
Terpene Coordinative Chain Transfer Polymerization: Understanding the Process through Kinetic Modeling

 , ,
, , 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Catalytic System
2.3. Polymerization System
2.4. Polymerizations
2.5. Determination of Conversion and Molecular Weight
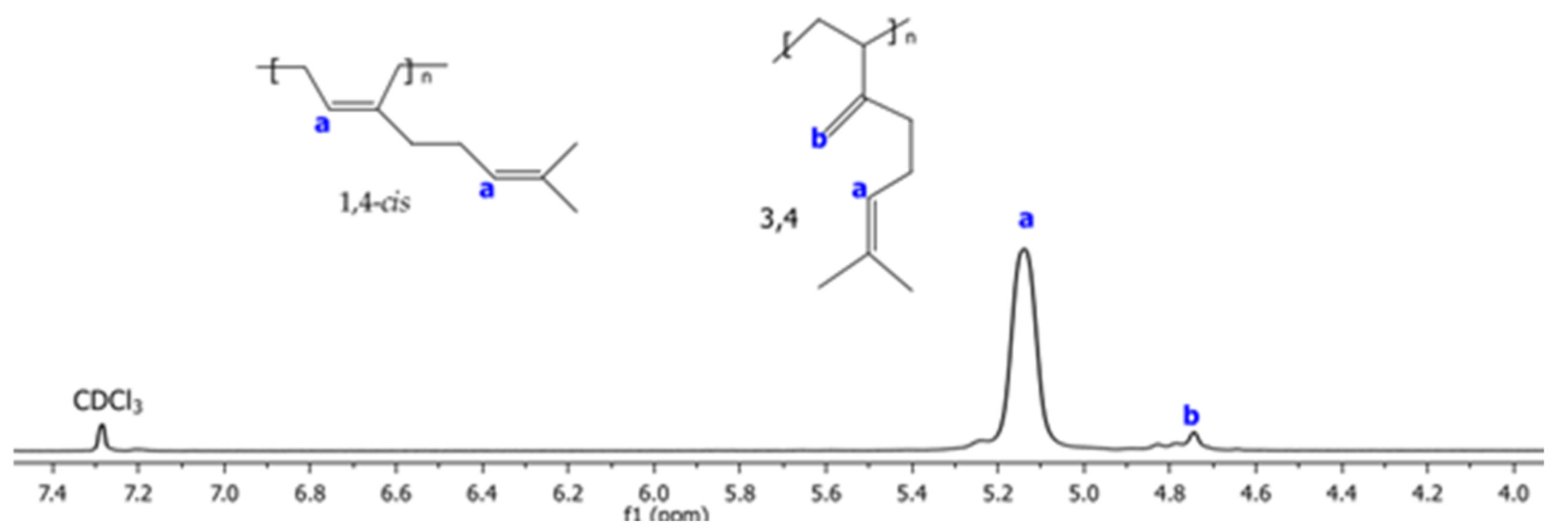
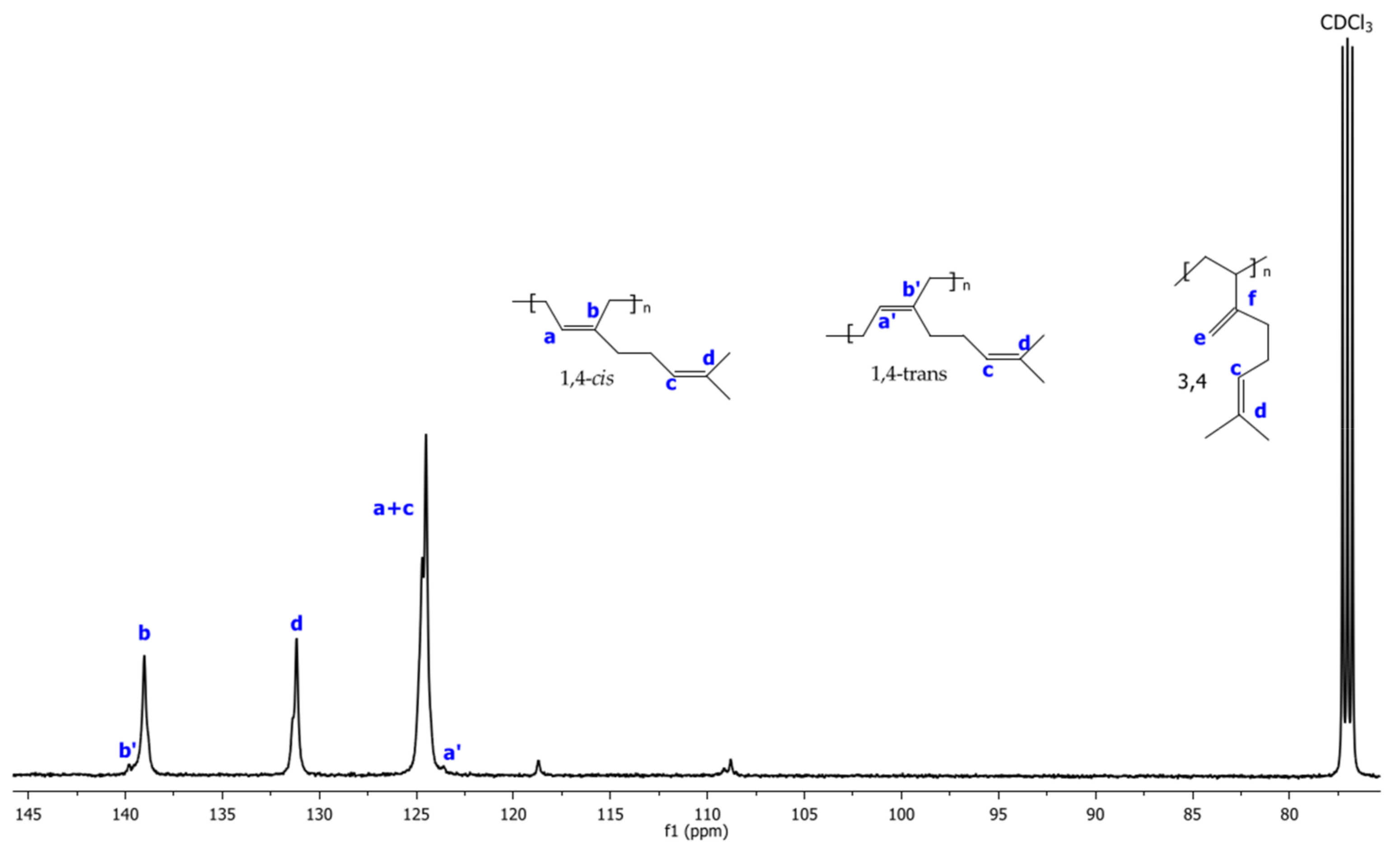
2.6. 1H and 13C NMR
3. Mathematical Modeling
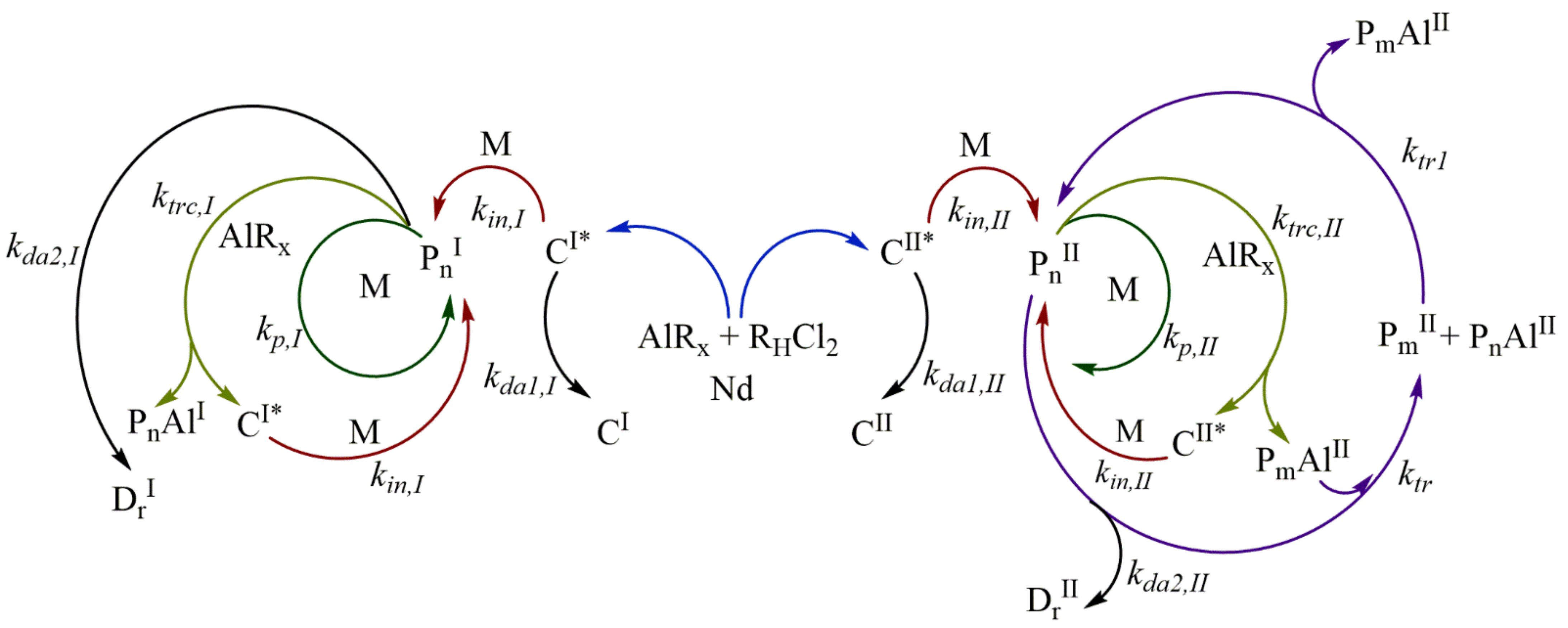
3.1. Reaction Mechanism
3.2. Population Balance Equations (PBEs)
3.3. Method of Moments
3.4. Optimization Strategy for the Parameter Estimation
3.5. Numerical Aspects and Equipment
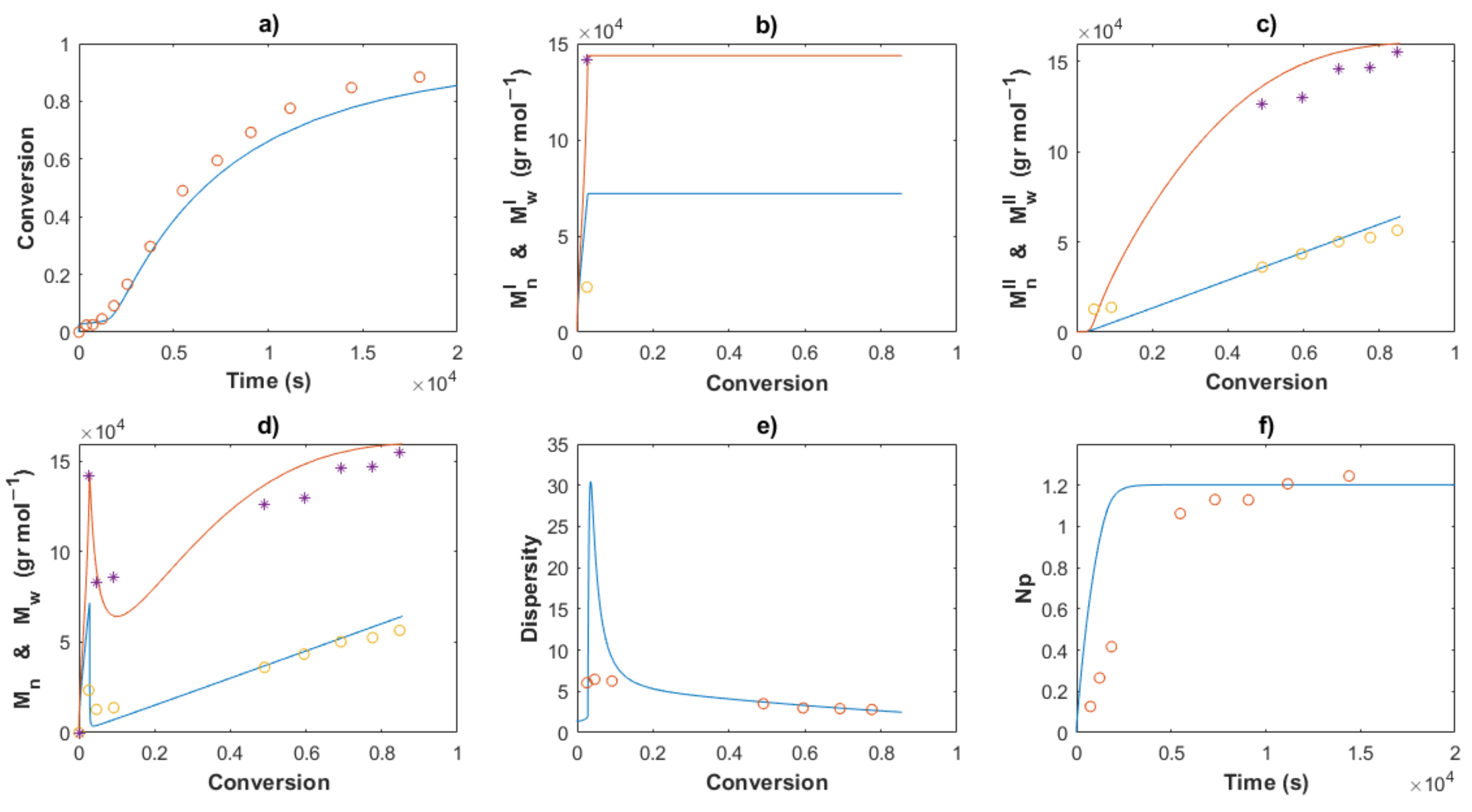
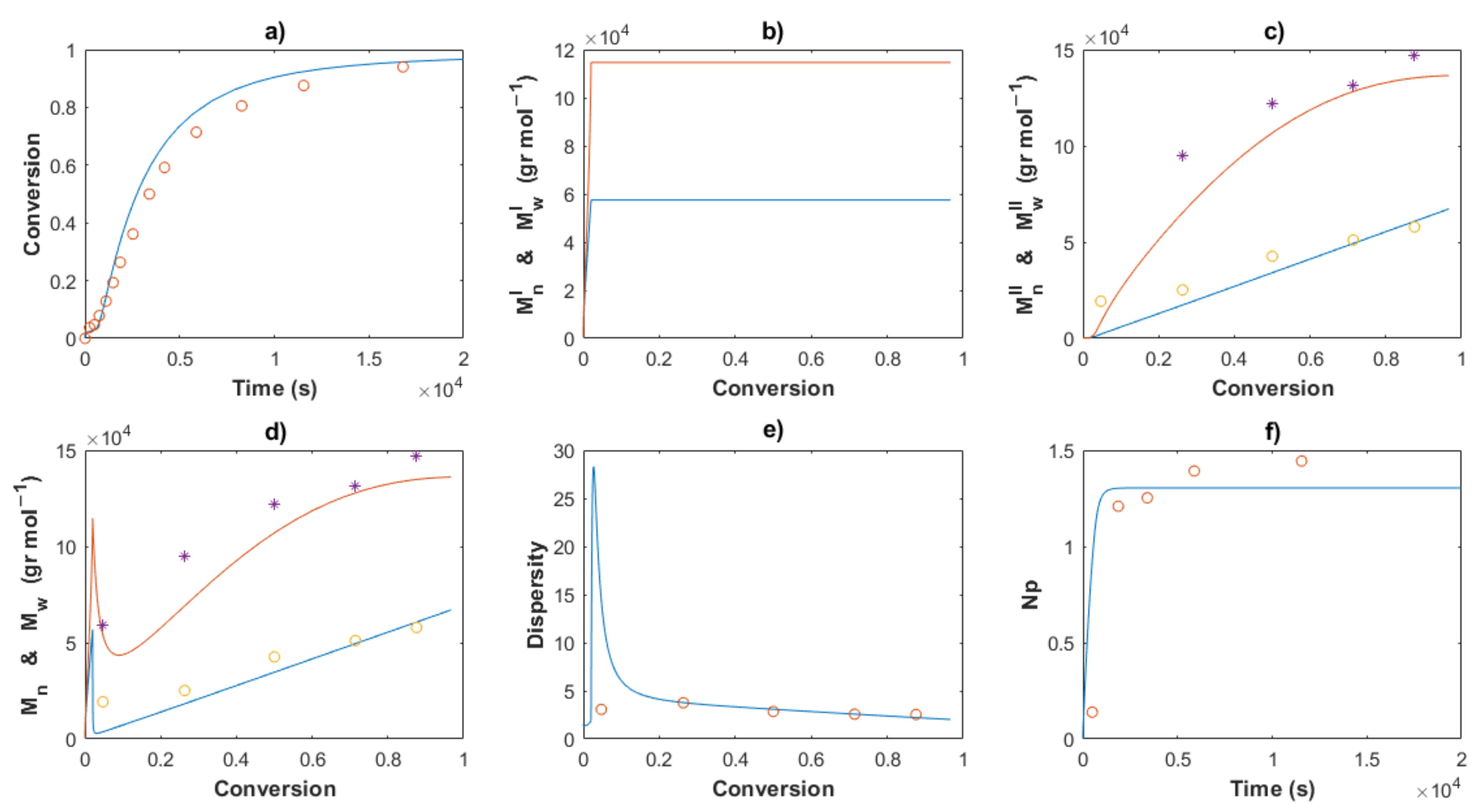
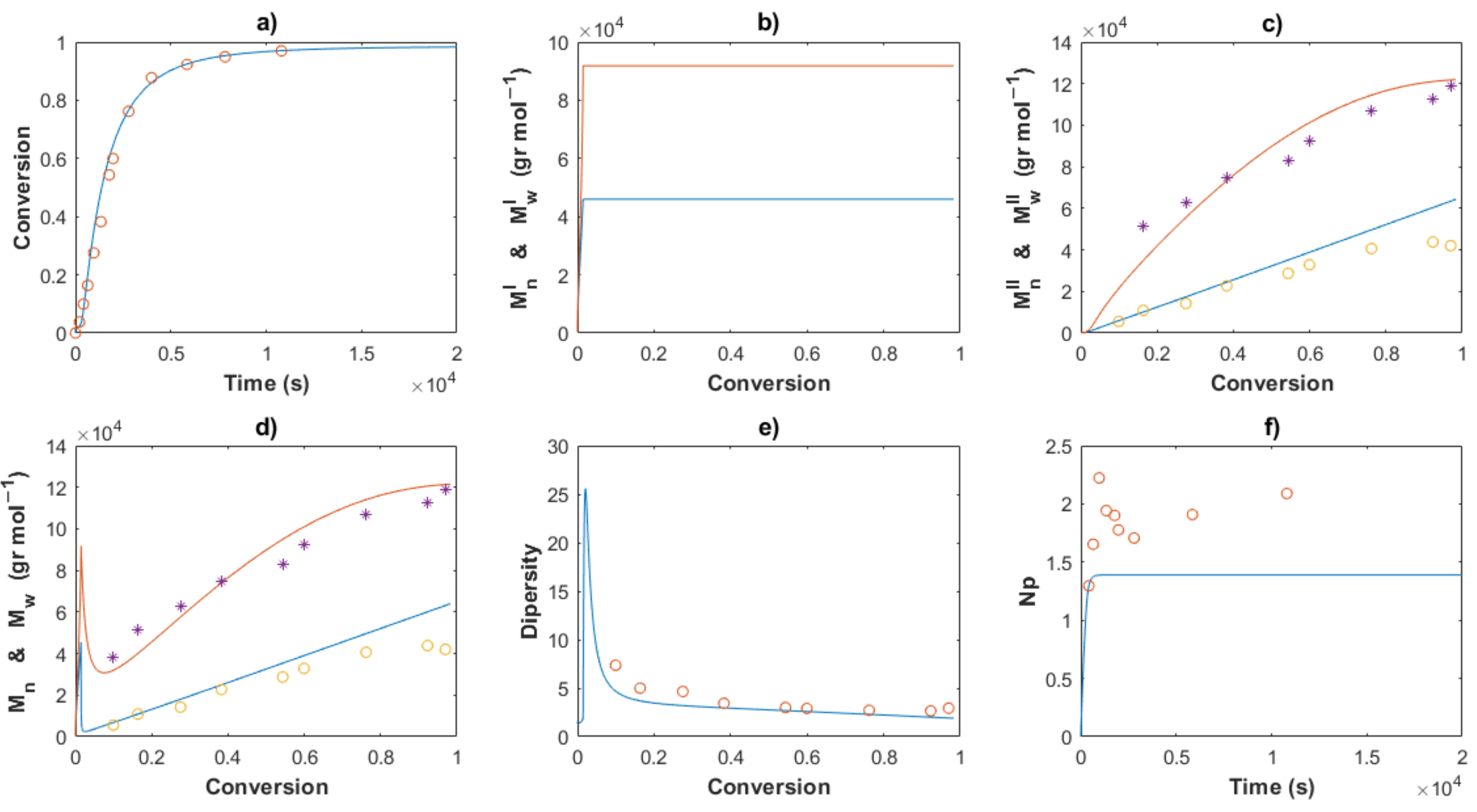
4. Results and Discussion
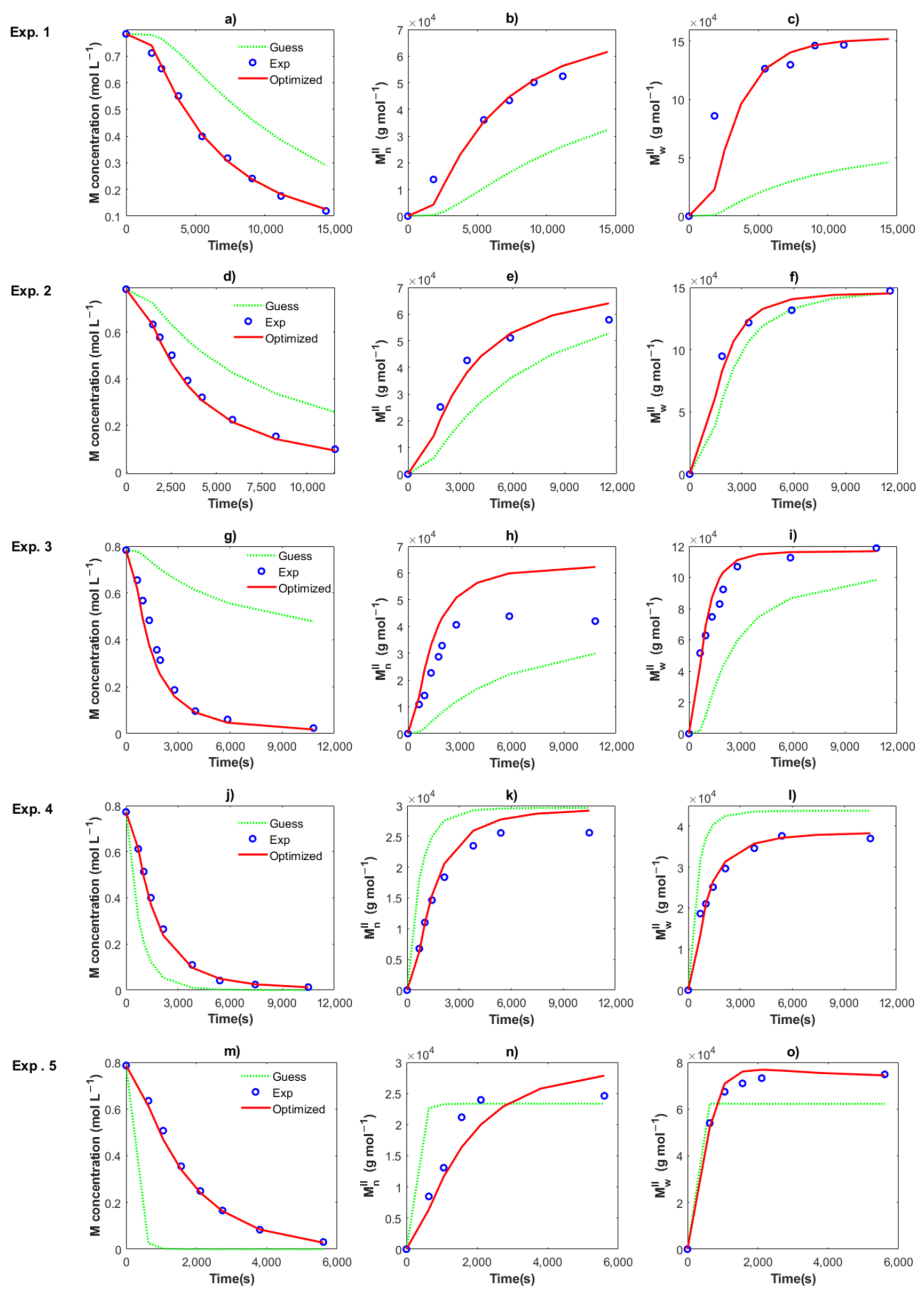
4.1. Polymerizations
4.2. Modeling and Simulations
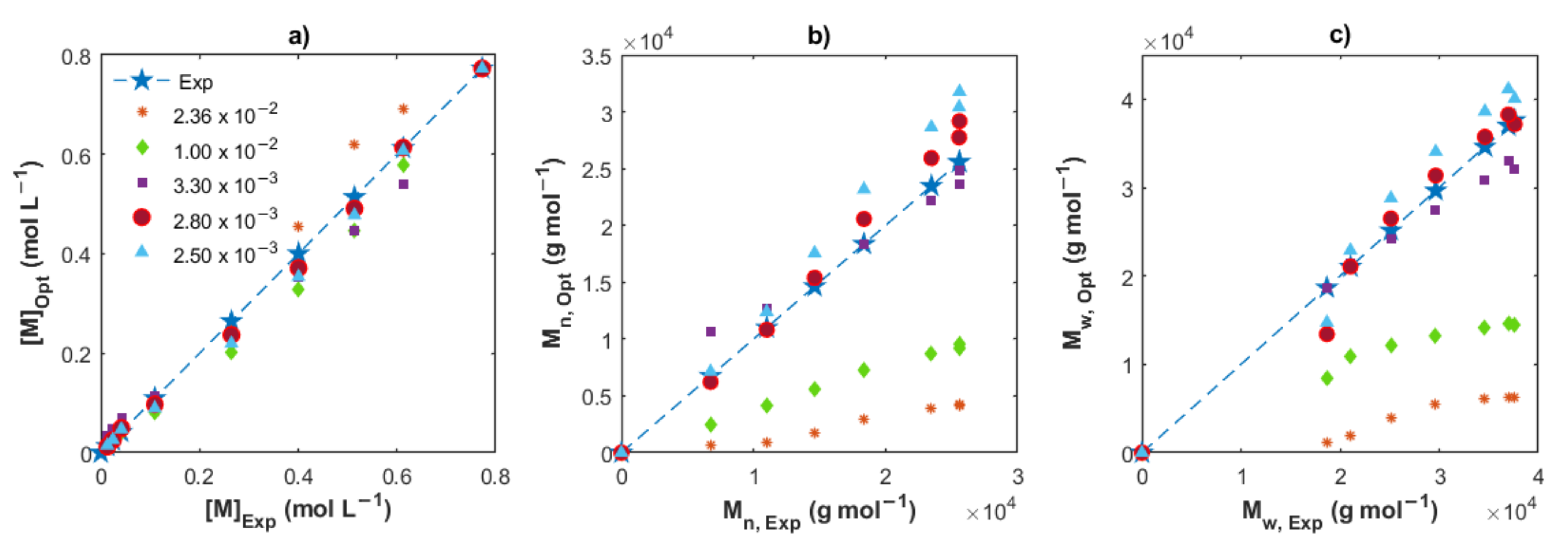
4.2.1. Estimation of [AlRx]0
4.2.2. Estimation of Kinetic Rate Constants
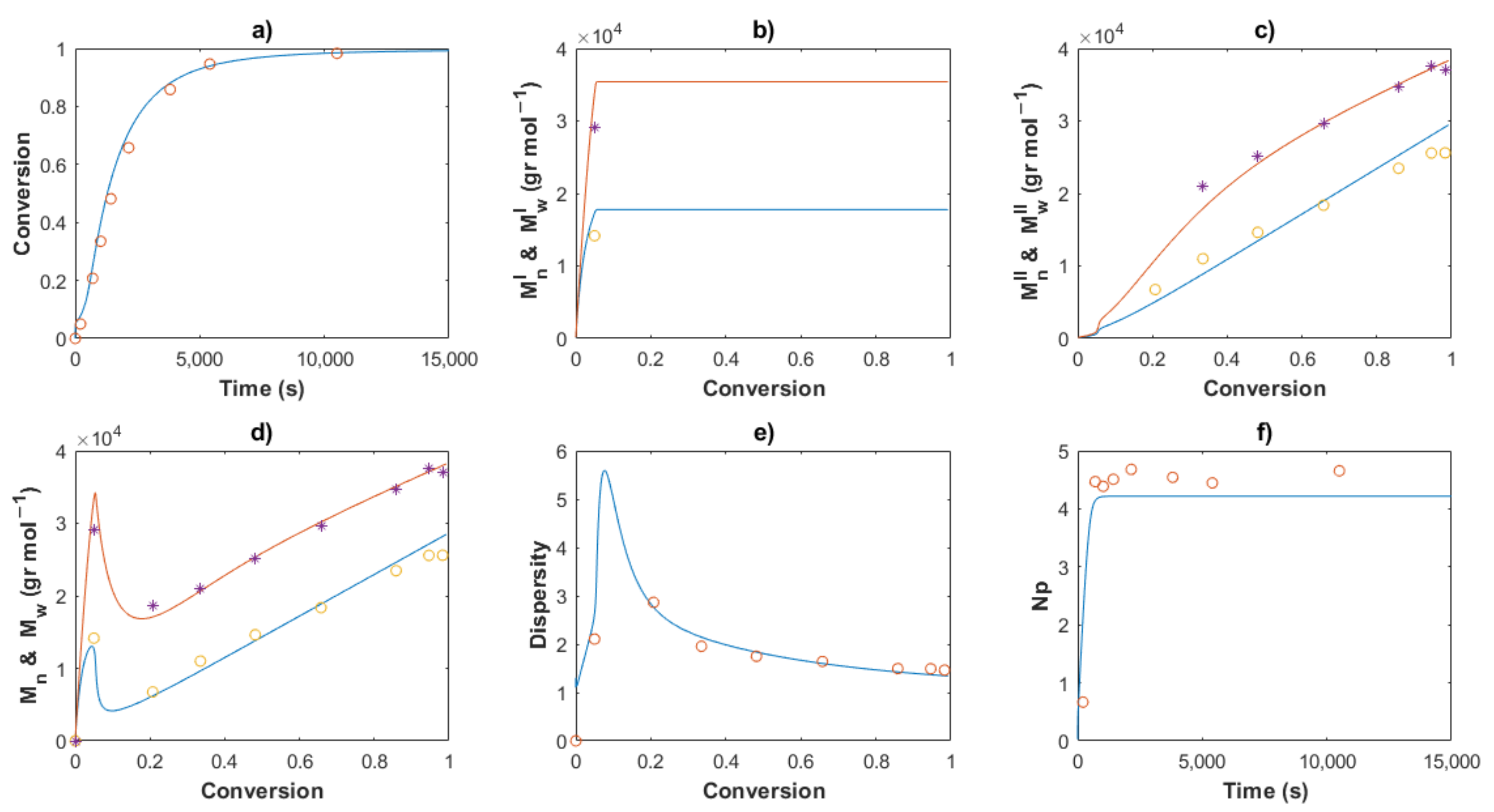
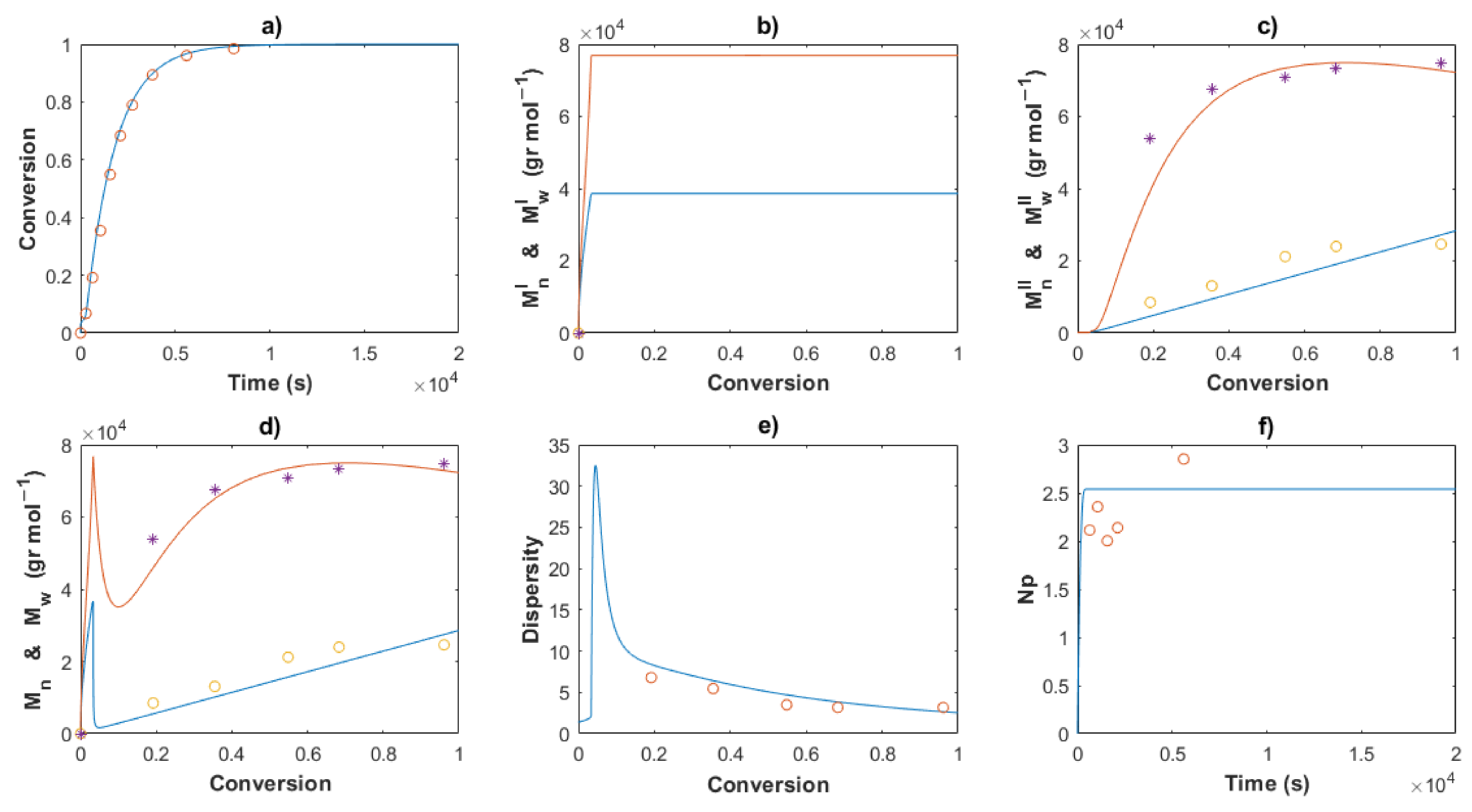
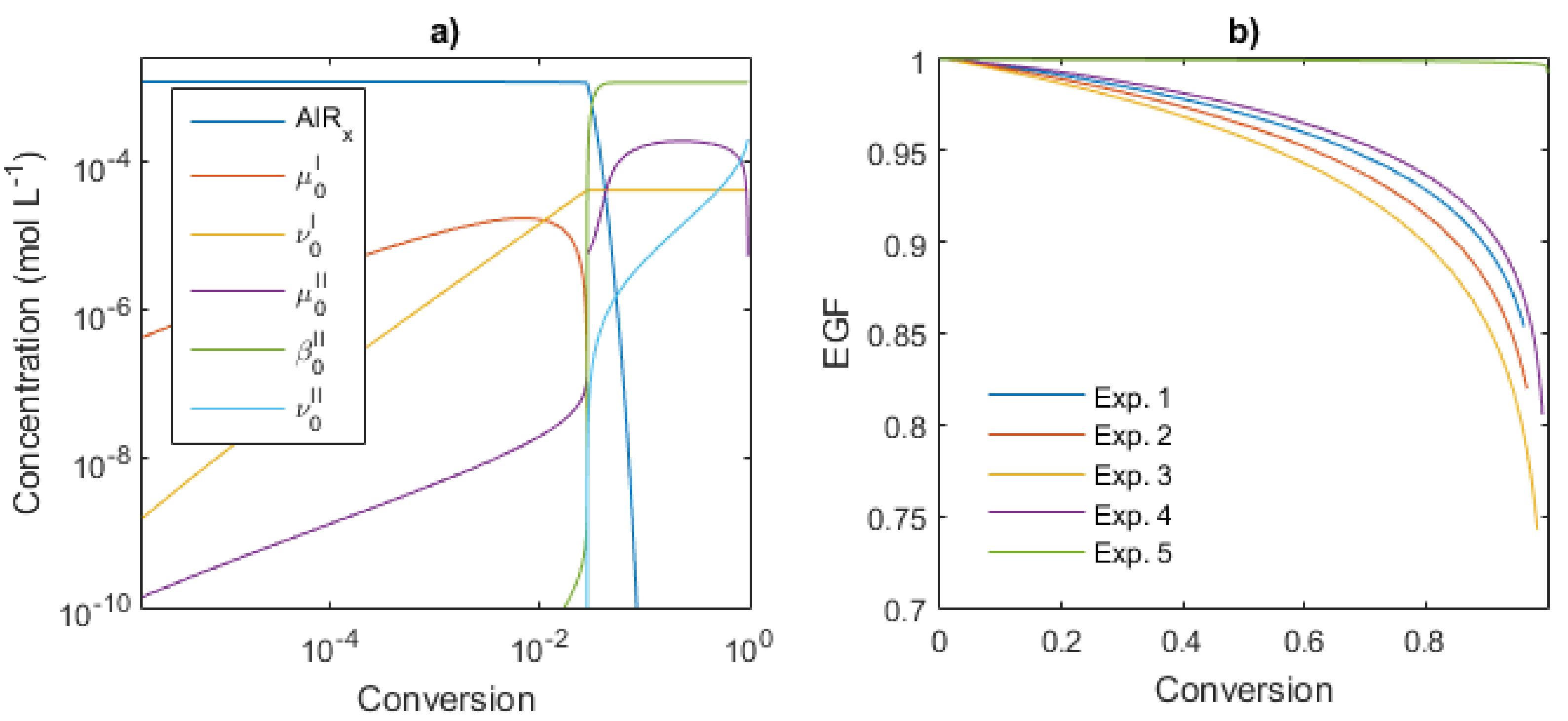
4.3. Kinetic Analysis
4.3.1. Polymer Chain Species
4.3.2. End-Group Functionality (EGF)
4.3.3. Molecular Weights and Dispersity
4.3.4. Stoichiometrical Analysis of the Polymerization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NdV3 | Neodymium versatate. |
| DIBAH | Diisobutylaluminum hydrade. |
| Me2SiCl2 | Dimethyldichlorosilane. |
| Np | Number of polymer chains per neodymium atom. |
| M | β-myrcene monomer. |
| C-hex | Cyclohexane. |
| [M]0 | β-myrcene monomer concentration at t = 0. |
| Nd | Neodymium catalyst. |
| [Nd]0 | Neodymium catalyst concentration at t = 0. |
| AlRx | Alkyl-aluminum cocatalyst (chain transfer agent during polymerization). |
| [AlRx]0 | Chain transfer agent concentration at t= 0. |
