
Preparation of High Mechanical Strength Chitosan Nanofiber/NanoSiO2/PVA Composite Scaffolds for Bone Tissue Engineering Using Sol–Gel Method

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
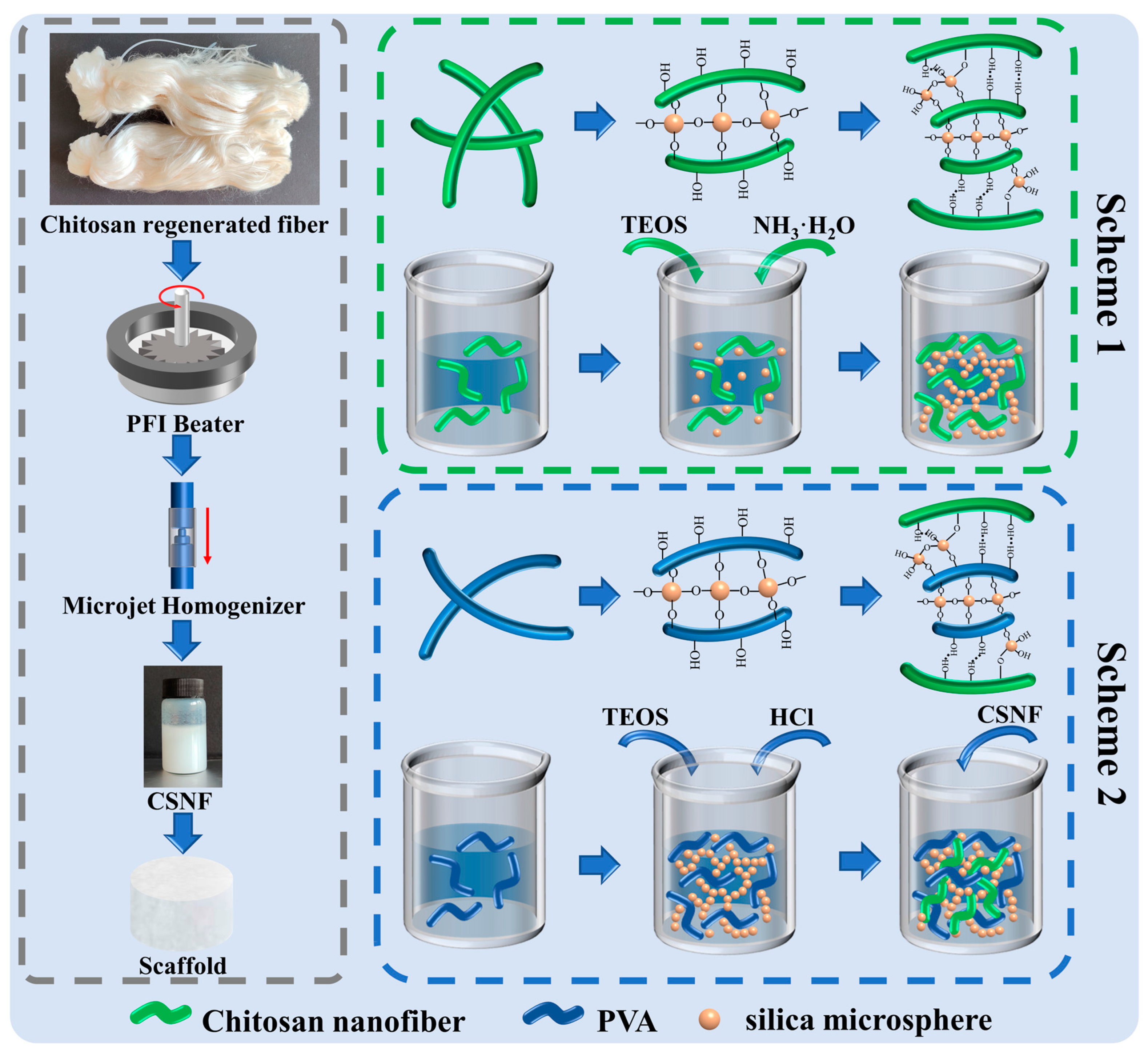
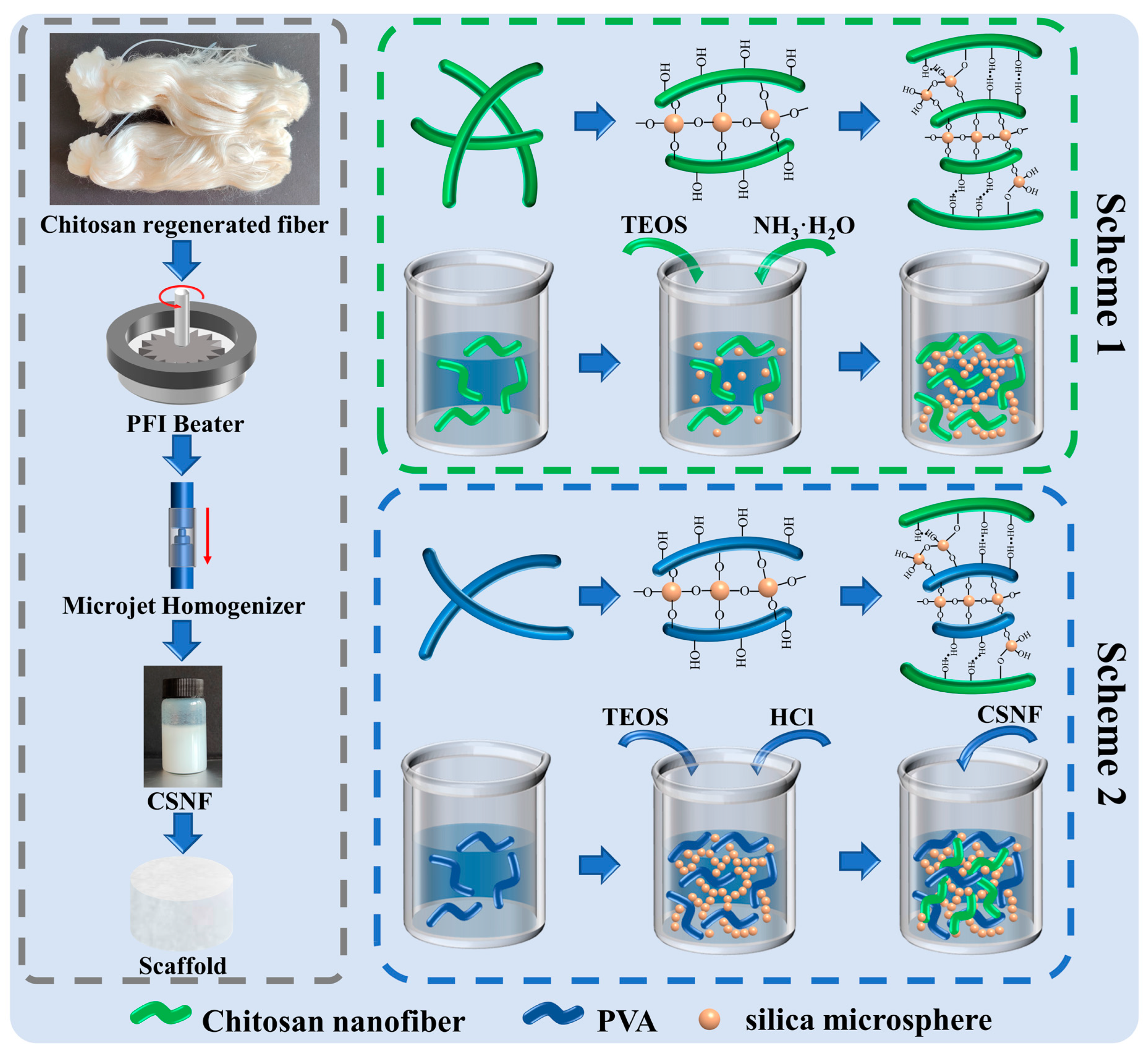
2.2. Preparation of CSNF
2.3. Preparation of P-A-T3 and F-A-T0/T3/T5 Scaffolds
- (1)
- Chitosan powder solution was made by weighing a certain amount of chitosan powder, adding deionized water and a certain amount of glacial acetic acid, and stirring with a magnetic stirrer for 3 h;
- (2)
- The homogenized CSNF was adjusted to a dispersion concentration of 2.5% (w/w) by centrifugation and the addition of deionized water;
- (3)
- A certain amount of TEOS was dissolved 1 mL of ethanol and then added to the dispersion of (1) and (2), which were then stirred for 1 h using a magnetic stirrer to ensure equitable mixing. Then, a certain amount of ammonia water (VNH3·H2O:VCSNF/CS dispersion = 1:10) was added and stirred for 24 h at 30 °C;
- (4)
- Pour the fully reacted dispersion into a 48-well plate, allowing it to stand for 3 h, and then freeze-dry the scaffold. Finally, soak then wash with deionized water three times, and freeze-dry again to achieve the final composite scaffolds.
