
Fabrication of Tissue-Engineered Cartilage Using Decellularized Scaffolds and Chondrocytes

Abstract
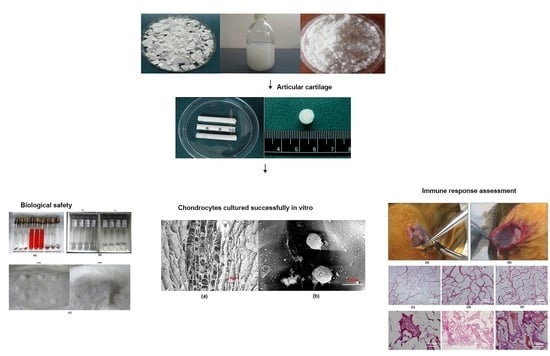
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
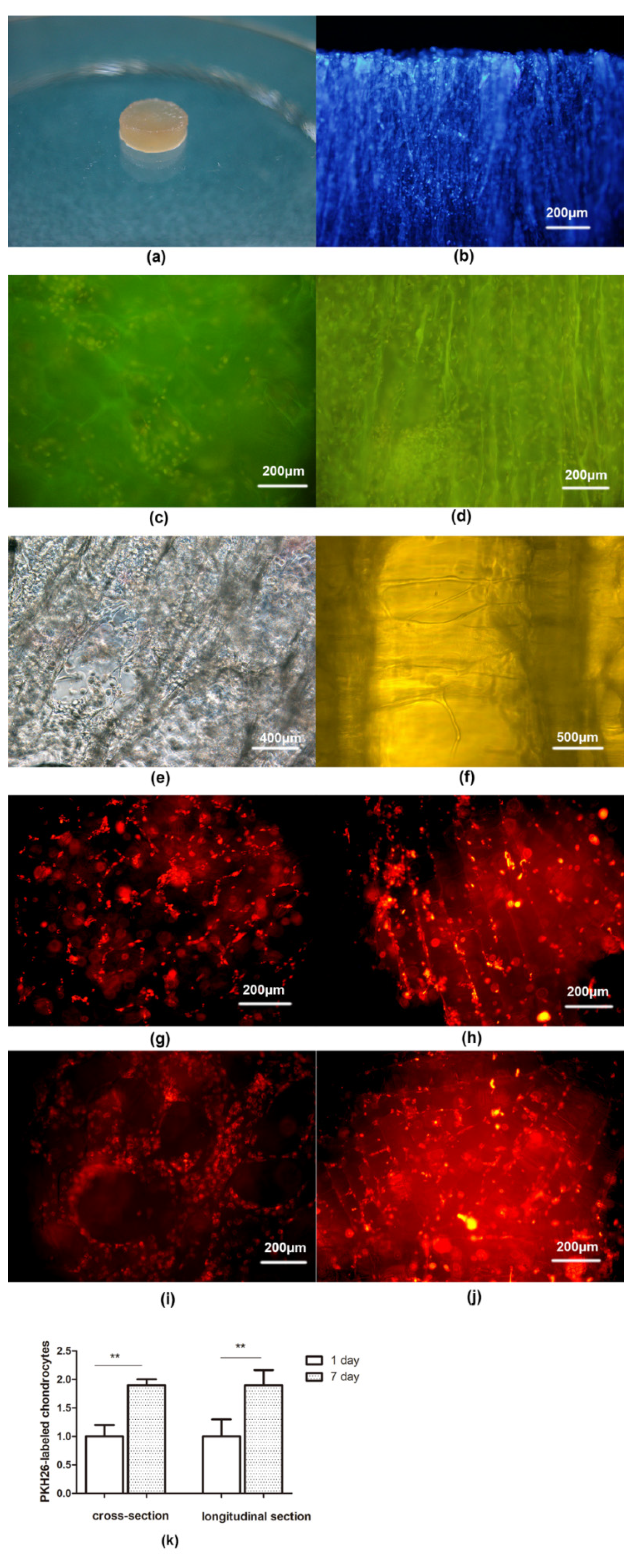
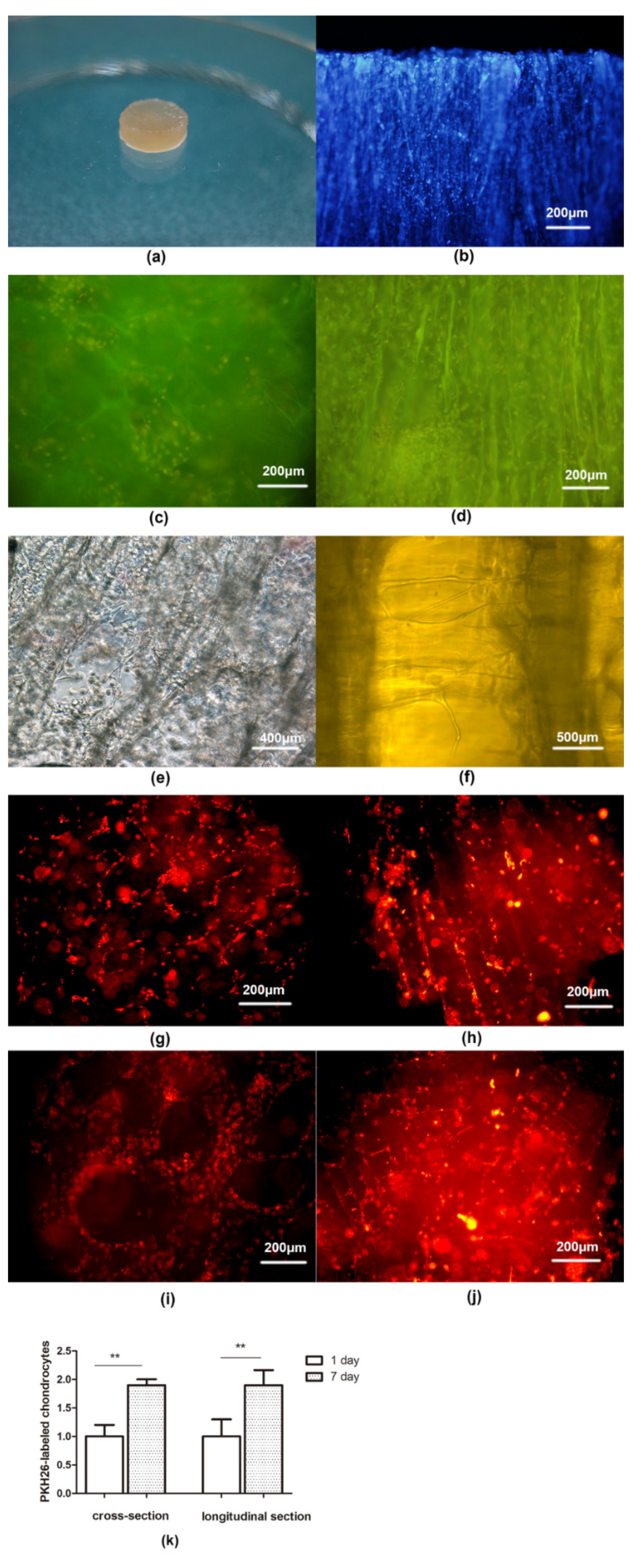{kind=link}
{kind=link}
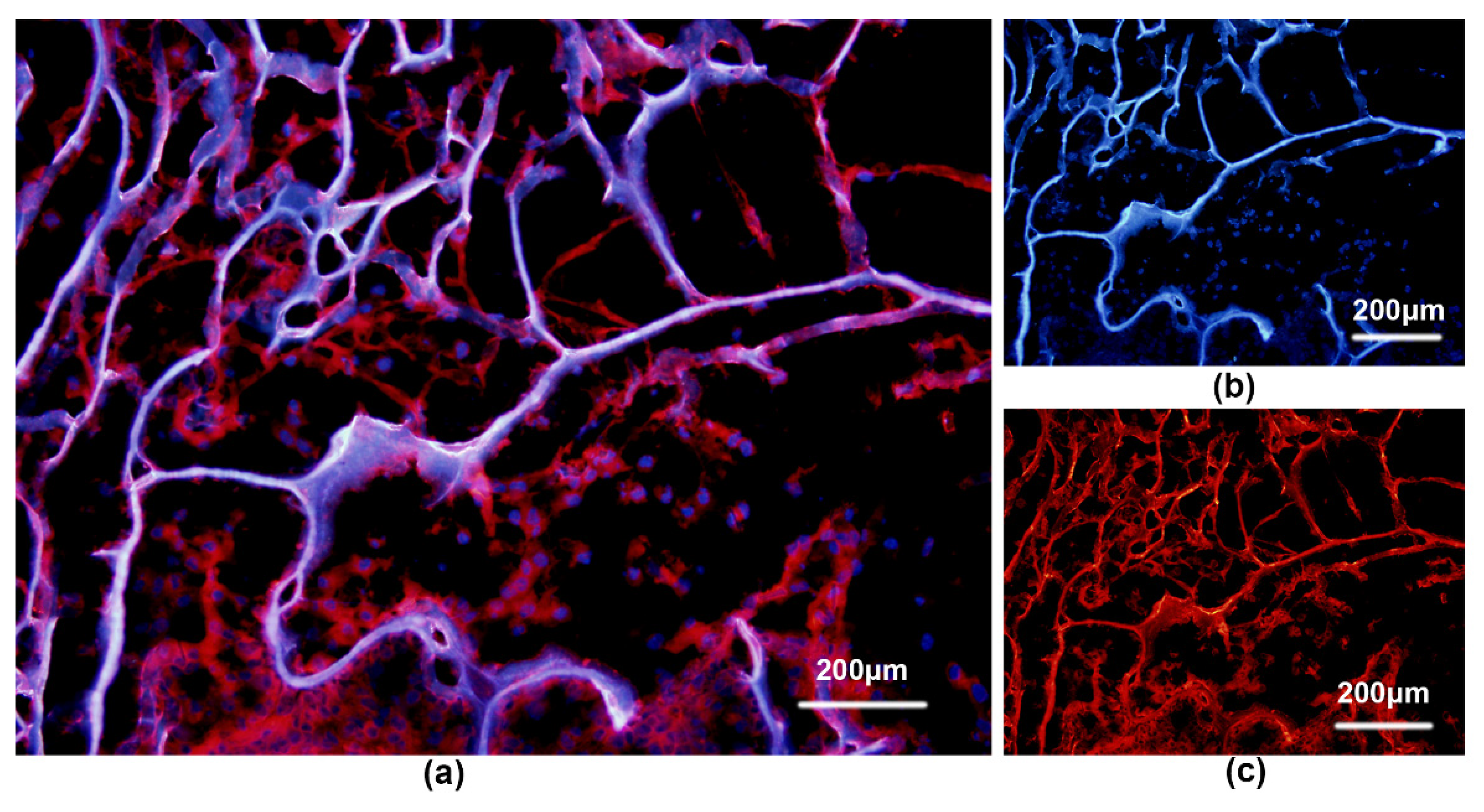{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals and Samples
2.2. Preparation of ACECM Scaffolds
2.3. Observation of ACECM and Decellularized Cartilage Scaffold
2.4. Biological Safety Analysis of ACECM Scaffold
2.5. Isolation and Culture of Porcine AC Chondrocytes
2.6. Inoculation of Porcine Articular Chondrocytes on Scaffolds
2.7. Examination of Cell–Scaffold Complexes
2.8. Analysis of Immunoreaction of ACECM Scaffolds
2.9. Statistical Analysis
3. Results
3.1. Characterization of ECM Slurry of AC
3.2. Characterization of Scaffolds
3.3. Characterization of Chondrocytes
3.4. Characterization and Biological Safety of ACECM-Derived Scaffolds
3.5. Immunoreaction of ACECM Scaffolds
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McCreery, K.P.; Calve, S.; Neu, C.P. Ontogeny informs regeneration: Explant models to investigate the role of the extracellular matrix in cartilage tissue assembly and development. Connect. Tissue Res. 2020, 61, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Ujam, A.B. Cartilage Tissue Engineering for Rhinoplasty; UCL (University College London): London, UK, 2020. [Google Scholar]
- Mall, N.; Harris, J.; Cole, B. Clinical Evaluation and Preoperative Planning of Articular Cartilage Lesions of the Knee. J. Am. Acad. Orthop. Surg. 2015, 23, 633–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, T.J.; Macgillivray, J.D. The technique of microfracture for the treatment of articular cartilage defects in the knee. Oper. Tech. Orthop. 2001, 11, 105–107. [Google Scholar] [CrossRef]