| RHCl2 | Halide donor. |
| [RHCl2]0 | Halide donor concentration at t= 0. |
| Mn | Number average molecular weight. |
| Mw | Weight average molecular weight. |
| Mn x | Number average molecular weight generated by the active centers of type x (x = I or II). |
| Mw x | Weight average molecular weight generated by the active centers of type x (x= I or II). |
| Dispersity index. | |
| MWD | Molecular Weight Distribution. |
| C I*, C II* | Active centers of type I and of type II, * denotes highly unstable species. |
| PnI, PnII | Type I and Type II propagating polymer chain of length n, active chains. |
| Pn Al I | Inactive chains for monomer insertion located on aluminum, dormant polymer of type I. |
| Pn Al II | Inactive chains for monomer insertion located on aluminum, dormant species of type II. |
| DnI, DnII | Type I and Type II inactive species, dead polymer. |
| kapp | Apparent rate constant of propagation. |
| R | Universal gas constant. |
| T | Absolute temperature. |
| A0 | Pre-exponential factor. |
| Ea | Activation energy. |
| kin,I | Initiation rate constant for type I species. |
| kin,II | Initiation rate constant for type II species. |
| kp,I | Propagation rate constant for type I species. |
| kp,II | Propagation rate constant for type II species. |
| ktrc,I | Chain transfer to cocatalyst rate constant for type I species. |
| ktrc,II | Chain transfer to cocatalyst rate constant for type II species. |
| kda1,I | Deactivation rate constant of type I active centers. |
| kda1,II | Deactivation rate constant of type II active centers. |
| kda1,I | Deactivation rate constant of the type I propagating polymer. |
| kda1,II | Deactivation rate constant of the type II propagating polymer. |
| ktr | Rate constant for the reversible transfer forward. |
| ktr1 | Rate constant for the reversible transfer backward. |