2.4. Preparation of F-B-T5-P0/P0.5/P1.5/P2.5 Scaffolds
- (1)
- Weigh a certain amount of PVA and add it into the deionized water, and continue heating and stirring at 80 °C for 5 h until the PVA is completely dissolved;
- (2)
- A certain amount of TEOS was dissolved in 1 mL of ethanol and then added to the PVA solution, which was then stirred for 1 h using a magnetic stirrer to ensure equitable mixing. Then, a certain amount of 10% (w/w) HCl (1 drop/10 mL PVA solution) was added and stirred for 12 h at 60 °C;
- (3)
- To the thoroughly reacted dispersion, an equal quality of 5.0% (w/w) CSNF dispersion was added and stirred for 12 h at 60 °C to ensure equitable mixing;
- (4)
- Pour the fully reacted dispersion into a 48-well plate, allowing it to stand for 3 h, and then freeze-dry the scaffold. Finally, soak then wash with deionized water three times, and freeze-dry again to achieve the final composite scaffolds.
2.5. Characterization of the Scaffolds
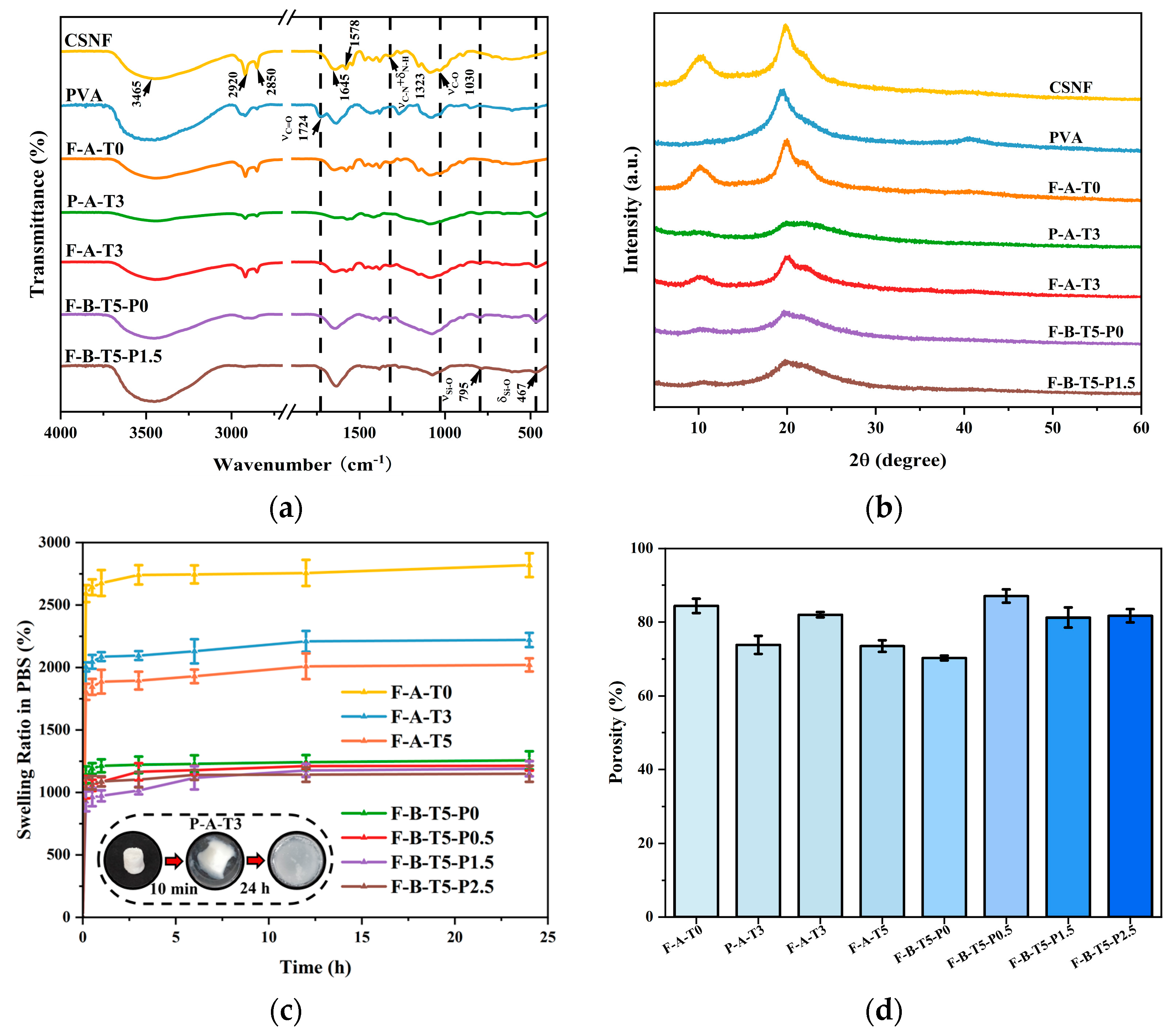
2.6. Porosity, Swelling, and Degradation Studies of Scaffolds
2.7. Compressive Strength of Scaffolds
2.8. MC3T3-E1 Cells Compatibility and Proliferation Assay
3. Results
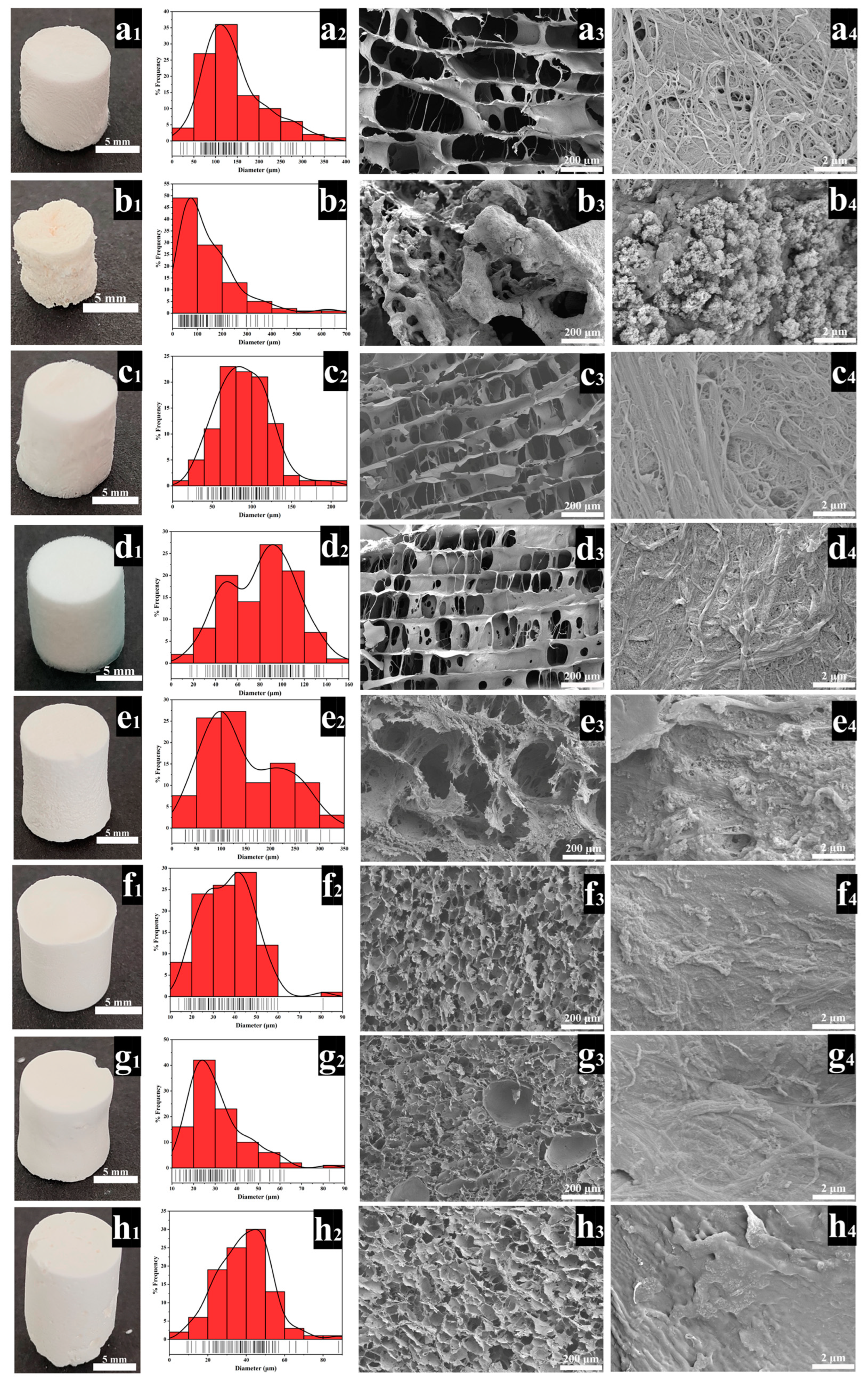
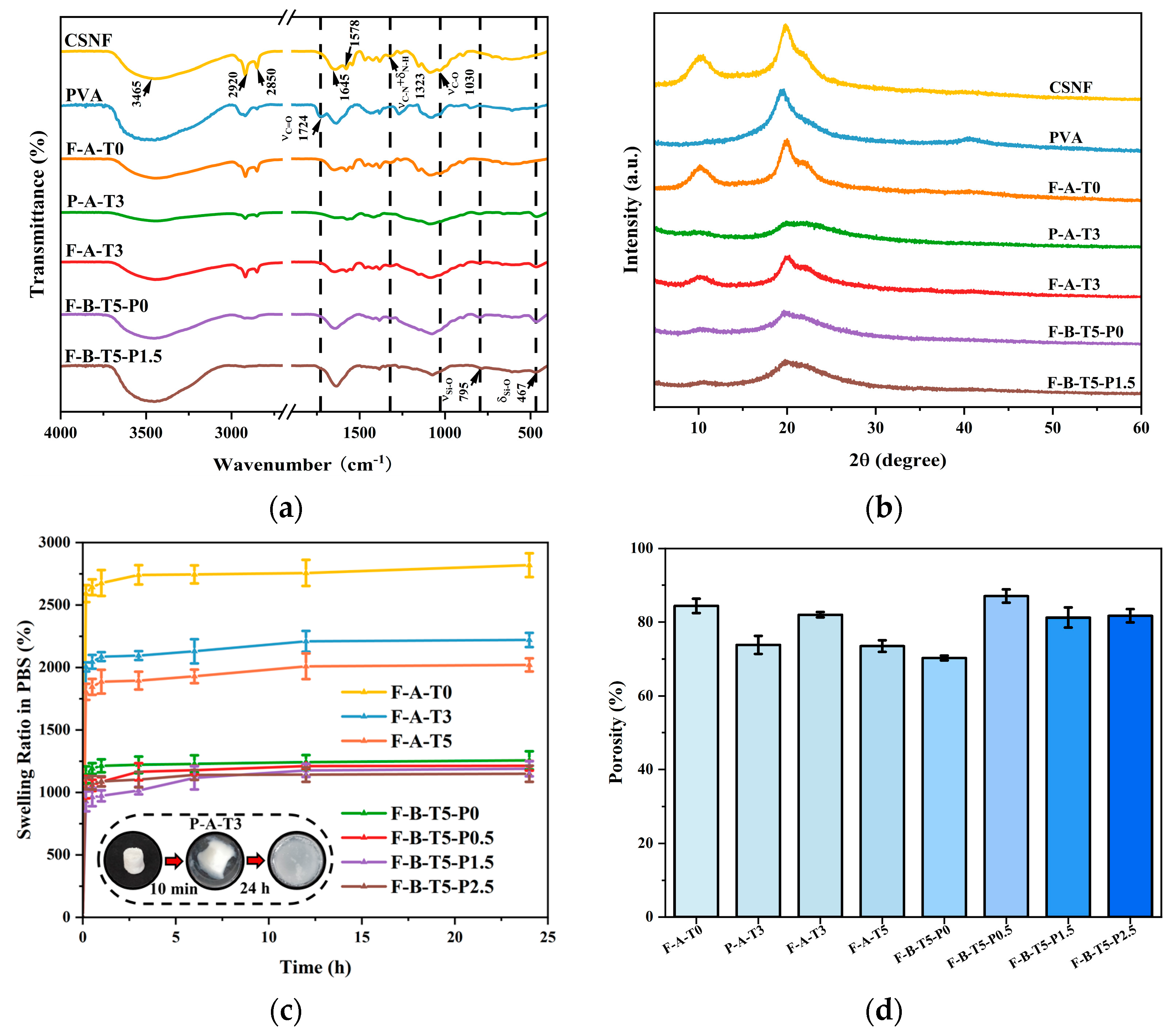
3.1. Structural Analyses of the Scaffolds
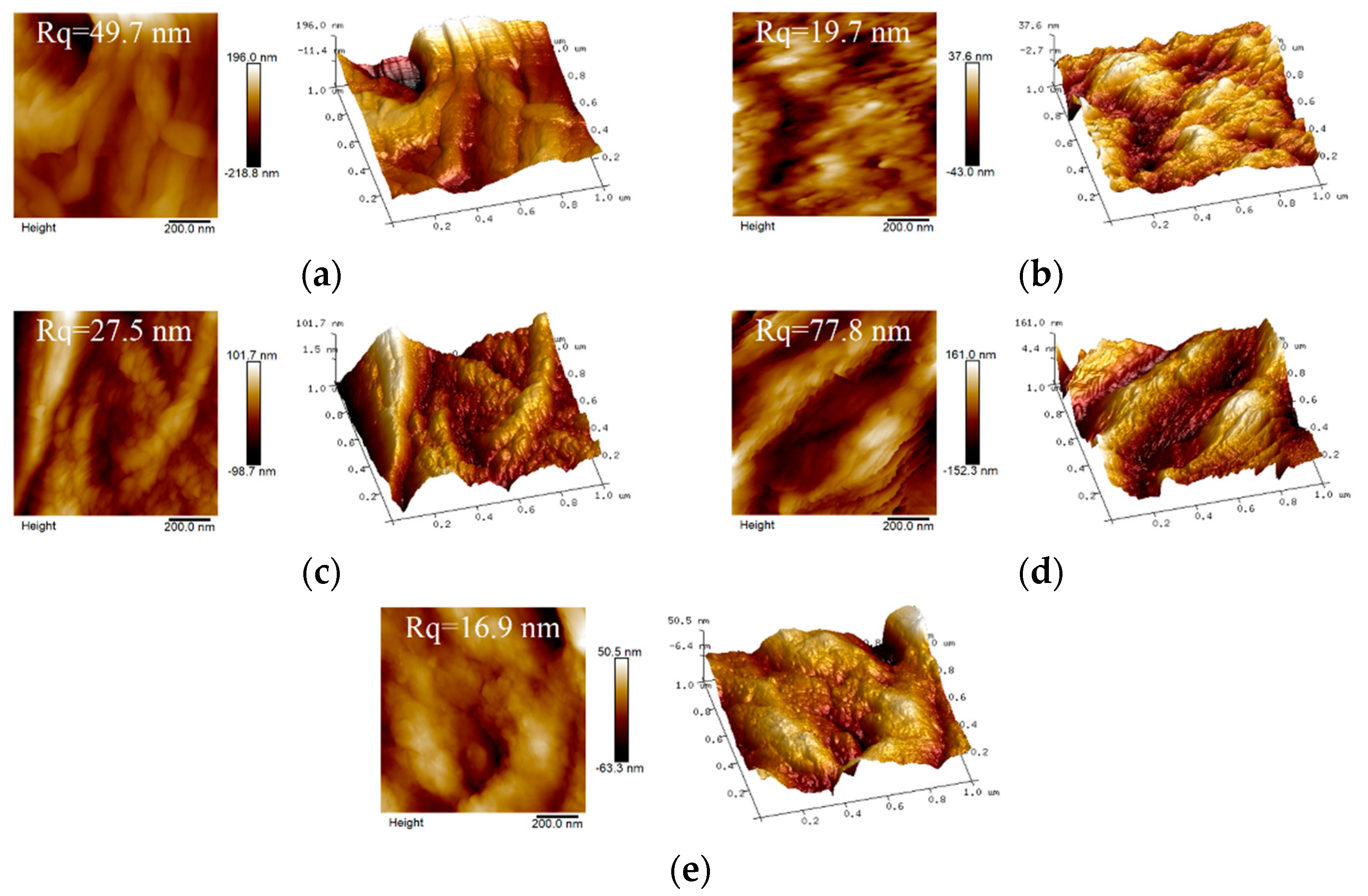
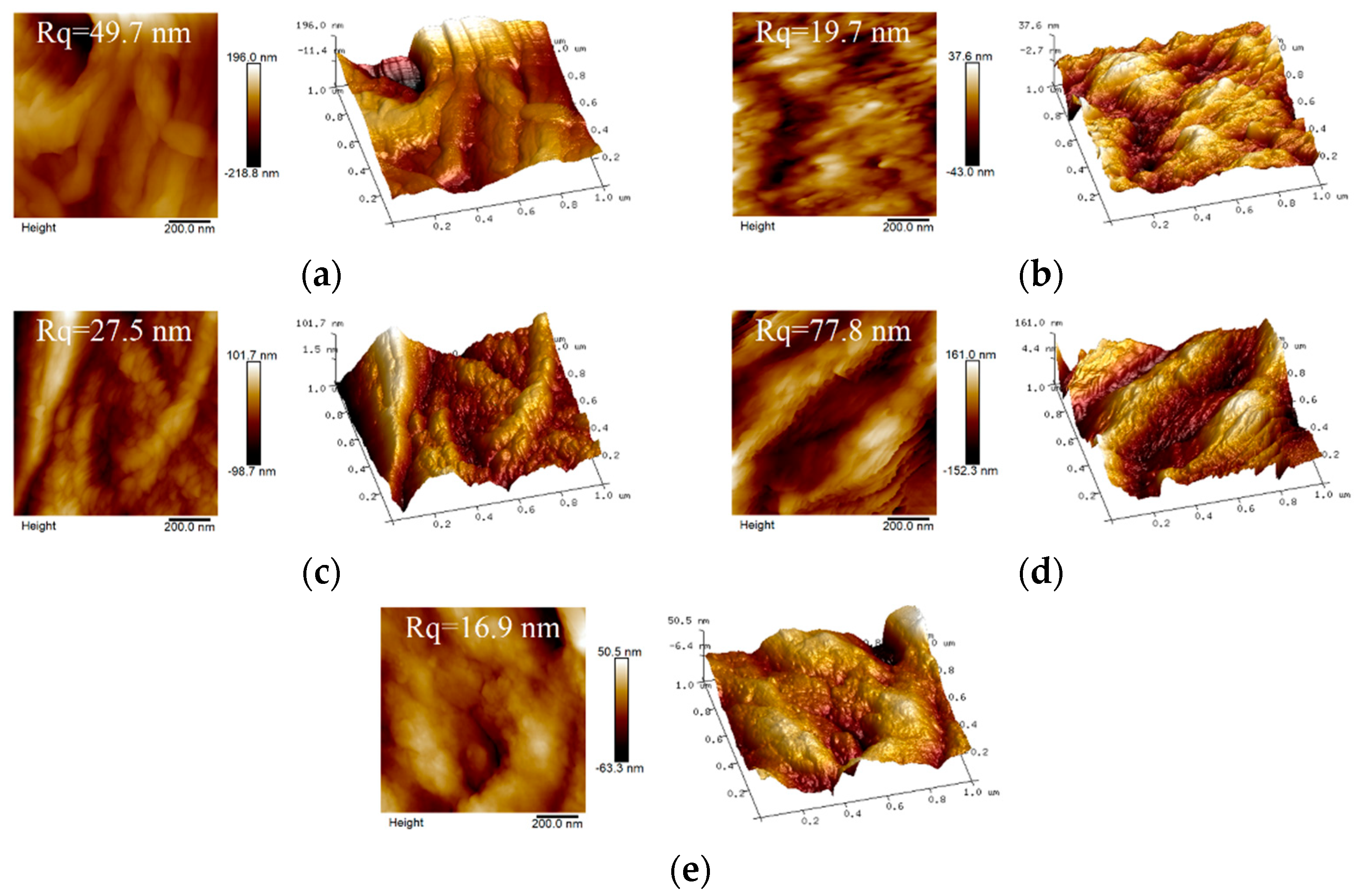
3.2. Characterization Analyses of the Scaffolds
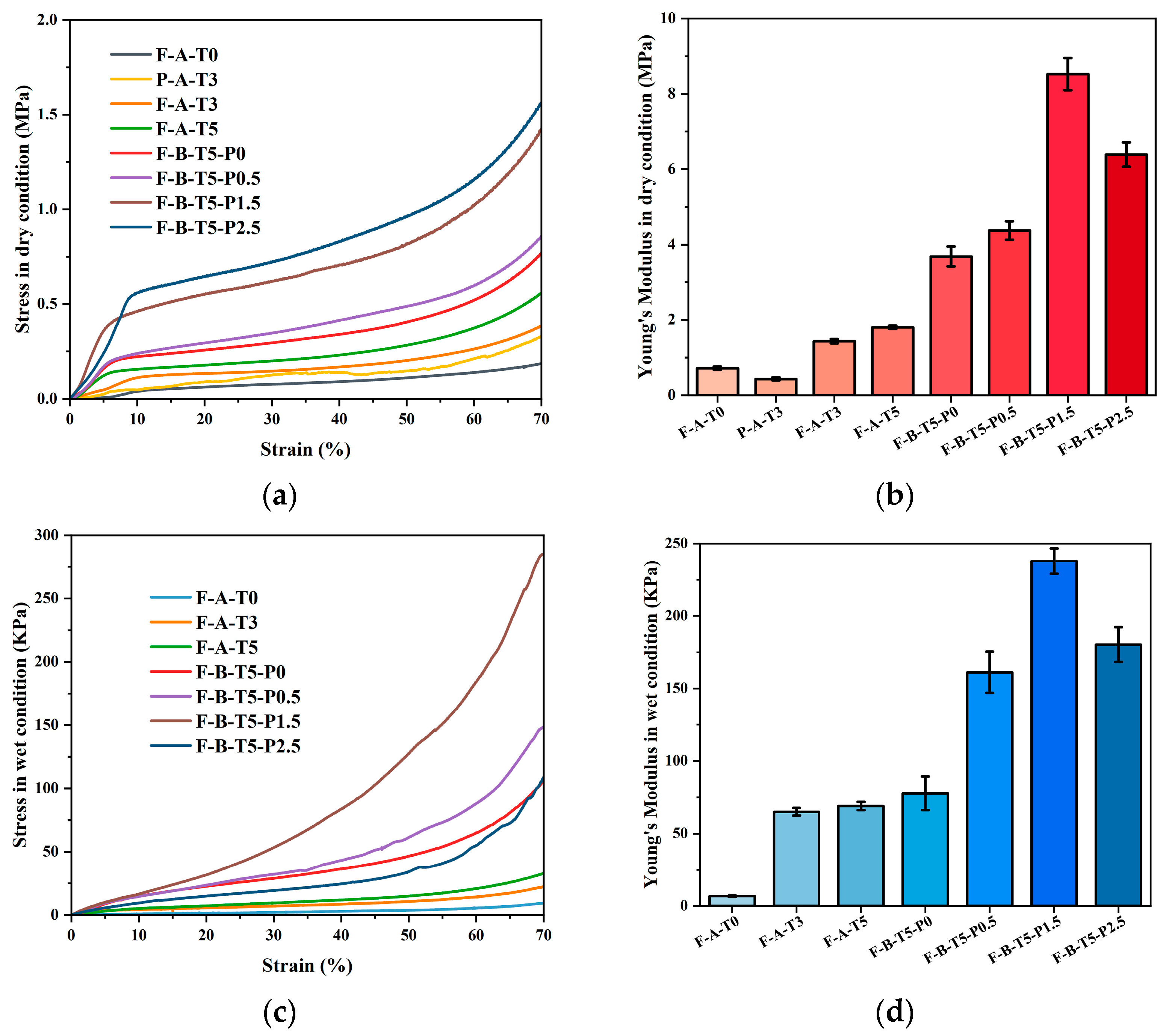
3.3. Compressive Strength of Scaffolds
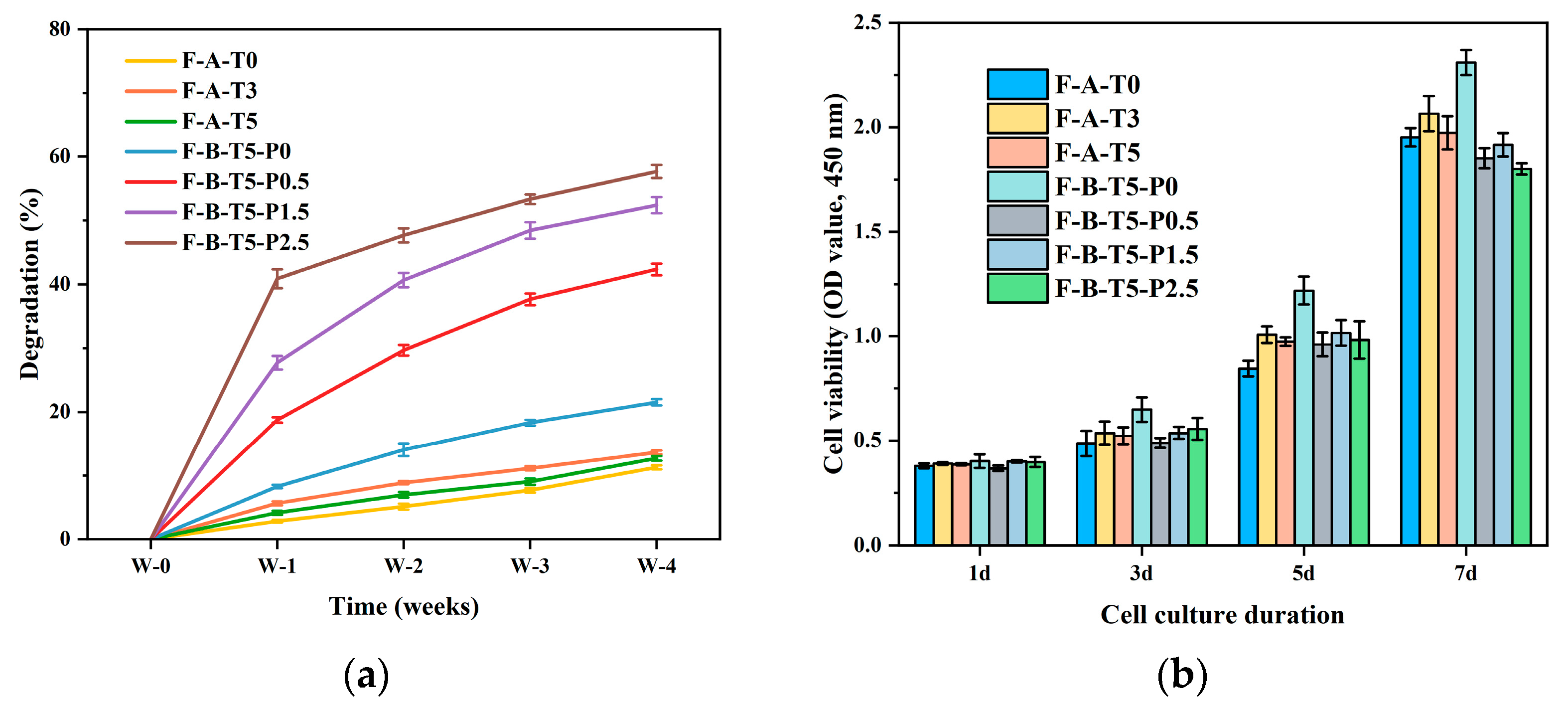
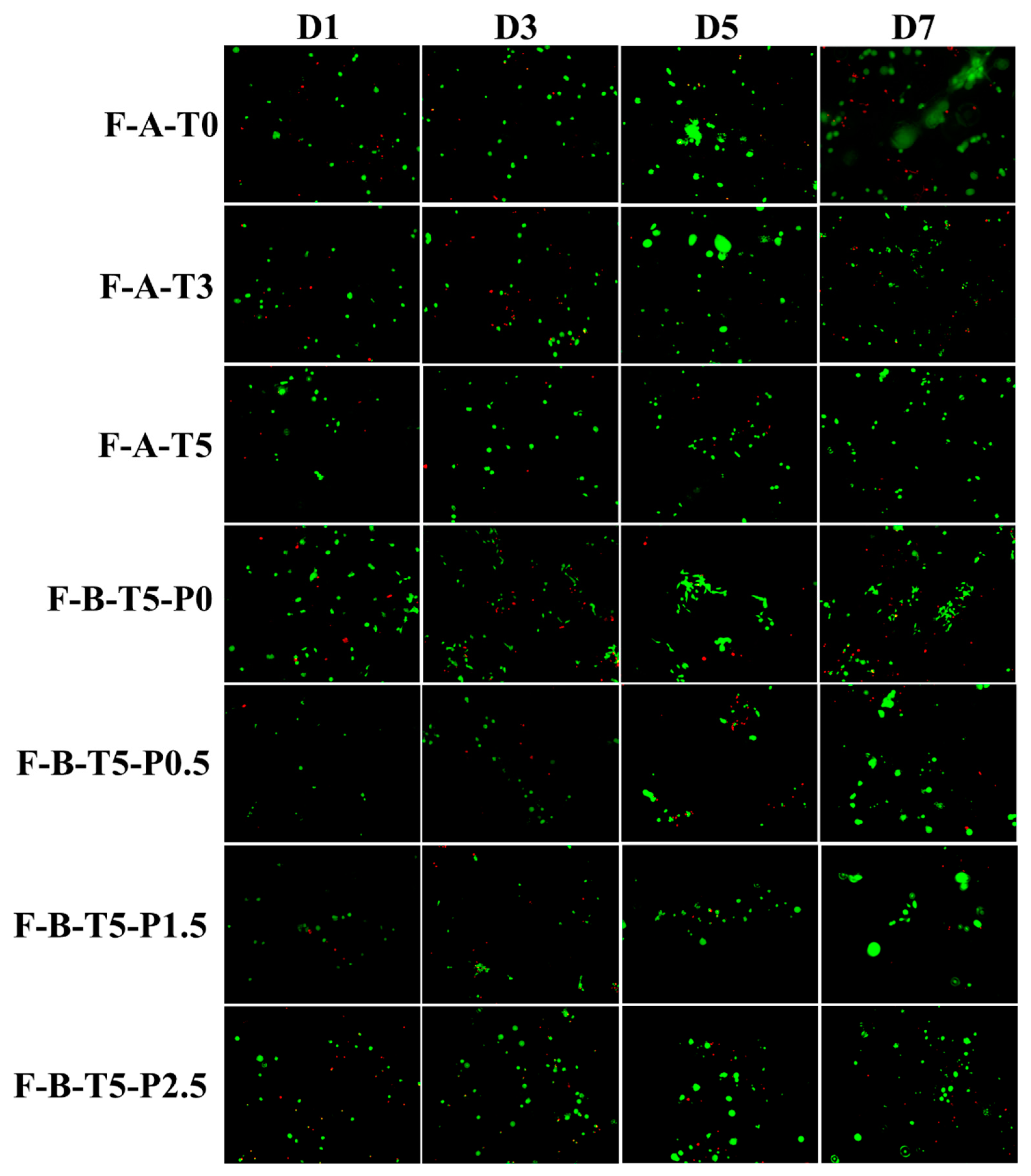
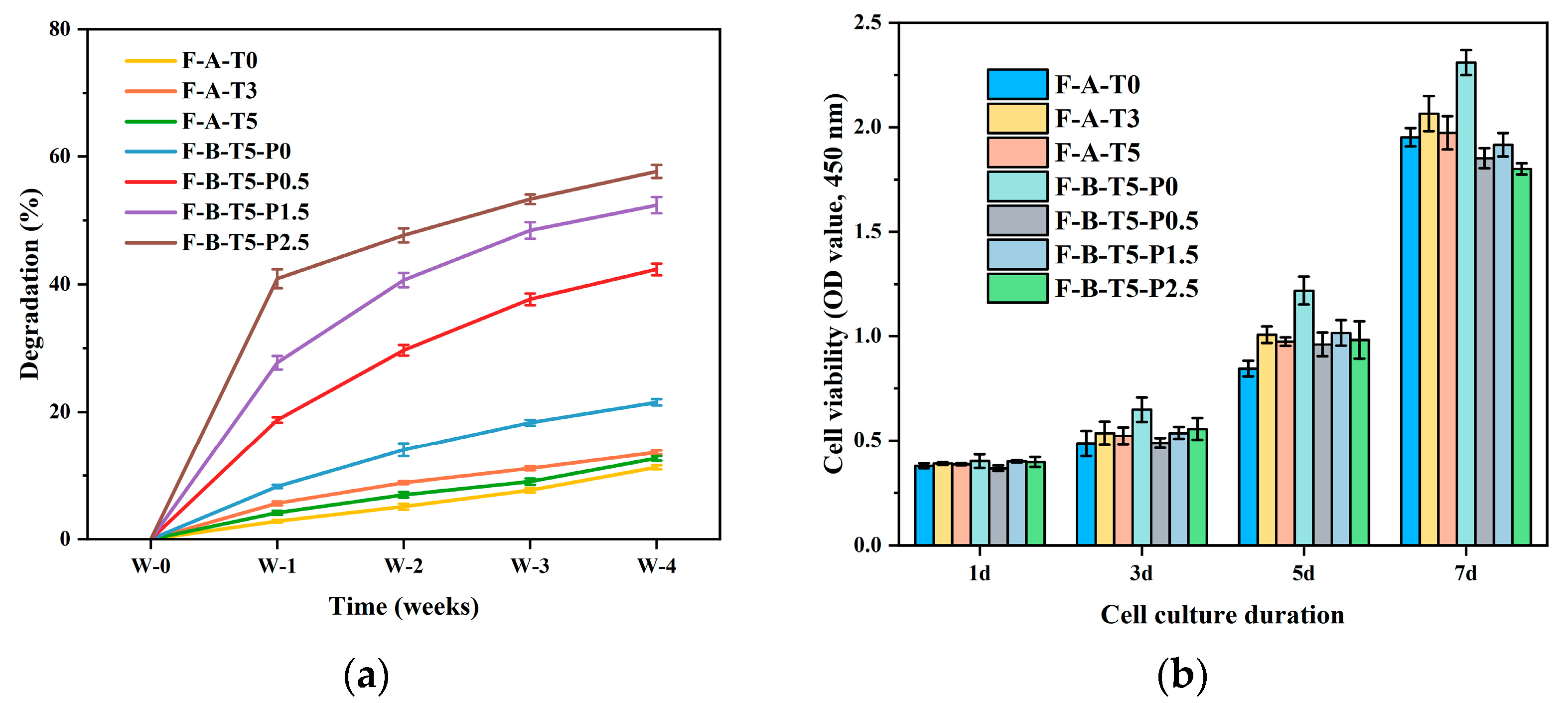
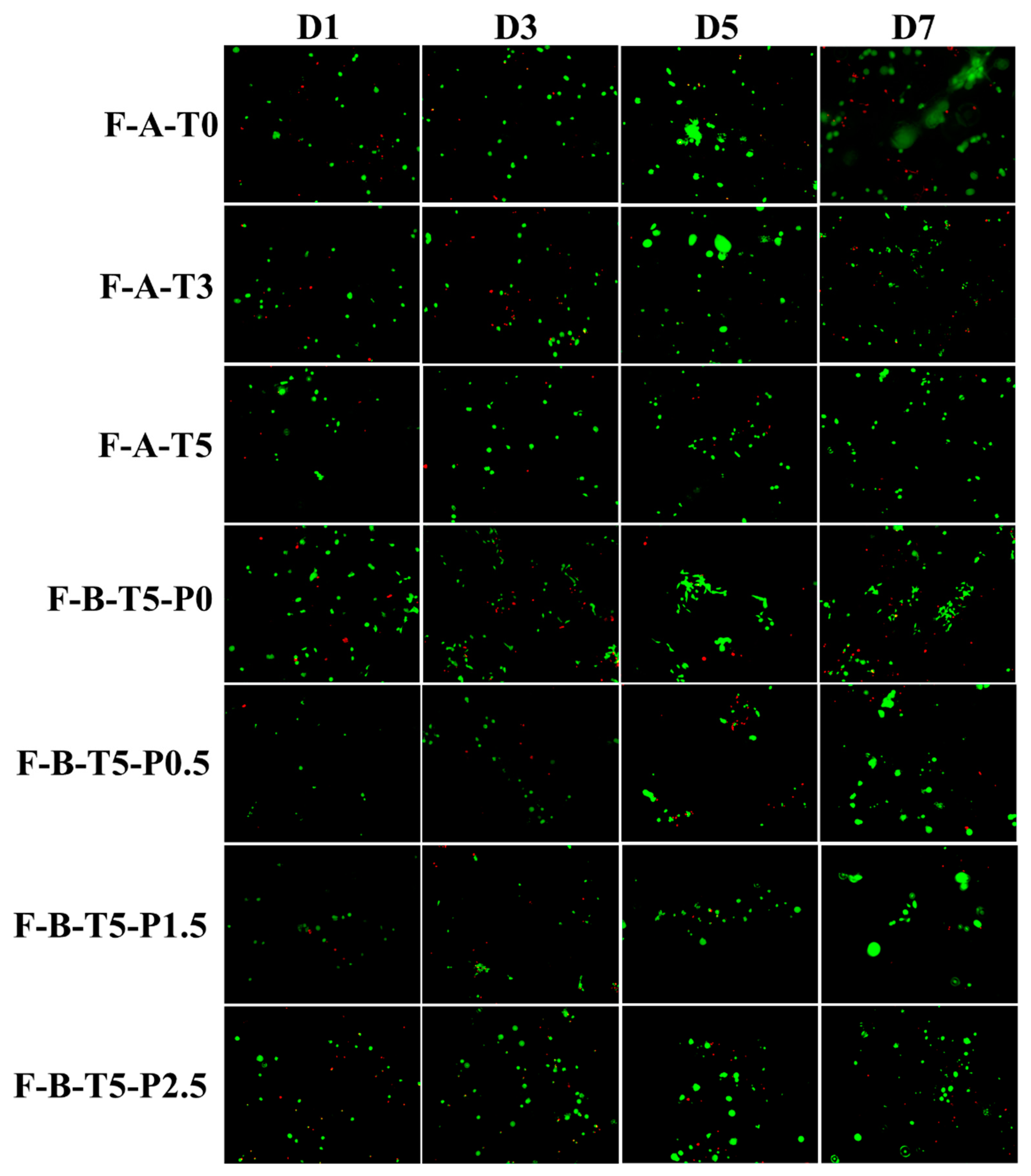
3.4. Degradation Rate and Cytocompatibility of Scaffolds
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Annamalai, R.T.; Hong, X.W.; Schott, N.G.; Tiruchinapally, G.; Levi, B.; Stegemann, J.P. Injectable osteogenic microtissues containing mesenchymal stromal cells conformally fill and repair critical-size defects. Biomaterials 2019, 208, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Koons, G.L.; Diba, M.; Mikos, A.G. Materials design for bone-tissue engineering. Nat. Rev. Mater. 2020, 5, 584–603. [Google Scholar] [CrossRef]