- Dou, Z.; Muder, D.; Baroncelli, M.; Bendre, A.; Gkourogianni, A.; Ottosson, L.; Vedung, T.; Nilsson, O. Rat perichondrium transplanted to articular cartilage defects forms articular-like, hyaline cartilage. Bone 2021, 151, 116035. [Google Scholar] [CrossRef]
- Zhao, Z. Research progress of osteochondral composite scaffolds in tissue engineering cartilage repair. J. Clin. Nurs. Res. 2019, 3, 5. [Google Scholar] [CrossRef]
- Simmons, P.; McElroy, T.; Allen, A.R. A bibliometric review of artificial extracellular matrices based on tissue engineering technology literature: 1990 through 2019. Materials 2020, 13, 2891. [Google Scholar] [CrossRef]
- Kwon, S.G.; Kwon, Y.W.; Lee, T.W.; Park, G.T.; Kim, J.H. Recent advances in stem cell therapeutics and tissue engineering strategies. Biomater. Res. 2018, 22, 36. [Google Scholar] [CrossRef] [Green Version]
- Eltom, A.; Zhong, G.; Muhammad, A. Scaffold techniques and designs in tissue engineering functions and purposes: A review. Adv. Mater. Sci. Eng. 2019, 2019, 3429527. [Google Scholar] [CrossRef] [Green Version]
- Rotter, N.; Aigner, J.; Naumann, A.; Hammer, C.; Sittinger, M. Behavior of tissue-engineered human cartilage after transplantation into nude mice. J. Mater. Sci. Mater. Med. 1999, 10, 689–693. [Google Scholar] [CrossRef]
- Jacob, J.; More, N.; Mounika, C.; Gondaliya, P.; Kalia, K.; Kapusetti, G. Smart Piezoelectric Nanohybrid of Poly(3-hydroxybutyrate-co-3-hydroxyvalerate) and Barium Titanate for Stimulated Cartilage Regeneration. ACS Appl. Bio Mater. 2019, 2, 4922–4931. [Google Scholar] [CrossRef]
- Jacob, J.; More, N.; Kalia, K.; Kapusetti, G. Piezoelectric smart biomaterials for bone and cartilage tissue engineering. Inflamm. Regen. 2018, 38, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.-G.; Tang, R.-F.; Qi, Y.-Y.; Chen, W.-P.; Xiong, Y.; Wu, L.-D. Restoration of cartilage defects using a superparamagnetic iron oxide-labeled adipose-derived mesenchymal stem cell and TGF-β3-loaded bilayer PLGA construct. Regen. Med. 2020, 16, 1735–1747. [Google Scholar] [CrossRef] [PubMed]