| k-th moment for the active polymer of type I. | |
| k-th moment for the dead polymer of type II. | |
| k-th moment for the active polymer of type II. | |
| k-th moment for the dormant polymer of type II. | |
| k-th moment for the dead polymer of type II. | |
| “Experimental” average number of polymer chains produced by single neodymium atom. | |
| Number average molecular weight obtained if each neodymium atom were to generate a polymer chain. | |
| Number average molecular weight experimentally determined by GPC. | |
| Number average molecular weight obtained by the model. | |
| EGF | End-Goup Functionality. |
| EGFII | End-Goup Functionality for polymer chains of Type II. |
| Rj2 | Coefficient of determination. |
| Sum of squared deviations between predicted values at the time i and the average experimental values for a specific characteristic j. | |
| SSE | Sum of squared errors. |
| S | Standard deviation. |
| Sum of squared deviations between predicted values at the time i and the experimental values at the time i for a specific characteristic j. | |
| Sum of squared deviations between predicted values at the time i and the average experimental values for a specific characteristic j. | |
| Log1M | The logarithm of the molecular weight corresponding to the slice retention time (averaged). |
| Normalized distribution of molecular weights, derivative of Weight fraction with respect to logarithm of molar Mass. |
References
- Jenkins, D.K. Butadiene polymerization with a rare earth compound using a magnesium alkyl cocatalyst: 1. Polymer 1985, 26, 147–151. [Google Scholar] [CrossRef]
- Liu, B.; Sun, G.; Li, S.; Liu, D.; Cui, D. Isoprene Polymerization with Iminophosphonamide Rare-Earth-Metal Alkyl Complexes: Influence of Metal Size on the Regio- and Stereoselectivity. Organometallics 2015, 34, 4063–4068. [Google Scholar] [CrossRef]