- Greenwald, A.S.; Boden, S.D.; Goldberg, V.M.; Khan, Y.; Laurencin, C.T.; Rosier, R.N.; Comm Biological, I. Bone-graft substitutes: Facts, fictions, and applications. J. Bone Joint Surg.-Am. Vol. 2001, 83A, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Bhumiratana, S.; Bernhard, J.C.; Alfi, D.M.; Yeager, K.; Eton, R.E.; Bova, J.; Shah, F.; Gimble, J.M.; Lopez, M.J.; Eisig, S.B. Tissue-engineered autologous grafts for facial bone reconstruction. Sci. Transl. Med. 2016, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Collins, M.N.; Ren, G.; Young, K.; Pina, S.; Reis, R.L.; Oliveira, J.M. Scaffold fabrication technologies and structure/function properties in bone tissue engineering. Adv. Funct. Mater. 2021, 31, 2010609. [Google Scholar] [CrossRef]
- Maquet, V.; Jerome, R. Design of macroporous biodegradable polymer scaffolds for cell transplantation. Mater. Sci. Forum 1997, 250, 15–42. [Google Scholar] [CrossRef]
- Agrawal, C.M.; Ray, R.B. Biodegradable polymeric scaffolds for musculoskeletal tissue engineering. J. Biomed. Mater. Res. 2001, 55, 141–150. [Google Scholar] [CrossRef]
- Kneser, U.; Schaefer, D.J.; Munder, B.; Klemt, C.; Andree, C.; Stark, G.B. Tissue engineering of bone. Minim. Invasive Ther. Allied Technol. 2004, 11, 107–116. [Google Scholar] [CrossRef]
- Salgado, A.J.; Coutinho, O.P.; Rui, L.R. Bone tissue engineering: State of the art and future trends. Macromol. Biosci. 2004, 4, 743–765. [Google Scholar] [CrossRef] [Green Version]
- Jodati, H.; Yılmaz, B.; Evis, Z. A review of bioceramic porous scaffolds for hard tissue applications: Effects of structural features. Ceram. Int. 2020, 46, 15725–15739. [Google Scholar] [CrossRef]
- Yu, J.H.; Xia, H.; Ni, Q.Q. A three-dimensional porous hydroxyapatite nanocomposite scaffold with shape memory effect for bone tissue engineering. J. Mater. Sci. 2018, 53, 4734–4744. [Google Scholar] [CrossRef]
