
Random and Diblock Thermoresponsive Oligo(ethylene glycol)-Based Copolymers Synthesized via Photo-Induced RAFT Polymerization

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
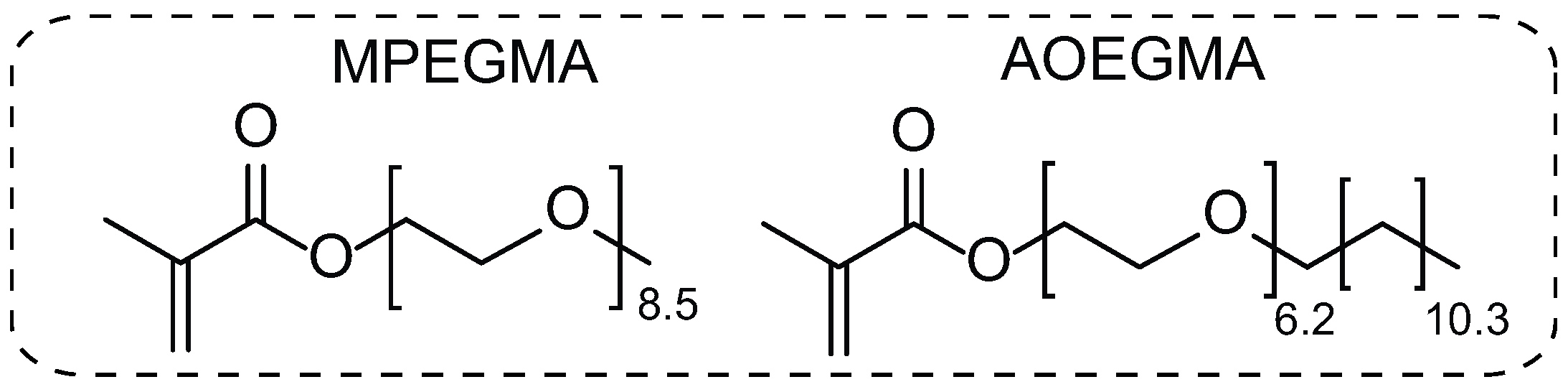