- Liu, L.; Song, Y.; Li, H. Carbene polymerization: Characterization of poly(carballyloxycarbene). Polym. Int. 2002, 51, 1047–1049. [Google Scholar] [CrossRef]
- Hollfelder, C.O.; Jende, L.N.; Diether, D. 1,3-Diene Polymerization Mediated by Homoleptic Tetramethylaluminates of the Rare-Earth Metals. Catalysts 2018, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Martins, N.; Bonnet, F.; Visseaux, M. Highly efficient cis-1,4 polymerisation of isoprene using simple homoleptic amido rare earth-based catalysts. Polymer 2014, 55, 5013–5016. [Google Scholar] [CrossRef]
- Göttker, S.I.; Kenyon, P.; Mecking, S. Coordinative Chain Transfer Polymerization of Butadiene with Functionalized Aluminum Reagents. Angew. Chem. Int. Ed. 2019, 58, 17777–17781. [Google Scholar] [CrossRef]
- Zheng, W.; Yan, N.; Zhu, Y.; Zhao, W.; Zhang, C.; Zhang, X.; Bai, C.; Hu, Y.; Zhang, X. Highly trans-1,4-stereoselective coordination chain transfer polymerization of 1,3-butadiene and copolymerization with cyclic esters by a neodymium-based catalyst system. Polym. Chem. 2015, 6, 6088–6095. [Google Scholar] [CrossRef]
- Wang, F.; Dong, B.; Liu, H.; Guo, J.; Zheng, W.; Zhang, C.; Zhao, L.; Bai, C.; Hu, Y.; Zhang, X. Synthesis of Block Copolymers Containing Polybutadiene Segments by Combination of Coordinative Chain Transfer Polymerization, Ring-Opening Polymerization, and Atom Transfer Radical Polymerization. Macromol. Chem. Phys. 2015, 216, 321–328. [Google Scholar] [CrossRef]
- Tang, Z.; Liang, A.; Liang, H.; Zhao, J.; Xu, L.; Zhang, J. Reversible Coordinative Chain Transfer Polymerization of Butadiene Using a Neodymium Phosphonate Catalyst. Macromol. Res. 2019, 27, 789–794. [Google Scholar] [CrossRef]