- Zhai, W.; Lu, H.; Chen, L.; Lin, X.; Huang, Y.; Dai, K.; Naoki, K.; Chen, G.; Chang, J. Silicate bioceramics induce angiogenesis during bone regeneration. Acta Biomater. 2012, 8, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Pipattanawarothai, A.; Suksai, C.; Srisook, K.; Trakulsujaritchok, T. Non-cytotoxic hybrid bioscaffolds of chitosan-silica: Sol-gel synthesis, characterization and proposed application. Carbohydr. Polym. 2017, 178, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Jindal, A.; Mondal, T.; Bhattacharya, J. An In vitro evaluation of zinc silicate fortified chitosan scaffolds for bone tissue engineering. Int. J. Biol. Macromol. 2020, 164, 4252–4262. [Google Scholar] [CrossRef]
- Owens, G.J.; Singh, R.K.; Foroutan, F.; Alqaysi, M.; Han, C.M.; Mahapatra, C.; Kim, H.W.; Knowles, J.C. Sol-gel based materials for biomedical applications. Prog. Mater. Sci. 2016, 77, 1–79. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Liang, J.; Cui, Y.; Xu, S.; Zhao, N. Fabrication of novel bioactive hydroxyapatite-chitosan-silica hybrid scaffolds: Combined the sol-gel method with 3D plotting technique. Carbohydr. Polym. 2018, 197, 183–193. [Google Scholar] [CrossRef]
- Salama, A.; Zeid, R.E.; Sakhawy, M.; Gendy, A. Carboxymethyl cellulose/silica hybrids as templates for calcium phosphate biomimetic mineralization. Int. J. Biol. Macromol. 2015, 74, 155–161. [Google Scholar] [CrossRef]
- Bartlett, D.H.; Azam, F. Chitin, cholera, and competence. Science 2005, 310, 1775–1777. [Google Scholar] [CrossRef]
- Alonso, B.; Belamie, E. Chitin—silica nanocomposites by self-assembly. Angew. Chem. 2010, 122, 8377–8380. [Google Scholar] [CrossRef]
- Kumar, A.; Han, S.S. PVA-based hydrogels for tissue engineering: A review. Int. J. Polym. Mater. Polym. Biomater. 2017, 66, 159–182. [Google Scholar] [CrossRef]
- Budnyak, T.M.; Pylypchuk, I.V.; Tertykh, V.A.; Yanovska, E.S.; Kolodynska, D. Synthesis and adsorption properties of chitosan-silica nanocomposite prepared by sol-gel method. Nanoscale Res. Lett. 2015, 10, 87. [Google Scholar] [CrossRef] [Green Version]