- Salati, M.; Khazai, J.; Tahmuri, A.; Samadi, A.; Taghizadeh, A.; Taghizadeh, M.; Zarrintaj, P.; Ramsey, J.; Habibzadeh, S.; Seidi, F.; et al. Agarose-Based Biomaterials: Opportunities and Challenges in Cartilage Tissue Engineering. Polymers 2020, 12, 1150. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.E.G.; Lardy, B.; Bossard, F.; Martínez, F.A.S.; Rinaudo, M. Chitosan based biomaterials for cartilage tissue engineering: Chondrocyte adhesion and proliferation. Food Hydrocoll. Health 2021, 1, 100018. [Google Scholar] [CrossRef]
- Jang, C.H.; Koo, Y.; Kim, G. ASC/chondrocyte-laden alginate hydrogel/PCL hybrid scaffold fabricated using 3D printing for auricle regeneration. Carbohydr. Polym. 2020, 248, 116776. [Google Scholar] [CrossRef]
- Massimino, L.C.; da Conceição Amaro Martins, V.; Vulcani, V.A.S.; de Oliveira, É.L.; Andreeta, M.B.; Bonagamba, T.J.; Klingbeil, M.F.G.; Mathor, M.B.; de Guzzi Plepis, A.M. Use of collagen and auricular cartilage in bioengineering: Scaffolds for tissue regeneration. Cell Tissue Bank. 2020, 5, 1–12. [Google Scholar] [CrossRef]
- Wang, X.; Wang, G.; Zingales, S.; Zhao, B. Biomaterials enabled cell-free strategies for endogenous bone regeneration. Tissue Eng. Part B Rev. 2018, 24, 463–481. [Google Scholar] [CrossRef]
- Goldberg-Bockhorn, E.; Wenzel, U.; Theodoraki, M.; Döscher, J.; Riepl, R.; Wigand, M.; Brunner, C.; Heßling, M.; Hoffmann, T.; Kern, J.; et al. Enhanced cellular migration and prolonged chondrogenic differentiation in decellularized cartilage scaffolds under dynamic culture conditions. J. Tissue Eng. Regen. Med. 2022, 16, 36–50. [Google Scholar] [CrossRef]
- Gong, D.; Yu, F.; Zhou, M.; Dong, W.; Yan, D.; Zhang, S.; Yan, Y.; Wang, H.; Tan, Y.; Chen, Y. Ex vivo and in vivo properties of an injectable hydrogel derived from acellular ear cartilage extracellular matrix. Front. Bioeng. Biotechnol. 2021, 9, 740635. [Google Scholar] [CrossRef]