- Wang, F.; Liu, H.; Zheng, W.; Guo, J.; Zhang, C.; Zhao, L.; Zhang, H.; Hu, Y.; Bai, C.; Zhang, X. Fully-reversible and semi-reversible coordinative chain transfer polymerizations of 1,3-butadiene with neodymium-based catalytic systems. Polymer 2013, 54, 6716–6724. [Google Scholar] [CrossRef]
- Valente, A.; Mortreux, A.; Visseauc, M.; Zinck, P. Coordinative Chain Transfer Polymerization. Chem. Rev. 2013, 113, 3836–3857. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, M.; Saeb, M.R.; Mohammadi, T.; Khorasani, M.M.; Stadler, F.J. A Perspective on Modeling and Characterization of Transformations in the Blocky Nature of Olefin Block Copolymers. Ind. Eng. Chem. Res. 2015, 54, 8867–8873. [Google Scholar] [CrossRef]
- Mohammadi, Y.; Ahmadi, M.; Saeb, M.R.; Khorasani, M.M.; Yang, P.; Stadler, F.J. A Detailed Model on Kinetics and Microstructure Evolution during Copolymerization of Ethylene and 1-Octene: From Coordinative Chain Transfer to Chain Shuttling Polymerization. Macromolecules 2014, 47, 4778–4789. [Google Scholar] [CrossRef]
- Mohammadi, Y.; Saeb, M.R.; Penlidis, A.; Jabbari, E.; Stadler, F.J.; Zinck, P.; Vivaldo-Lima, E. Toward Olefin Multiblock Copolymers with Tailored Properties: A Molecular Perspective. Macromol. Theory Simul. 2021, 30, 2100003. [Google Scholar] [CrossRef]
- Ahmadi, M.; Nasresfahani, A. Realistic Representation of Kinetics and Microstructure Development during Chain Shuttling Polymerization of Olefin Block Copolymers. Macromol. Theory Simul. 2015, 24, 311–321. [Google Scholar] [CrossRef]
- Cavalcante de Sá, M.C.; Córdova, A.M.T.; Díaz de León-Gómez, R.E.; Pinto, J.C. Modeling of Isoprene Solution Coordinative Chain Transfer Polymerization. Macromol. React. Eng. 2021, 15, 2100005. [Google Scholar] [CrossRef]
- Zhang, M.; Karjala, T.W.; Jain, P.; Villa, C. Theoretical Modeling of Average Block Structure in Chain-Shuttling α–Olefin Copolymerization Using Dual Catalysts. Macromolecules 2013, 46, 4847–4853. [Google Scholar] [CrossRef]