- Anju, V.T.; Paramanantham, P.; Sruthil, L.; Sharan, A.; Siddhardha, B. Antimicrobial photodynamic activity of toluidine blue-carbon nanotube conjugate against Pseudomonas aeruginosa and Staphylococcus aureus—Understanding the mechanism of action. Photodiagnosis Photodyn. Ther. 2019, 27, 305–316. [Google Scholar] [CrossRef]
- Hu, J.; Feng, K.; Liu, X.; Ma, P.X. Chondrogenic and osteogenic differentiations of human bone marrow-derived mesenchymal stem cells on a nanofibrous scaffold with designed pore network. Biomaterials 2009, 30, 5061–5067. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ma, P.X. Phase separation, pore structure, and properties of nanofibrous gelatin scaffolds. Biomaterials 2009, 30, 4094–4103. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Tanaka, M.; Kinoshita, T.; Higuchi, M.; Tan, T. Nanofibrous scaffold from self-assembly of β-sheet peptides containing phenylalanine for controlled release. J. Control. Release 2010, 142, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Han, W.; Chen, H.; Mei, T.; Rong, Z.; Shi, Y.; Cha, Z.; Zhou, C. Preparation, structure and crystallinity of chitosan nano-fibers by a solid–liquid phase separation technique. Carbohydr. Polym. 2011, 83, 1541–1546. [Google Scholar] [CrossRef]
- Chen, V.J.; Ma, P.X. Nano-fibrous poly(L-lactic acid) scaffolds with interconnected spherical macropores. Biomaterials 2004, 25, 2065–2073. [Google Scholar] [CrossRef]
- Zhao, J.; Han, W.; Mei, T.; Huan, S.; Rong, Z.; Hao, W.; Cha, Z.; Zhou, C. Preparation and properties of biomimetic porous nanofibrous poly(L-lactide) scaffold with chitosan nanofiber network by a dual thermally induced phase separation technique. Mater. Sci. Eng. C 2012, 32, 1496–1502. [Google Scholar] [CrossRef] [PubMed]
- Jayakumar, R.; Menon, D.; Manzoor, K.; Nair, S.V.; Tamura, H. Biomedical applications of chitin and chitosan based nanomaterials—A short review. Carbohydr. Polym. 2010, 82, 227–232. [Google Scholar] [CrossRef]