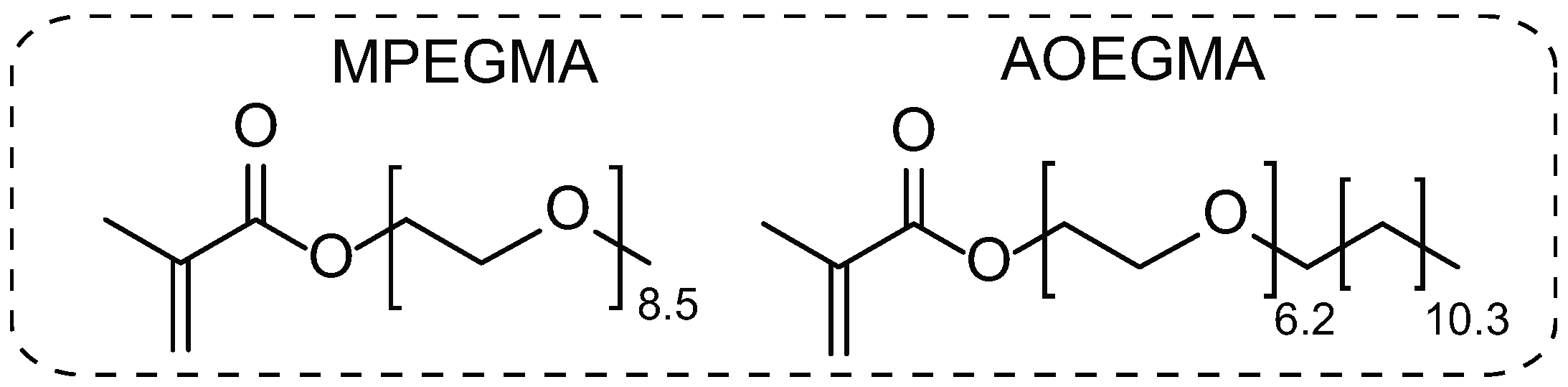
2.2. Synthesis of AOEGMA
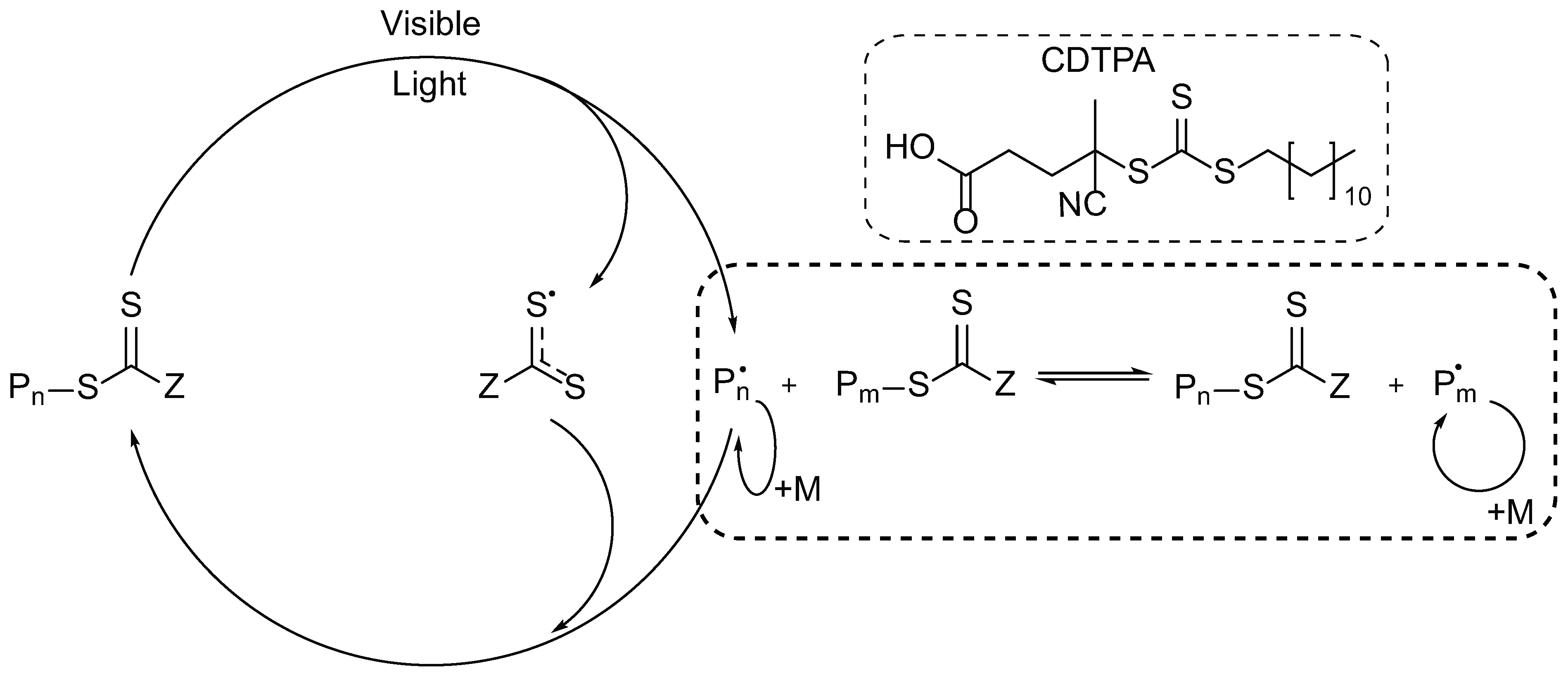
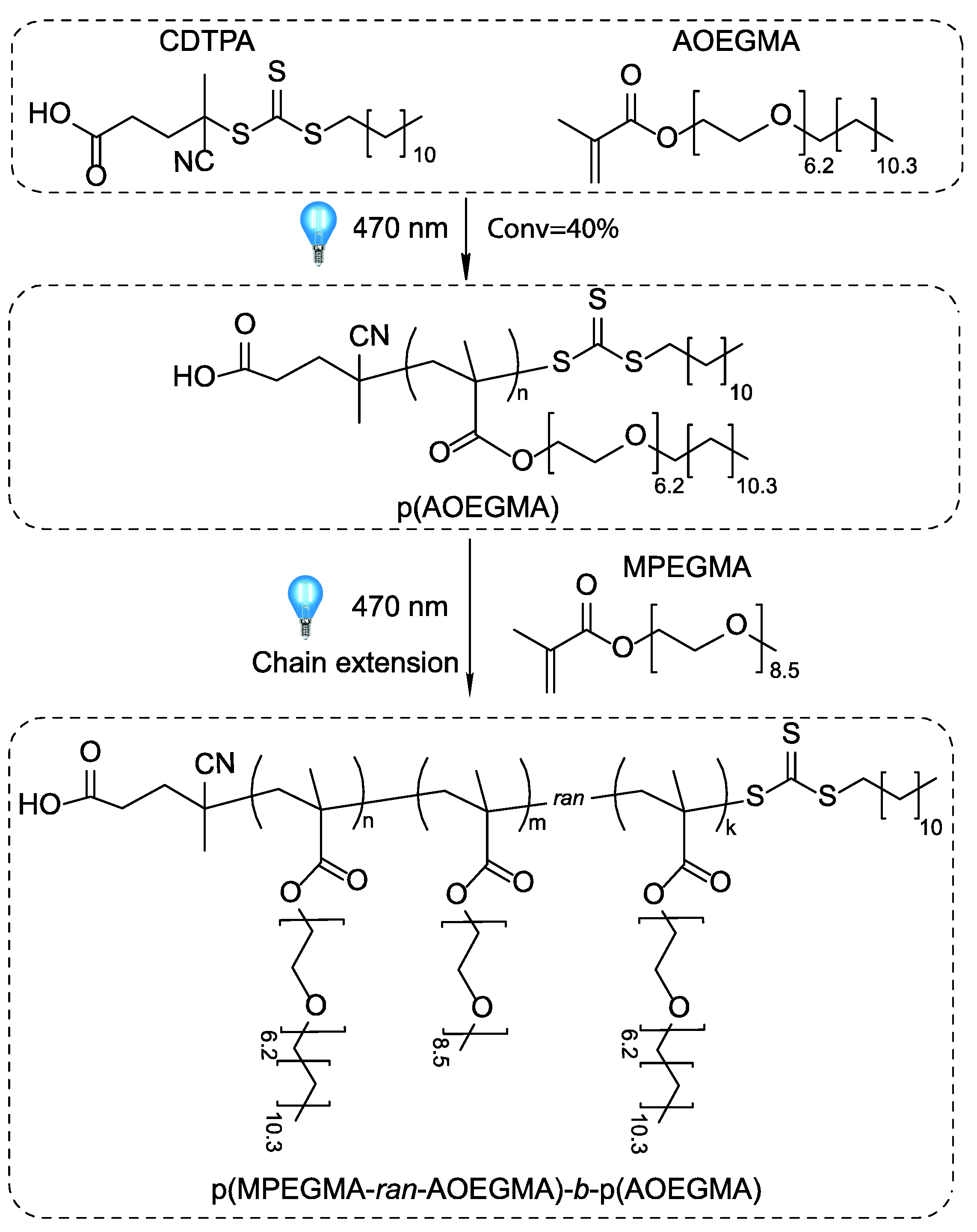