- Yao, J.; Lu, S.B.; Peng, J.; Guo, Q.Y.; Zhang, L.; Huang, J.X.; Wang, A.Y.; Xu, W.J. Preparation of Articular Cartilage Extracellular Matrix Derived Oriented Scaffold for Cartilage Tissue Engineering. Zhongguo Zuzhi Gongcheng Yanjiu 2009, 13, 432–436. [Google Scholar]
- Zhang, J.; Lu, S.; Huang, J.; Yuan, M.; Zhao, B.; Sun, M.; Cui, X. Preparation of acellular matrix of human articular cartilage. Orthop. J. China 2005, 13, 276–277. [Google Scholar]
- Zhang, J.; Liu, Z.; Li, Y.; You, Q.; Yang, J.; Jin, Y.; Zou, G.; Tang, J.; Ge, Z.; Liu, Y. FGF2: A key regulator augmenting tendon-to-bone healing and cartilage repair. Regen. Med. 2020, 15, 2129–2142. [Google Scholar] [CrossRef] [PubMed]
- Won Lee, G.; Thangavelu, M.; Joung Choi, M.; Yeong Shin, E.; Sol Kim, H.; Seon Baek, J.; Woon Jeong, Y.; Eun Song, J.; Carlomagno, C.; Miguel Oliveira, J. Exosome mediated transfer of miRNA-140 promotes enhanced chondrogenic differentiation of bone marrow stem cells for enhanced cartilage repair and regeneration. J. Cell. Biochem. 2020, 121, 3642–3652. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yang, X.; Feng, Z.; Wang, P.; Zhu, W.; Cui, S. Catalase enhances viability of human chondrocytes in culture by reducing reactive oxygen species and counteracting tumor necrosis factor-α-induced apoptosis. Cell. Physiol. Biochem. 2018, 49, 2427–2442. [Google Scholar] [CrossRef] [PubMed]
- Boumediene, K.; Hammad, M.; Dugué, J.; Veyssière, A.; Baugé, C. Tissue engineering of different cartilage types: A review of different approaches and recent advances. Int. J. Adv. Med. Biotechnol.—IJAMB 2019, 2, 14–16. [Google Scholar] [CrossRef] [Green Version]
- Moutos, F.T.; Freed, L.E.; Guilak, F. A biomimetic three-dimensional woven composite scaffold for functional tissue engineering of cartilage. Nat. Mater. 2007, 6, 162. [Google Scholar] [CrossRef]