- Mori, S.; Barth, H.G. Approaches to Molecular Weight Calibration. In Size Exclusion Chromatography, 1st ed.; Springer: Berlin/Heidelberg, Germany, 1999; pp. 95–113. [Google Scholar]
- González-Villa, J.; Saldívar-Guerra, E.; Díaz de León-Gómez, R.E.; López-González, H.R.; Infante-Martínez, J.R. Kinetics of the Anionic Homopolymerizations of ß–Myrcene and 4–Methylstyrene in Cyclohexane Initiated by n–Butyllithium. J. Polym. Sci. Part A Polym. Chem. 2019, 57, 2157–2165. [Google Scholar] [CrossRef]
- Hattam, P.; Gauntlett, S.; Mays, J.W.; Hadjichristidis, N.; Young, R.N.; Fetters, L.J. Conformational Characteristics of Some Model Polydienes and Polyolefins. Macromolecules 1991, 24, 6199–6209. [Google Scholar] [CrossRef]
- Friebe, L.; Nuyken, O.; Obrecht, W. Polymerization in Solution. In Neodymium Based Ziegler Catalysts-Fundamental Chemistry, 1st ed.; Nuyken, O., Ed.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 12–92. [Google Scholar]
- Manuiko, G.V.; Salakhov, I.I.; Aminova, G.A.; Akhmetov, I.G.; Dyakonov, G.S.; Bronskaya, V.V.; Demidova, E.V. Mathematical Modeling of 1,3 Butadiene Polymerization over a Neodymium Based Catalyst in a Batch Reactor with Account Taken of the Multisite Nature of the Catalyst and Chain Transfer to the Polymer. Theor. Found. Chem. Eng. 2010, 44, 139–149. [Google Scholar] [CrossRef]
- Quirk, R.P.; Kells, A.M.; Yunlu, K.; Cuif, J.P. Butadiene Polymerization using Neodymium Versatate-Based Catalyst: Catalyst Optimization and Effects of Water and Excess Versatic Acid. Polymer 2000, 41, 5903–5908. [Google Scholar] [CrossRef]
- Wilson, D.J.; Jenkins, D.K. Butadiene polymerization using ternary neodymium-based catalyst systems—The effect of catalyst component addition order. Polym. Bull. 1992, 27, 407–411. [Google Scholar] [CrossRef]