2.3. Photoiniferter RAFT Polymerization
2.4. Characterization Techniques
2.4.1. General Methods
2.4.2. Dynamic (DLS) and Static (SLS) Light Scattering
2.4.3. Turbidimetry
3. Results and Discussion
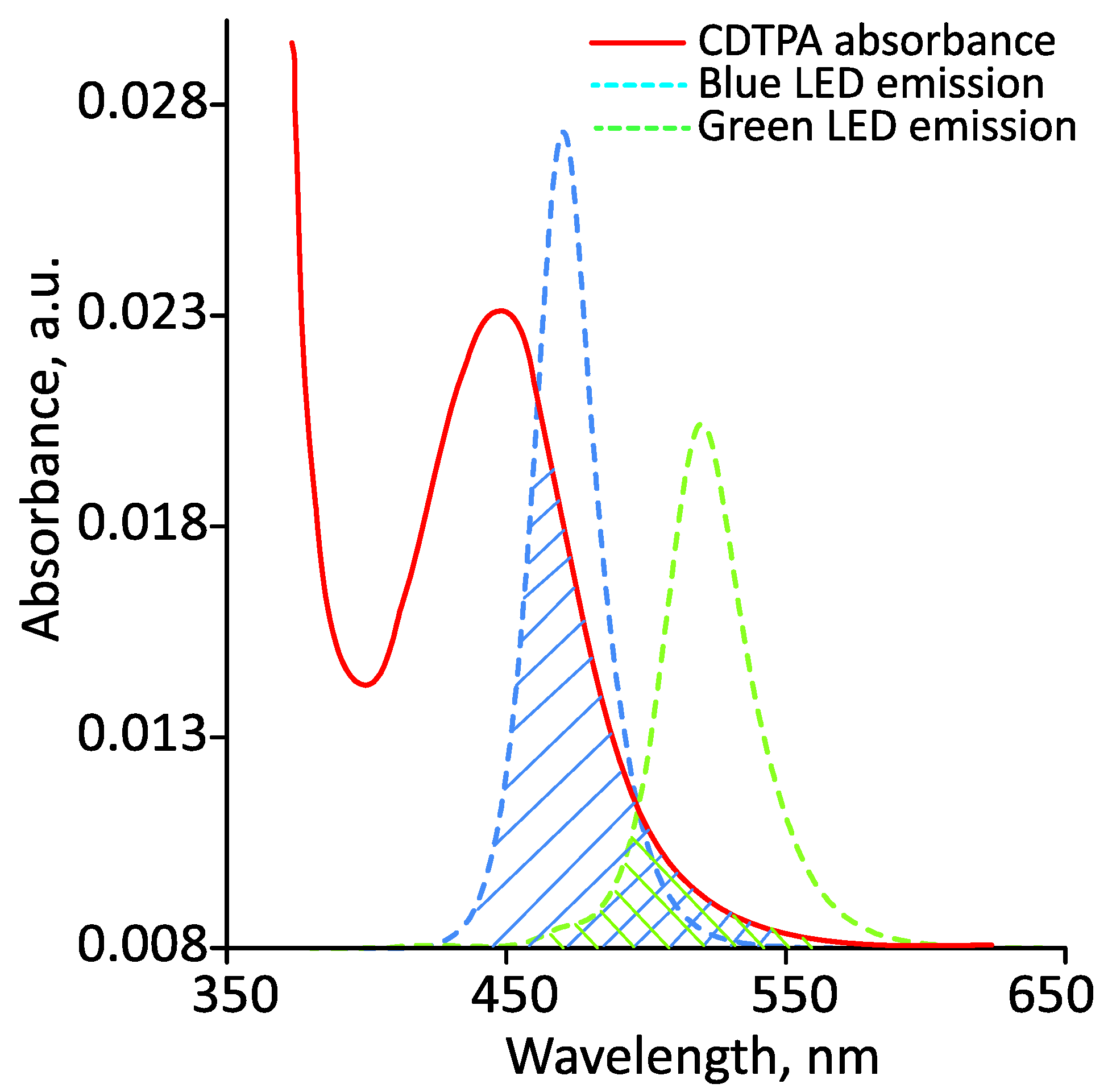