- Manzari-Tavakoli, A.; Tarasi, R.; Sedghi, R.; Moghimi, A.; Niknejad, H. Fabrication of nanochitosan incorporated polypyrrole/alginate conducting scaffold for neural tissue engineering. Sci. Rep. 2020, 10, 22012. [Google Scholar] [CrossRef]
- Irawan, V.; Sung, T.-C.; Higuchi, A.; Ikoma, T. Collagen scaffolds in cartilage tissue engineering and relevant approaches for future development. Tissue Eng. Regen. Med. 2018, 15, 673–697. [Google Scholar] [CrossRef]
- Yue, H.; Pathak, J.L.; Zou, R.; Qin, L.; Liao, T.; Hu, Y.; Kuang, W.; Zhou, L. Fabrication of chondrocytes/chondrocyte-microtissues laden fibrin gel auricular scaffold for microtia reconstruction. J. Biomater. Appl. 2021, 35, 838–848. [Google Scholar] [CrossRef]
- Li, T.-T.; Zhang, Y.; Ren, H.-T.; Peng, H.-K.; Lou, C.-W.; Lin, J.-H. Two-step strategy for constructing hierarchical pore structured chitosan–hydroxyapatite composite scaffolds for bone tissue engineering. Carbohydr. Polym. 2021, 260, 117765. [Google Scholar] [CrossRef]
- Yazdani, M.; Shahdadfar, A.; Jackson, C.J.; Utheim, T.P. Hyaluronan-based hydrogel scaffolds for limbal stem cell transplantation: A review. Cells 2019, 8, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beninatto, R.; Barbera, C.; De Lucchi, O.; Borsato, G.; Serena, E.; Guarise, C.; Pavan, M.; Luni, C.; Martewicz, S.; Galesso, D. Photocrosslinked hydrogels from coumarin derivatives of hyaluronic acid for tissue engineering applications. Mater. Sci. Eng. C 2019, 96, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Sachlos, E.; Gotora, D.; Czernuszka, J.T. Collagen scaffolds reinforced with biomimetic composite nano-sized carbonate-substituted hydroxyapatite crystals and shaped by rapid prototyping to contain internal microchannels. Tissue Eng. 2006, 12, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Statham, P.; Jones, E.; Jennings, L.M.; Fermor, H.L. Reproducing the biomechanical environment of the chondrocyte for cartilage tissue engineering. Tissue Eng. Part B Rev. 2022, 28, 405–420. [Google Scholar] [CrossRef]
