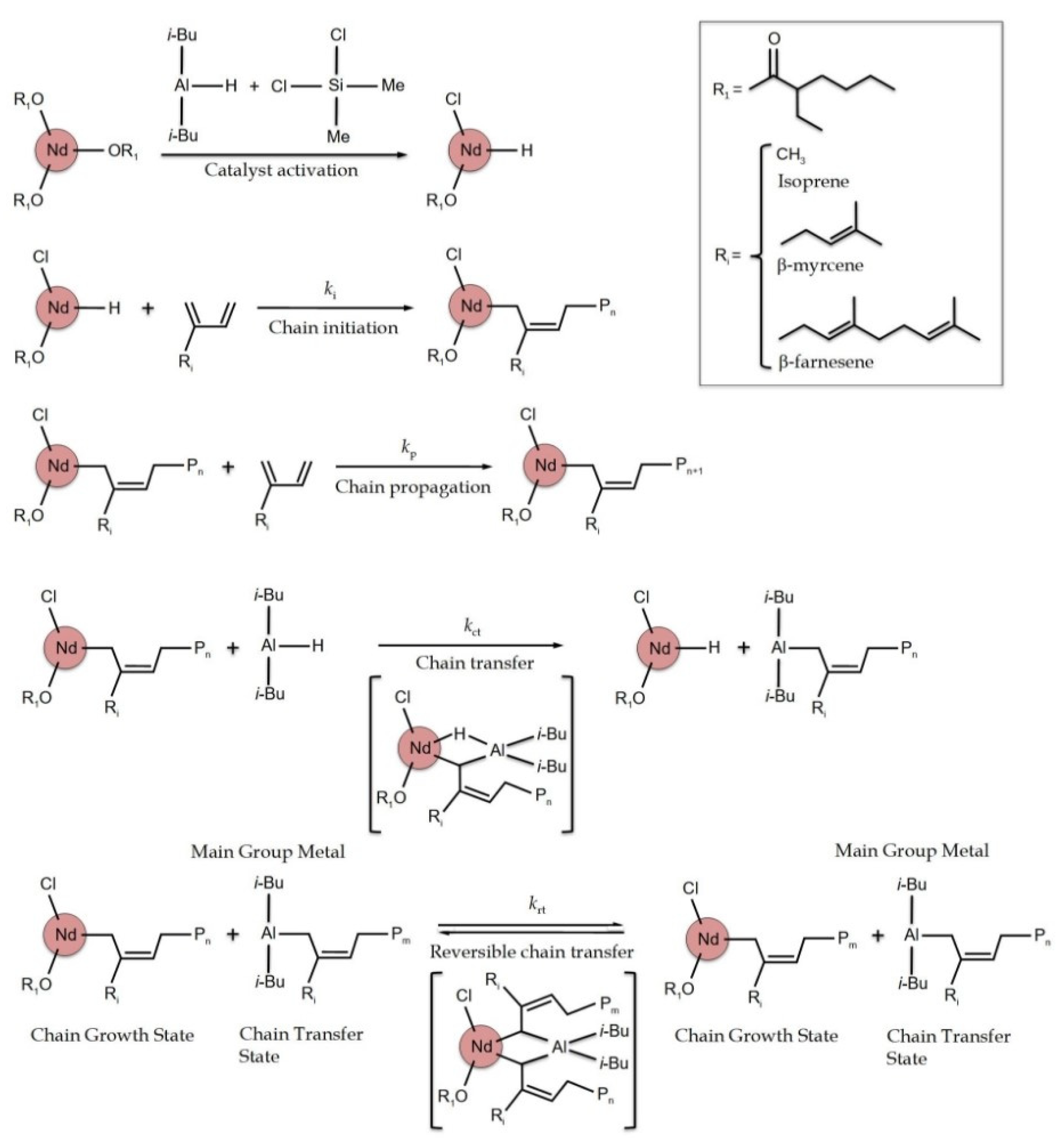


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
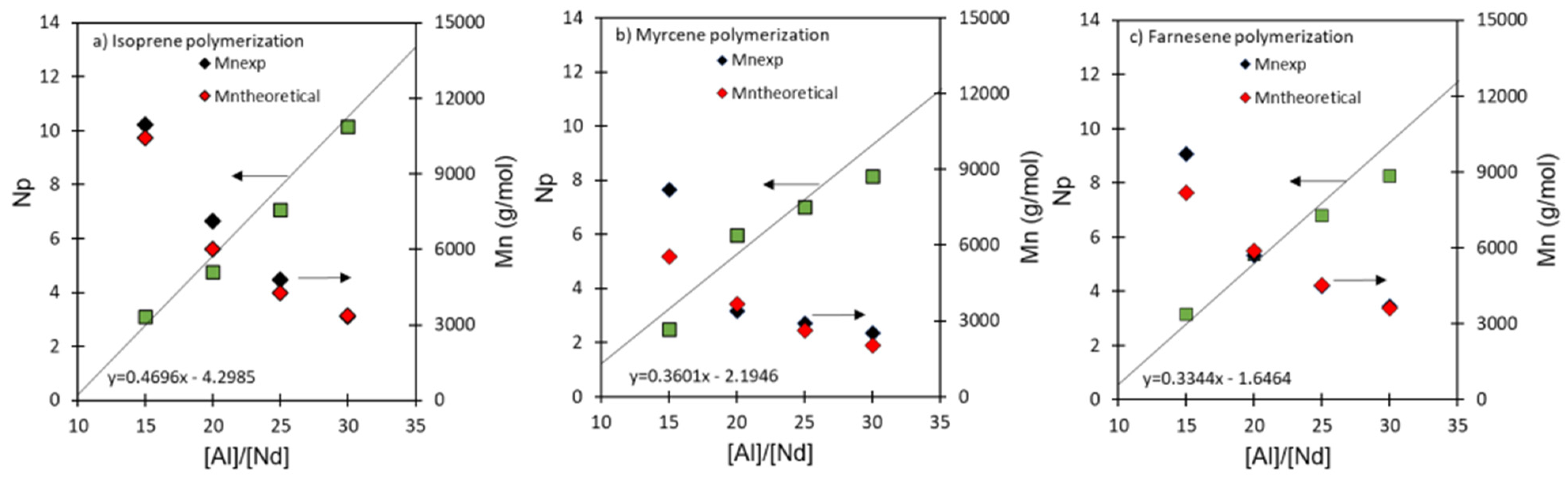{kind=link}
| Entry | [Nd]:[Al]:[Cl] | [Nd]0 (μmol) | Time (min) | Yield a (%) | Ð b | ||
|---|---|---|---|---|---|---|---|
| Ip-1 d | 1:20:01 | 1.73 | 75 | 99 | 2600 | 1.3 | 2.7 |
| Ip-2 d | 1:20:03 | 1.73 | 45 | 93 | 8000 | 1.8 | 2.3 |
| My-1 e | 1:20:01 | 0.15 | 240 | 96 | 2900 | 1.6 | 6.1 |
| My-2 e | 1:20:03 | 0.15 | 240 | 96 | 8300 | 3 | 2.5 |
| Fa-1 e | 1:20:01 | 0.09 | 240 | 99 | 7600 | 2.3 | 4 |
| Fa-2 e | 1:20:03 | 0.09 | 240 | 99 | 24,000 | 4.1 | 1.3 |
| Entry a | [Nd]:[Al]:[Cl] | T (°C) | Time (min) | Yield b (%) | Ð c | Isoprene Structure (%) e | (°C) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3,4 | 1,4-trans | 1,4-cis | |||||||||
| Ip-3 | 1:15:01 | 60 | 90 | 84 | 10,900 | 1.8 | 2.6 | 3 | 15.5 | 81.5 | −68.2 |
| Ip-4 | 1:20:01 | 60 | 90 | 90 | 7100 | 1.5 | 4.1 | 3 | 22.3 | 74.7 | −69.4 |
| Ip-5 | 1:25:01 | 60 | 75 | 92 | 4800 | 1.4 | 6.6 | 3.2 | 27.5 | 69.3 | −69.2 |
| Ip-6 | 1:30:01 | 60 | 60 | 97 | 3300 | 1.4 | 9.9 | 3 | 27 | 70 | −70.5 |
| Ip-7 | 1:15:01 | 70 | 80 | 87 | 9700 | 1.5 | 3.2 | 3 | 26.5 | 70.5 | −66.4 |
| Ip-8 | 1:20:01 | 70 | 80 | 93 | 6400 | 1.4 | 4.9 | 4 | 32 | 64 | −68.3 |
| Ip-9 | 1:25:01 | 70 | 70 | 94 | 3800 | 1.4 | 8.2 | 3 | 34 | 63 | −69.3 |
| Ip-10 | 1:30:01 | 70 | 55 | 96 | 3600 | 1.3 | 8.2 | 3 | 32 | 65 | −70.3 |