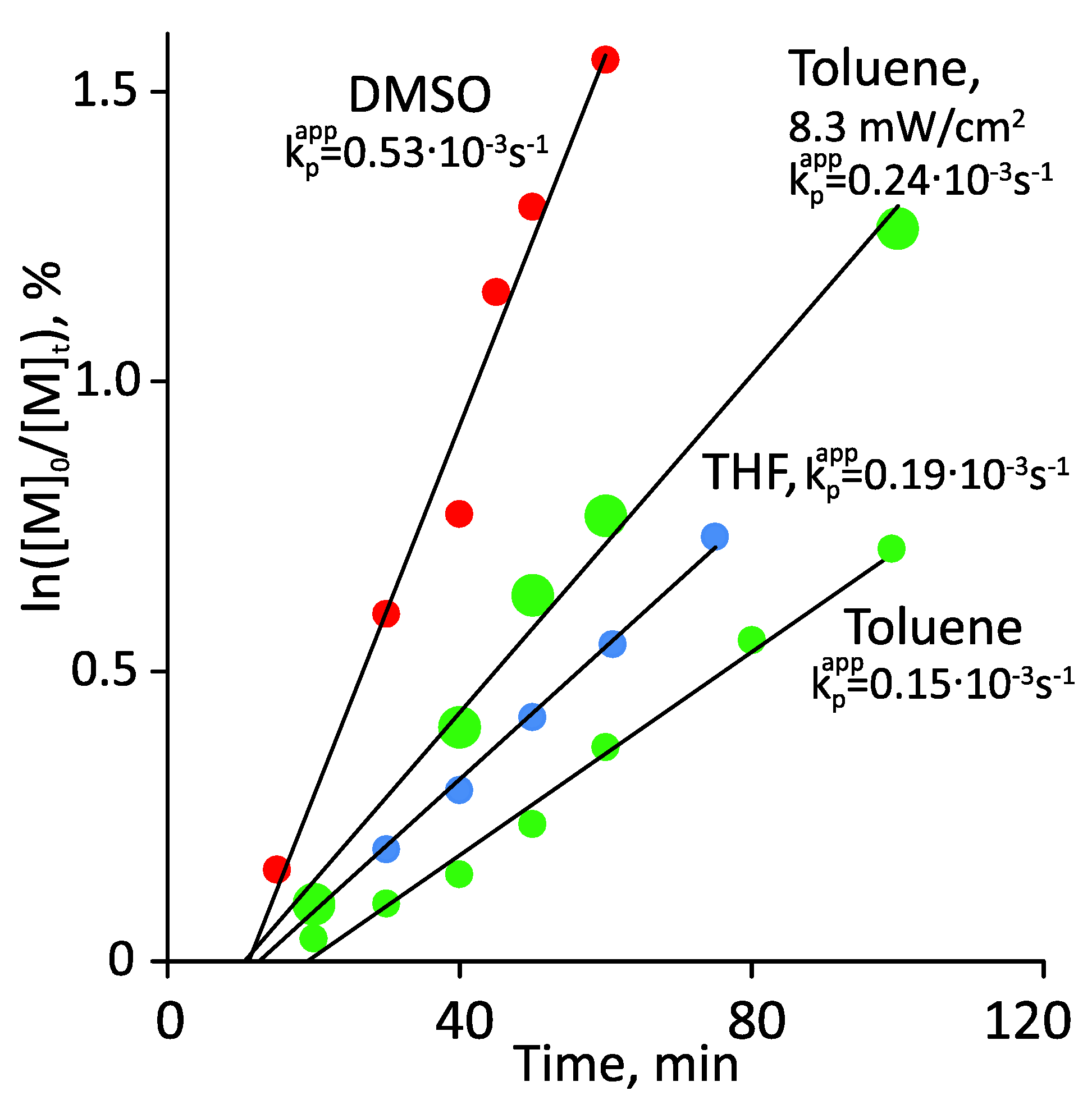
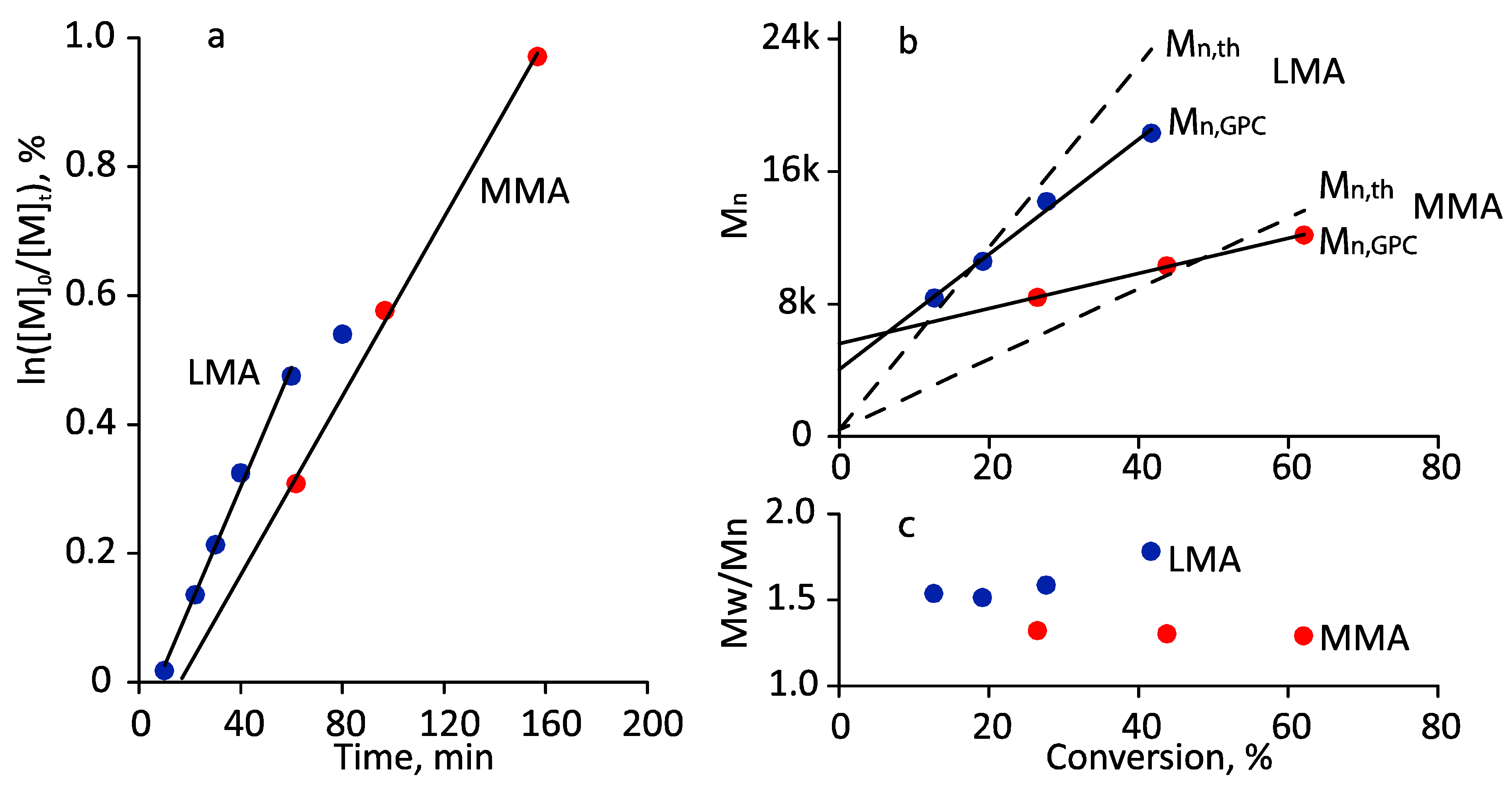
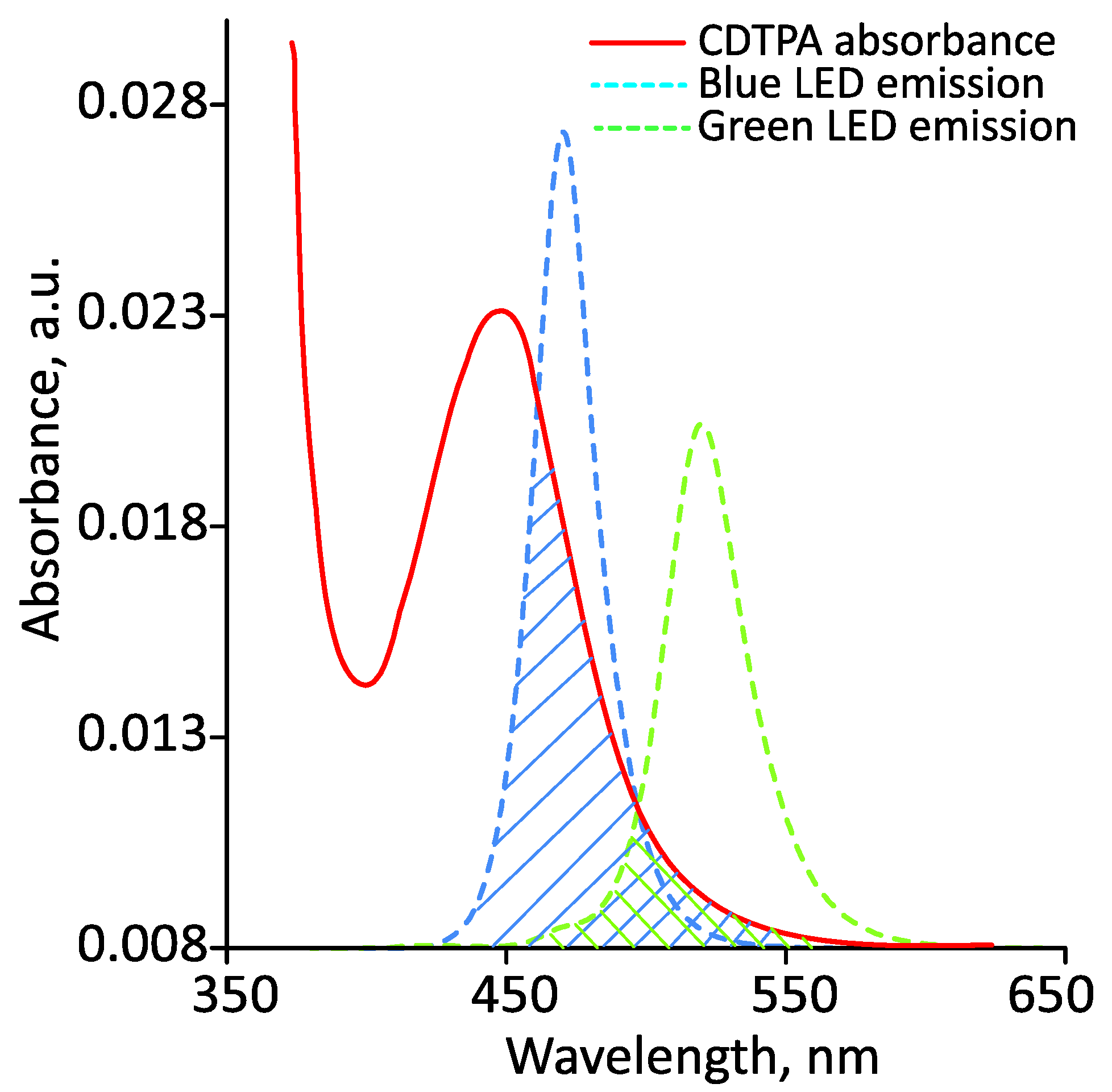