- Ogawa, R.; Mizuno, H.; Watanabe, A.; Migita, M.; Shimada, T.; Hyakusoku, H. Osteogenic and chondrogenic differentiation by adipose-derived stem cells harvested from GFP transgenic mice. Biochem. Biophys. Res. Commun. 2004, 313, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Antmen, E.; Vrana, N.; Hasirci, V. The role of biomaterials and scaffolds in immune responses in regenerative medicine: Macrophage phenotype modulation by biomaterial properties and scaffold architectures. Biomater. Sci. 2021, 9, 8090–8110. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hu, C.; Feng, Y.; Li, D.; Ai, T.; Huang, Y.; Chen, X.; Huang, L.; Tan, J. Osteoimmunomodulatory effects of biomaterial modification strategies on macrophage polarization and bone regeneration. Regen. Biomater. 2020, 7, 233–245. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, L.; Shang, X.; Liu, B.; Chen, W.; Zhang, Y.; Liu, S.; Sui, X.; Wang, A.; Guo, Q. Fabrication of Tissue-Engineered Cartilage Using Decellularized Scaffolds and Chondrocytes. Polymers 2022, 14, 2848. https://doi.org/10.3390/polym14142848
Lu L, Shang X, Liu B, Chen W, Zhang Y, Liu S, Sui X, Wang A, Guo Q. Fabrication of Tissue-Engineered Cartilage Using Decellularized Scaffolds and Chondrocytes. Polymers. 2022; 14(14):2848. https://doi.org/10.3390/polym14142848
Chicago/Turabian StyleLu, Liang, Xifu Shang, Bin Liu, Weijian Chen, Yu Zhang, Shuyun Liu, Xiang Sui, Aiyuan Wang, and Quanyi Guo. 2022. "Fabrication of Tissue-Engineered Cartilage Using Decellularized Scaffolds and Chondrocytes" Polymers 14, no. 14: 2848. https://doi.org/10.3390/polym14142848
APA StyleLu, L., Shang, X., Liu, B., Chen, W., Zhang, Y., Liu, S., Sui, X., Wang, A., & Guo, Q. (2022). Fabrication of Tissue-Engineered Cartilage Using Decellularized Scaffolds and Chondrocytes. Polymers, 14(14), 2848. https://doi.org/10.3390/polym14142848