- Fellows, C.M.; Jones, R.G.; Keddie, D.J.; Luscombe, C.K.; Matson, J.B.; Moad, G.; Matyjaszewski, K.; Merna, J.; Nakano, T.; Penczek, S.; et al. Terminology for Chain Polymerization (IUPAC Recommendations 2021). Pure Appl. Chem. 2022. under review. [Google Scholar]
- Soares, J.B.P.; McKenna, T.F.L.; Cheng, C.P. Coordination Polymerization. In Polymer Reaction Engineering; Asua, J.M., Ed.; Blackwell Publishing Ltd.: Oxford, UK, 2007; pp. 29–117. [Google Scholar]
- Saldívar-Guerra, E.; Vivaldo-Lima, E. Introduction to Polymers and Polymer Types. In Handbook of Polymer Synthesis, Characterization, and Processing; Saldívar-Guerra, E., Vivaldo-Lima, E., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 1–14. [Google Scholar]
- Soares, J.B.P.; McKenna, T.F.L. Polyolefin Microstructural Modeling. In Polyolefin Reaction Engineering; Wiley-VCH: Weinheim, Germany, 2012; pp. 187–269. [Google Scholar]
- Ray, W.H. On the Mathematical Modeling of Polymerization Reactors. J. Macromol. Sci. Part C 1972, 8, 1–56. [Google Scholar] [CrossRef]
- Zhong, M.; Matyjaszewski, K. How Fast Can a CRP Be Conducted with Preserved Chain End Functionality? Macromolecules 2011, 44, 2668–2677. [Google Scholar] [CrossRef]
- Valencia, L.; Enríquez-Medrano, F.; López-González, R.; Quiñonez-Ángulo, P.; Saldívar-Guerra, E.; Díaz-Elizondo, J.; Zapata-González, I.; Díaz de León, R. Ethylene Polymerization via Zirconocene Catalysts and Organoboron Activators: An Experimental and Kinetic Modeling Study. Processes 2021, 9, 162. [Google Scholar] [CrossRef]
- Shampine, L.F.; Reichelt, M.W. The MATLAB ODE Suite. SIAM J. Sci. Comput. 1997, 18, 1–22. [Google Scholar] [CrossRef] [Green Version]
- MathWorks®. Fmincon. Available online: https://la.mathworks.com/help/optim/ug/fmincon.html?lang=en (accessed on 28 January 2022).