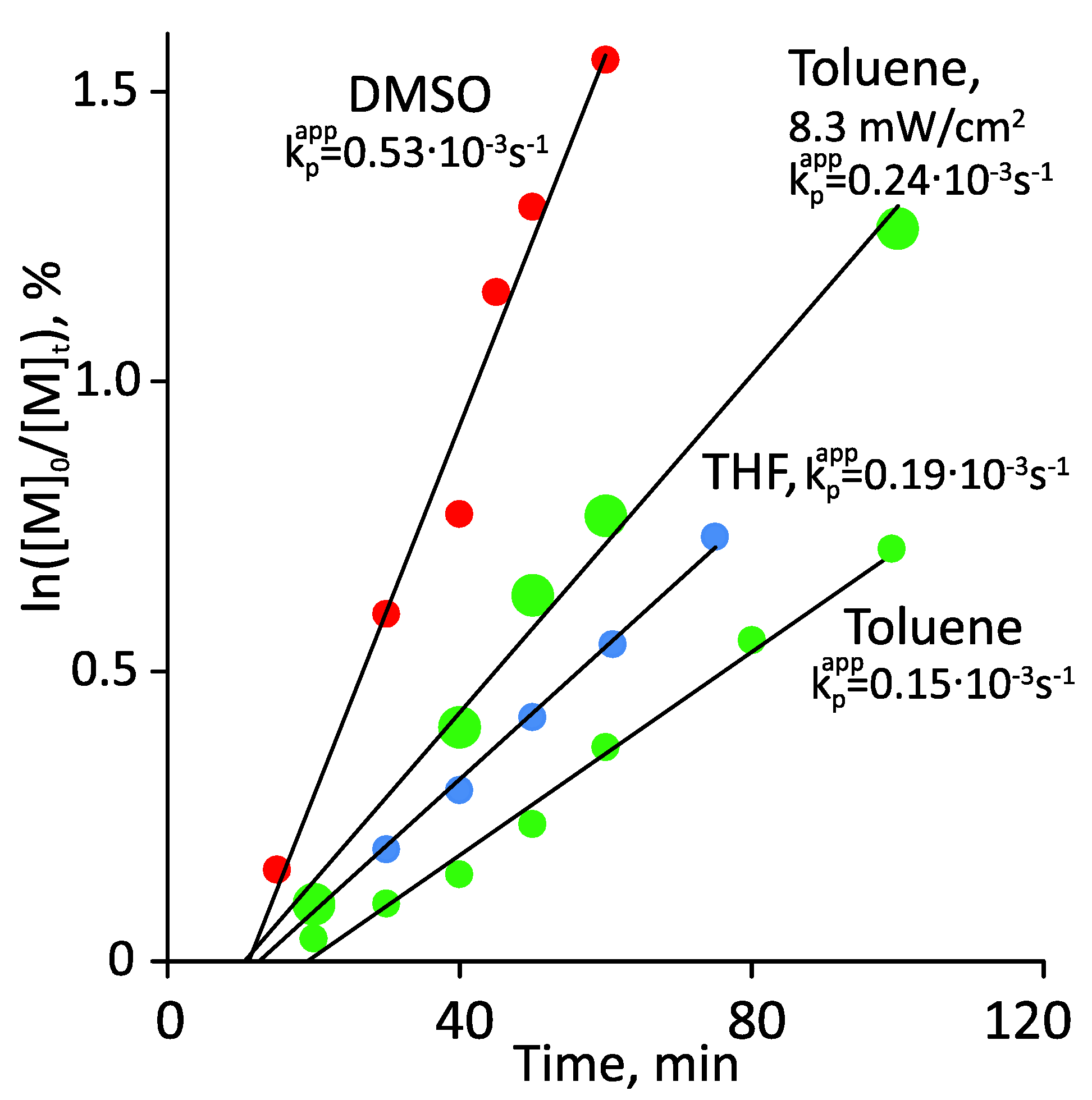
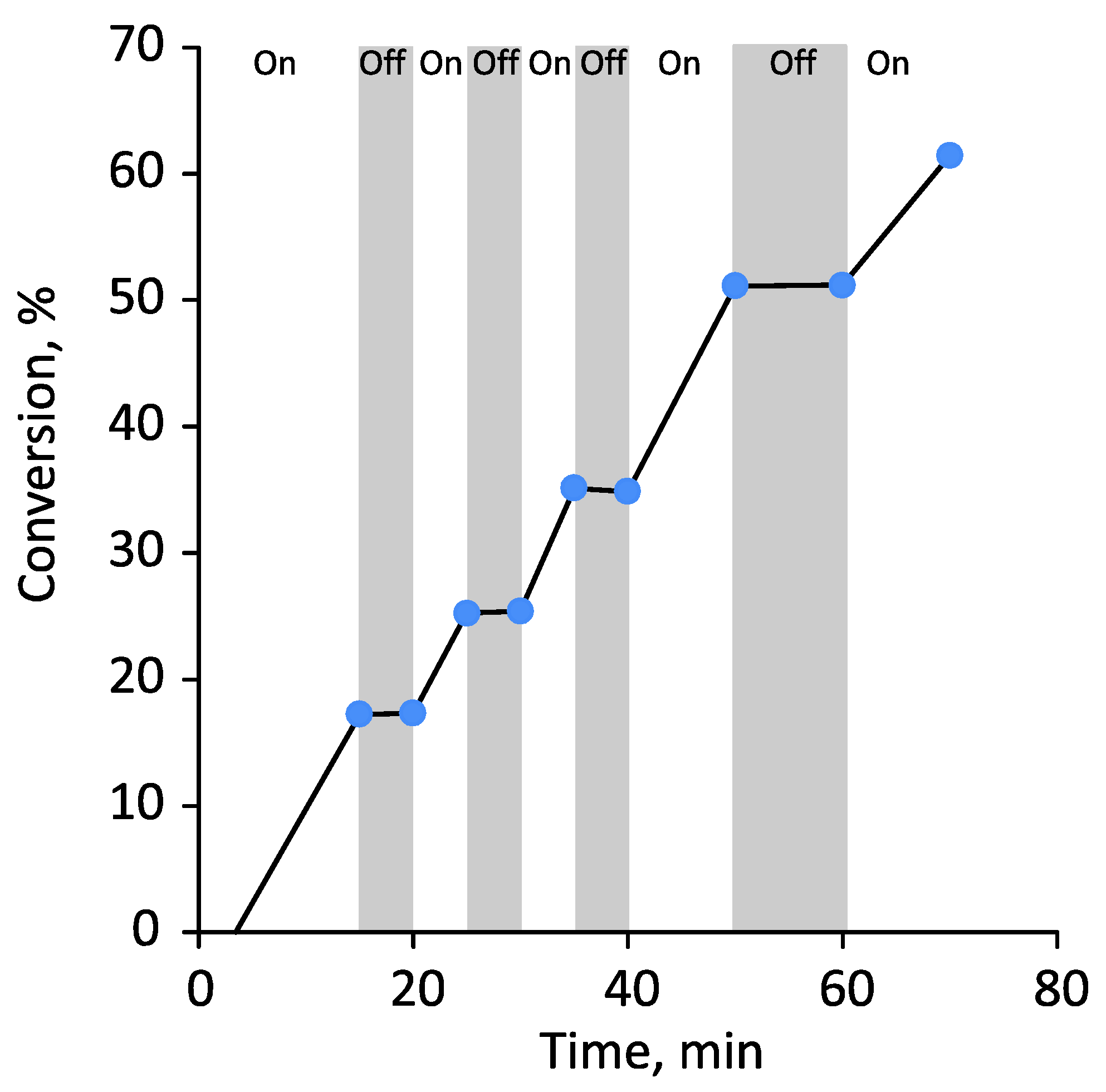
3.1. Photoiniferter RAFT