| Entry a | [Nd]:[Al]:[Cl] | T (°C) | Time (min) | Yield b (%) | Ð c | β-myrcene Structure (%) e | (°C) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3,4 | 1,4-trans | 1,4-cis | |||||||||
| My-3 | 1:15:1 | 60 | 80 | 85 | 8200 | 1.5 | 2.2 | 6.3 | 3.4 | 90.3 | −68.4 |
| My-4 | 1:20:1 | 60 | 75 | 90 | 4000 | 1.5 | 4.6 | 6.3 | 1.6 | 92.1 | −72.2 |
| My-5 | 1:25:1 | 60 | 60 | 88 | 2900 | 1.8 | 7.3 | 6.1 | 5.6 | 87.4 | −73.7 |
| My-6 | 1:30:1 | 60 | 45 | 95 | 2500 | 1.8 | 6.9 | 9.7 | 16.8 | 73.5 | −78.0 |
| My-7 | 1:15:1 | 70 | 75 | 86 | 7100 | 1.3 | 2.5 | 13.0 | 4.1 | 82.9 | −69.4 |
| My-8 | 1:20:1 | 70 | 65 | 89 | 3400 | 1.5 | 5.2 | 8.0 | 2.3 | 89.7 | −72.1 |
| My-9 | 1:25:1 | 70 | 60 | 93 | 2500 | 1.6 | 7.6 | 8.1 | 8.7 | 83.2 | −74.3 |
| My-10 | 1:30:1 | 70 | 45 | 94 | 2300 | 1.5 | 7.5 | 10.0 | 18.8 | 71.2 | −77.0 |
| Entry a | [Nd]:[Al]:[Cl] | T (°C) | Time (min) | Yield b (%) | Ð c | β-farnesene Structure (%) e | (°C) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3,4 | 1,4-trans | 1,4-cis | |||||||||
| Fa-3 | 1:15:1 | 60 | 60 | 92 | 9700 | 2.9 | 3.2 | 9.4 | 1.2 | 89.4 | −75.1 |
| Fa-4 | 1:20:1 | 60 | 60 | 97 | 5700 | 2.4 | 5.4 | 3.1 | 2.2 | 94.7 | −76.6 |
| Fa-5 | 1:25:1 | 60 | 40 | 99 | 4500 | 2.0 | 6.8 | 3.4 | 4.7 | 91.8 | −76.9 |
| Fa-6 | 1:30:1 | 60 | 30 | 99 | 3700 | 2.0 | 8.3 | 4.7 | 8.5 | 86.8 | −78.4 |
| Fa-7 | 1:15:1 | 70 | 40 | 99 | 8900 | 2.0 | 3.4 | 4.0 | 1.0 | 95.0 | −75.2 |