- Saad, Q.; Muhammad, Z.; Shariq, N.; Zohaib, K.; Altaf, S.; Shehriar, H.; Ihtesham, R. Electrospinning of chitosan-based solutions for tissue engineering and regenerative medicine. Int. J. Mol. Sci. 2018, 19, 407. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.H.; Zhao, G.L.; Wang, J.M.; Xie, C.; Liang, W.Q.; Chen, K.B.; Wen, Y.; Li, X.F. Organic Solvent-Free Preparation of Chitosan Nanofibers with High Specific Surface Charge and Their Application in Biomaterials. ACS Appl. Mater. Interfaces 2021, 13, 12347–12358. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Ito, S.; Obata, A.; Mizuno, T.; Jones, J.R.; Kasuga, T. Fabrication and in vitro characterization of electrospun poly (γ-glutamic acid)-Silica hybrid scaffolds for bone regeneration. Polymer 2016, 91, 106–117. [Google Scholar] [CrossRef]
- Lukanina, K.I.; Grigoriev, T.E.; Krasheninnikov, S.V.; Mamagulashvilli, V.G.; Kamyshinsky, R.A.; Chvalun, S.N. Multi-hierarchical tissue-engineering ECM-like scaffolds based on cellulose acetate with collagen and chitosan fillers. Carbohydr. Polym. 2018, 191, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Saiz, E.; Rahaman, M.N.; Tomsia, A.P. Bioactive glass scaffolds for bone tissue engineering: State of the art and future perspectives. Mater. Sci. Eng. C 2011, 31, 1245–1256. [Google Scholar] [CrossRef] [Green Version]
- Fu, Q.; Rahaman, M.N.; Dogan, F.; Bal, B.S. Freeze casting of porous hydroxyapatite scaffolds. I. Processing and general microstructure. J. Biomed. Mater. Res. B Appl. Biomater. 2010, 86B, 125–135. [Google Scholar] [CrossRef]
- John, L.; Ba Tt Rukiewicz, M.; Sobota, P.; Brykner, R.; Cwynar-Zajaac, T.; Dzifgiel, P. Non-cytotoxic organic-inorganic hybrid bioscaffolds: An efficient bedding for rapid growth of bone-like apatite and cell proliferation. Mater. Sci. Eng. 2012, 32, 1849–1858. [Google Scholar] [CrossRef]
- Marchessault, R.H.; Pearson, F.G.; Liang, C.Y. Infrared spectra of crystalline polysaccharides: VI. Effect of orientation on the tilting spectra of chitin films. Biochim. Biophys. Acta 1960, 45, 499–507. [Google Scholar] [CrossRef]