- Díaz de León, G.R.E.; Enríquez-M, F.J.; Maldonado, T.H.; Mendoza, C.R.; Reyes, A.K.; López, G.H.R.; Olivares, R.J.L.; Lugo, U.L.E. Synthesis and characterization of high cis-polymyrcene using neodymium-based catalysts. Can. J. Chem. Eng. 2016, 94, 823–832. [Google Scholar] [CrossRef]
- Friebe, L.; Nuyken, O.; Windisch, H.; Obrecht, W. Polymerization of 1,3-Butadiene Initiated by Neodymium Versatate/Diisobutylaluminium Hydride/Ethylaluminium Sesquichloride: Kinetics and Conclusions about the Reaction Mechanism. Macromol. Chem. Phys. 2002, 203, 1055–1064. [Google Scholar] [CrossRef]
- Hustad, P.D.; Kuhlman, R.L.; Carnahan, E.M.; Wenzel, T.T.; Arriola, D.J. An Exploration of the Effects of Reversibility in Chain Transfer to Metal in Olefin Polymerization. Macromolecules 2008, 41, 4081–4089. [Google Scholar] [CrossRef]
- Fan, C.; Bai, C.; Cai, H.; Dai, Q.; Zhang, X.; Wang, F. Preparation of High cis-1,4 Polyisoprene with Narrow Molecular Weight Distribution via Coordinative Chain Transfer Polymerization. J. Polym. Sci. A Polym. Chem. 2010, 48, 4768–4774. [Google Scholar] [CrossRef]
- Georges, S.; Touré, A.O.; Visseaux, M.; Zinck, P. Coordinative Chain Transfer Copolymerization and Terpolymerization of Conjugated Dienes. Macromolecules 2014, 47, 4538–4547. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Exp. 1 | Exp. 2 | Exp. 3 | Exp. 4 | Exp. 5 |
|---|---|---|---|---|---|
| Temperature (°C) | 50 | 60 | 70 | 60 | 60 |
| [M]0/[Nd]0 | 660 | 885 | 533 | ||
| M/C-hex (wt.%) | 16 | 15.5 | 16 | ||
| [M]0 (mol L−1) | 7.83 × 10−1 | 7.73 × 10−1 | 7.86 × 10−1 | ||
| [Nd]0 (mol L−1) | 11.85 × 10−4 | 8.74 × 10−4 | 14.75 × 10−4 | ||
| [AlRx]0 (mol L−1) | 2.36 × 10−2 | 1.74 × 10−2 | 2.93 × 10−2 | ||
| [RHCl2]0 (mol L−1) | 11.81 × 10−4 | 8.71 × 10−4 | 14.67 × 10−4 | ||
| [AlRx]0/[Nd]0 | 20 | 20 | 20 | ||
| [RHCl2]0/[Nd]0 | 1 | 1 | 1 | ||
| Experiment | Exp. 1 | Exp. 2 | Exp. 3 | Exp. 4 | Exp. 5 |
|---|---|---|---|---|---|
| kin,I (L mol−1 s−1) | 20 | 27 | 36.45 | 80 | 15 |
| kp, I (L mol−1 s−1) | 50,000 | 55,000 | 60,500 | 90 | 20,000 |
| ktrc,I (L mol−1 s−1) | 45,000 | 58,500 | 75,465 | 120 | 10,000 |
| kda1,I (s−1) | 100 | 150 | 222 | 0.16 | 40 |
| kda2,I (s−1) | 20 | 32 | 50.2 | 0.20 | 20 |
| kin,II (L mol−1 s−1) | 2 × 10−3 | 4.2 × 10−3 | 8.8 × 10−3 | 1.6 × 10−2 | 3.2 × 10−2 |
| kp,II (L mol−1 s−1) | 0.75 | 1.2 | 1.7 | 1.02 | 3.0 |
| kda1,II (s−1) | 7.00 × 10−4 | 1.45 × 10−3 | 1.80 × 10−3 | 1.25 × 10−10 | 6 × 10−3 |
| kda2,II (s−1) | 4.60 × 10−5 | 1.00 × 10−4 | 1.70 × 10−4 | 14 × 10−4 | 7 × 10−6 |
| ktrc,II (L mol−1 s−1) | 200 | 280 | 384 | 30 | 500 |
| ktr (L mol−1 s−1) | 0.44 | 0.75 | 1 | 2.3 | 1.55 |
| Exp. 1 | Exp. 2 | Exp. 3 | Exp. 4 | Exp. 5 | |
|---|---|---|---|---|---|
| R2M | 0.98 | 0.97 | 0.98 | 0.99 | 0.99 |
| SM | 0.04 | 0.05 | 0.04 | 0.03 | 0.03 |
| R2Mn | 0.88 | 0.91 | 0.81 | 0.87 | 0.91 |
| SMn | 8349.90 | 8065.40 | 10,426.00 | 3743.00 | 3048.70 |
| R2Mw | 0.79 | 0.87 | 0.61 | 0.97 | 0.97 |
| SMw | 13,406.00 | 11,761.00 | 17,168.00 | 1973.80 | 2118.90 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ubaldo-Alarcón, A.; Soriano-Corral, F.; Córdova, T.; Zapata-González, I.; Díaz-de-León, R. Terpene Coordinative Chain Transfer Polymerization: Understanding the Process through Kinetic Modeling. Polymers 2022, 14, 2352. https://doi.org/10.3390/polym14122352
Ubaldo-Alarcón A, Soriano-Corral F, Córdova T, Zapata-González I, Díaz-de-León R. Terpene Coordinative Chain Transfer Polymerization: Understanding the Process through Kinetic Modeling. Polymers. 2022; 14(12):2352. https://doi.org/10.3390/polym14122352
Chicago/Turabian StyleUbaldo-Alarcón, Andrés, Florentino Soriano-Corral, Teresa Córdova, Iván Zapata-González, and Ramón Díaz-de-León. 2022. "Terpene Coordinative Chain Transfer Polymerization: Understanding the Process through Kinetic Modeling" Polymers 14, no. 12: 2352. https://doi.org/10.3390/polym14122352
APA StyleUbaldo-Alarcón, A., Soriano-Corral, F., Córdova, T., Zapata-González, I., & Díaz-de-León, R. (2022). Terpene Coordinative Chain Transfer Polymerization: Understanding the Process through Kinetic Modeling. Polymers, 14(12), 2352. https://doi.org/10.3390/polym14122352