| Fa-8 | 1:20:1 | 70 | 40 | 99 | 6500 | 2.1 | 4.7 | 3.0 | 1.5 | 95.5 | −77.6 |
| Fa-9 | 1:25:1 | 70 | 30 | 99 | 4800 | 1.9 | 6.4 | 3.8 | 7.2 | 89.0 | −81.6 |
| Fa-10 | 1:30:1 | 70 | 30 | 99 | 3800 | 2.6 | 8.0 | 4.8 | 5.2 | 90.0 | −76.9 |
| Fa-11 g | 1:25:1 | 50 | 85 | 92 | 10,500 | 2.5 | 4.9 | - | - | - | - |
| Fa-12 g | 1:25:1 | 60 | 70 | 96 | 8200 | 1.8 | 6.2 | - | - | - | - |
| Fa-13 g | 1:25:1 | 70 | 60 | 97 | 7800 | 1.7 | 6.6 | - | - | - | - |
| Fa-14 g | 1:25:1 | 80 | 60 | 85 | 7100 | 1.6 | 7.2 | - | - | - | - |
| Entry | T (K) | (L/mol min) | (L/mol min) | ||
|---|---|---|---|---|---|
| Ip-5 | 333 | 7.6 | 5.2 | 4400 | 3.2 |
| Ip-9 | 343 | 9.6 | 9060 | ||
| My-5 | 333 | 8.4 | 6.3 | 2005 | 4.6 |
| My-9 | 343 | 11.1 | 2197 | ||
| Fa-12 | 333 | 20.5 | 10.5 | 3554 | 3.7 |
| Fa-13 | 343 | 33.3 | 4188 |
| First Step | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Time (min) | Yield (%) | Đ a | (g/mol) | (L/mol-min) | (L/mol-min) | |
| Ip-11 | 30 | 99 | 1.4 | 2600 | 36,700 | 37.2 | 7.5 |
| My-11 | 95 | 99 | 1.5 | 8000 | 9970 | 21.6 | 4.1 |
| Fa-15 | 95 | 99 | 1.6 | 17,900 | 8150 | 39 | 2.8 |
| Second Step | |||||||
| Entry | Time (min) | Yield (%) | Đ a | a (g/mol) | (L/mol-min) | (L/mol-min) | b |
| Ip-11 | 80 | 144 | 1.3 | 3800 | 9080 | 6.0 | 8.5 |
| My-11 | 150 | 132 | 1.5 | 10,700 | 2300 | 5.1 | 4.5 |