- Bryaskova, R.; Pencheva, D.; Kale, G.M.; Lad, U.; Kantardjiev, T. Synthesis, characterisation and antibacterial activity of PVA/TEOS/Ag-Np hybrid thin films. J. Colloid Interface Sci. 2010, 349, 77–85. [Google Scholar] [CrossRef]
- Georgieva, N.; Bryaskova, R.; Tzoneva, R. New Polyvinyl alcohol-based hybrid materials for biomedical application. Mater. Lett. 2012, 88, 19–22. [Google Scholar] [CrossRef]
- Dodda, J.M.; Bělský, P.; Chmelař, J.; Remiš, T.; Smolná, K.; Tomáš, M.; Kullová, L.; Kadlec, J. Comparative study of PVA/SiO2 and PVA/SiO2/glutaraldehyde (GA) nanocomposite membranes prepared by single-step solution casting method. J. Mater. Sci. 2015, 50, 6477–6490. [Google Scholar] [CrossRef]
- Abdelfattah, A.M.E.; Saeid, A.M.M.; Mahmoud, S.A.; Fathy, K.H.; Mohamed, E.G. Chitosan nanoparticles extracted from shrimp shells, application for removal of Fe(II) and Mn(II) from aqueous phases. Sep. Sci. Technol. 2018, 53, 2870–2881. [Google Scholar] [CrossRef]
- Muzzarelli, C.; Francescangeli, O.; Tosi, G.; Muzzarelli, R.A.A. Susceptibility of dibutyryl chitin and regenerated chitin fibres to deacylation and depolymerization by lipases. Carbohydr. Polym. 2004, 56, 137–146. [Google Scholar] [CrossRef]
- Kruyt, M.C.; Bruijn, J.; Wilson, C.E.; Oner, F.C.; Dhert, W. Viable Osteogenic Cells Are Obligatory for Tissue-Engineered Ectopic Bone Formation in Goats. Tissue Eng. 2003, 9, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Zeng, Y.P.; Jiang, D. Preparation of porous TiO2 by a novel freeze casting. Ceram. Int. 2009, 35, 1267–1270. [Google Scholar] [CrossRef]
- Zuo, K.H. Properties of microstructure-controllable porous yttria-stabilized ziroconia ceramics fabricated by freeze casting. Int. J. Appl. Ceram. Technol. 2010, 5, 198–203. [Google Scholar] [CrossRef]
- Polo Corrales, L.; Latorre Esteves, M.; Ramirez Vick, J.E. Scaffold design for bone regeneration. J. Nanosci. Nanotechnol. 2014, 14, 15–56. [Google Scholar] [CrossRef] [Green Version]