| Fa-15 | 135 | 144 | 1.5 | 22,200 | 3710 | 16.4 | 3.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Córdova, T.; Enríquez-Medrano, F.J.; Cartagena, E.M.; Villanueva, A.B.; Valencia, L.; Álvarez, E.N.C.; González, R.L.; Díaz-de-León, R. Coordinative Chain Transfer Polymerization of Sustainable Terpene Monomers Using a Neodymium-Based Catalyst System. Polymers 2022, 14, 2907. https://doi.org/10.3390/polym14142907
Córdova T, Enríquez-Medrano FJ, Cartagena EM, Villanueva AB, Valencia L, Álvarez ENC, González RL, Díaz-de-León R. Coordinative Chain Transfer Polymerization of Sustainable Terpene Monomers Using a Neodymium-Based Catalyst System. Polymers. 2022; 14(14):2907. https://doi.org/10.3390/polym14142907
Chicago/Turabian StyleCórdova, Teresa, Francisco Javier Enríquez-Medrano, Eduardo Martínez Cartagena, Arnulfo Banda Villanueva, Luis Valencia, Edgar Nazareo Cabrera Álvarez, Ricardo López González, and Ramón Díaz-de-León. 2022. "Coordinative Chain Transfer Polymerization of Sustainable Terpene Monomers Using a Neodymium-Based Catalyst System" Polymers 14, no. 14: 2907. https://doi.org/10.3390/polym14142907
APA StyleCórdova, T., Enríquez-Medrano, F. J., Cartagena, E. M., Villanueva, A. B., Valencia, L., Álvarez, E. N. C., González, R. L., & Díaz-de-León, R. (2022). Coordinative Chain Transfer Polymerization of Sustainable Terpene Monomers Using a Neodymium-Based Catalyst System. Polymers, 14(14), 2907. https://doi.org/10.3390/polym14142907