- Kharkar, P.M.; Kiick, K.L.; Kloxin, A.M. Designing degradable hydrogels for orthogonal control of cell microenvironments. Chem. Soc. Rev. 2013, 42, 7335–7372. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Kim, H.W. Emerging properties of hydrogels in tissue engineering. J. Tissue Eng. 2018, 9, 4. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Code | Chitosan, 2.5% (w/w) | Hydrolysis Type | TEOS Concentration% (w/w) | PVA Concentration% (w/w) |
|---|---|---|---|---|
| Types of Chitosan | ||||
| P-A-T3 | poder | NH4OH (A) | 3 | — |
| F-A-T0 | fiber | NH4OH (A) | 0 | — |
| F-A-T3 | fiber | NH4OH (A) | 3 | — |
| F-A-T5 | fiber | NH4OH (A) | 5 | — |
| F-B-T5-P0 | fiber | HCl (B) | 5 | 0 |
| F-B-T5-P0.5 | fiber | HCl (B) | 5 | 0.5 |
| F-B-T5-P1.5 | fiber | HCl (B) | 5 | 1.5 |
| F-B-T5-P2.5 | fiber | HCl (B) | 5 | 2.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, W.; Zhang, S.; Xie, C.; Wan, X.; Li, X.; Chen, K.; Zhao, G. Preparation of High Mechanical Strength Chitosan Nanofiber/NanoSiO2/PVA Composite Scaffolds for Bone Tissue Engineering Using Sol–Gel Method. Polymers 2022, 14, 2083. https://doi.org/10.3390/polym14102083
Ma W, Zhang S, Xie C, Wan X, Li X, Chen K, Zhao G. Preparation of High Mechanical Strength Chitosan Nanofiber/NanoSiO2/PVA Composite Scaffolds for Bone Tissue Engineering Using Sol–Gel Method. Polymers. 2022; 14(10):2083. https://doi.org/10.3390/polym14102083
Chicago/Turabian StyleMa, Wei, Sihan Zhang, Chong Xie, Xing Wan, Xiaofeng Li, Kebing Chen, and Guanglei Zhao. 2022. "Preparation of High Mechanical Strength Chitosan Nanofiber/NanoSiO2/PVA Composite Scaffolds for Bone Tissue Engineering Using Sol–Gel Method" Polymers 14, no. 10: 2083. https://doi.org/10.3390/polym14102083
APA StyleMa, W., Zhang, S., Xie, C., Wan, X., Li, X., Chen, K., & Zhao, G. (2022). Preparation of High Mechanical Strength Chitosan Nanofiber/NanoSiO2/PVA Composite Scaffolds for Bone Tissue Engineering Using Sol–Gel Method. Polymers, 14(10), 2083. https://doi.org/10.3390/polym14102083