Homopolymerization in Different Solvents
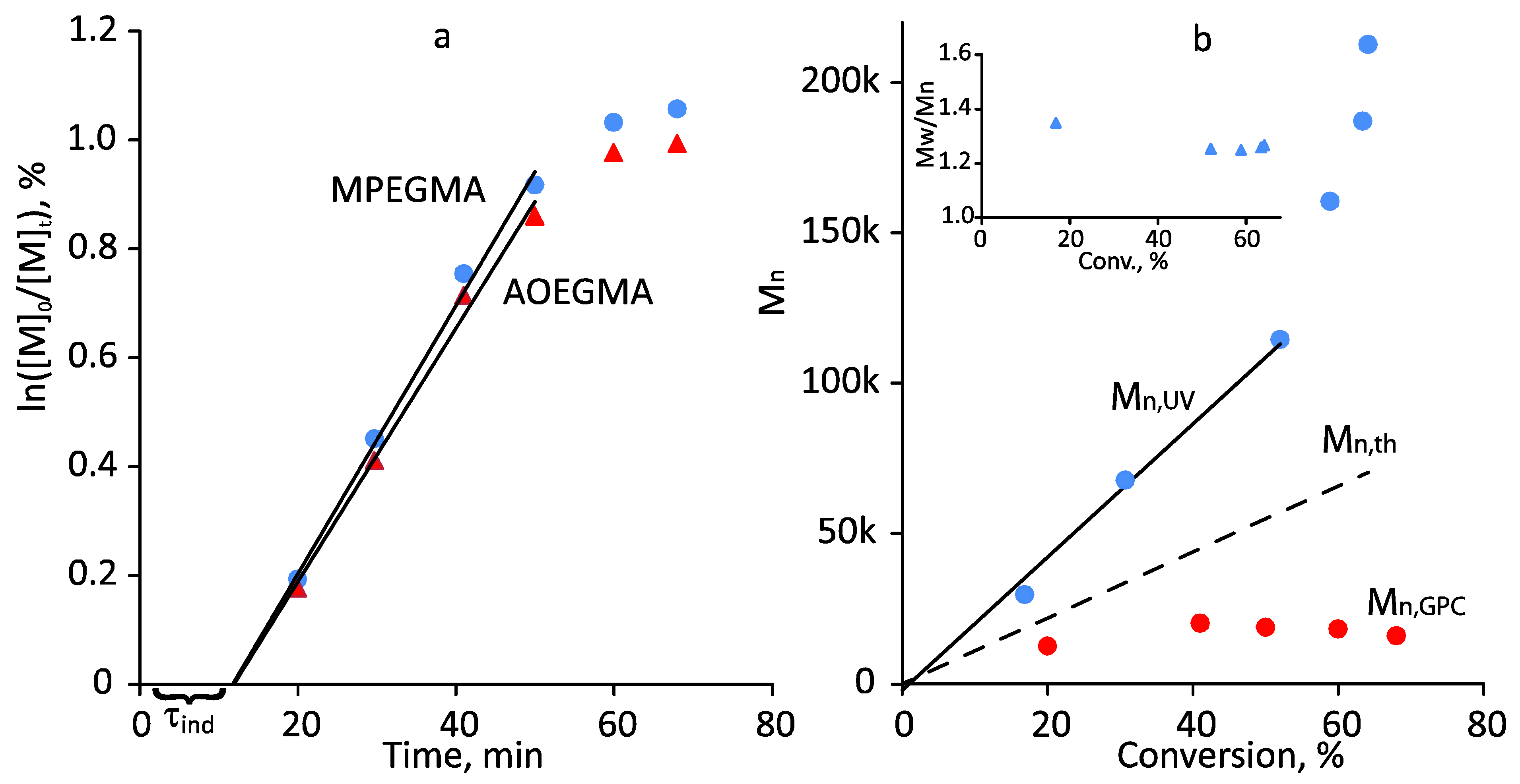
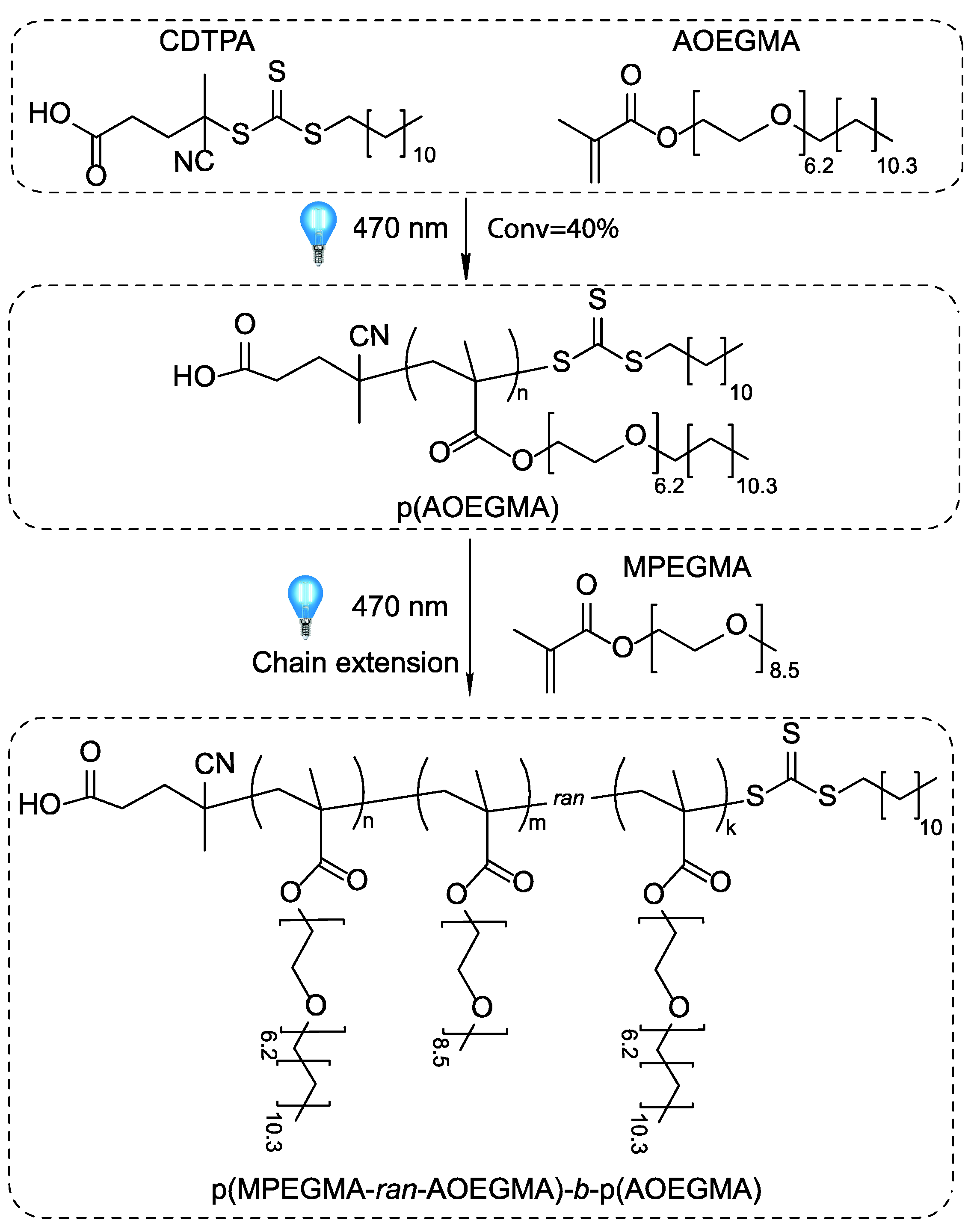
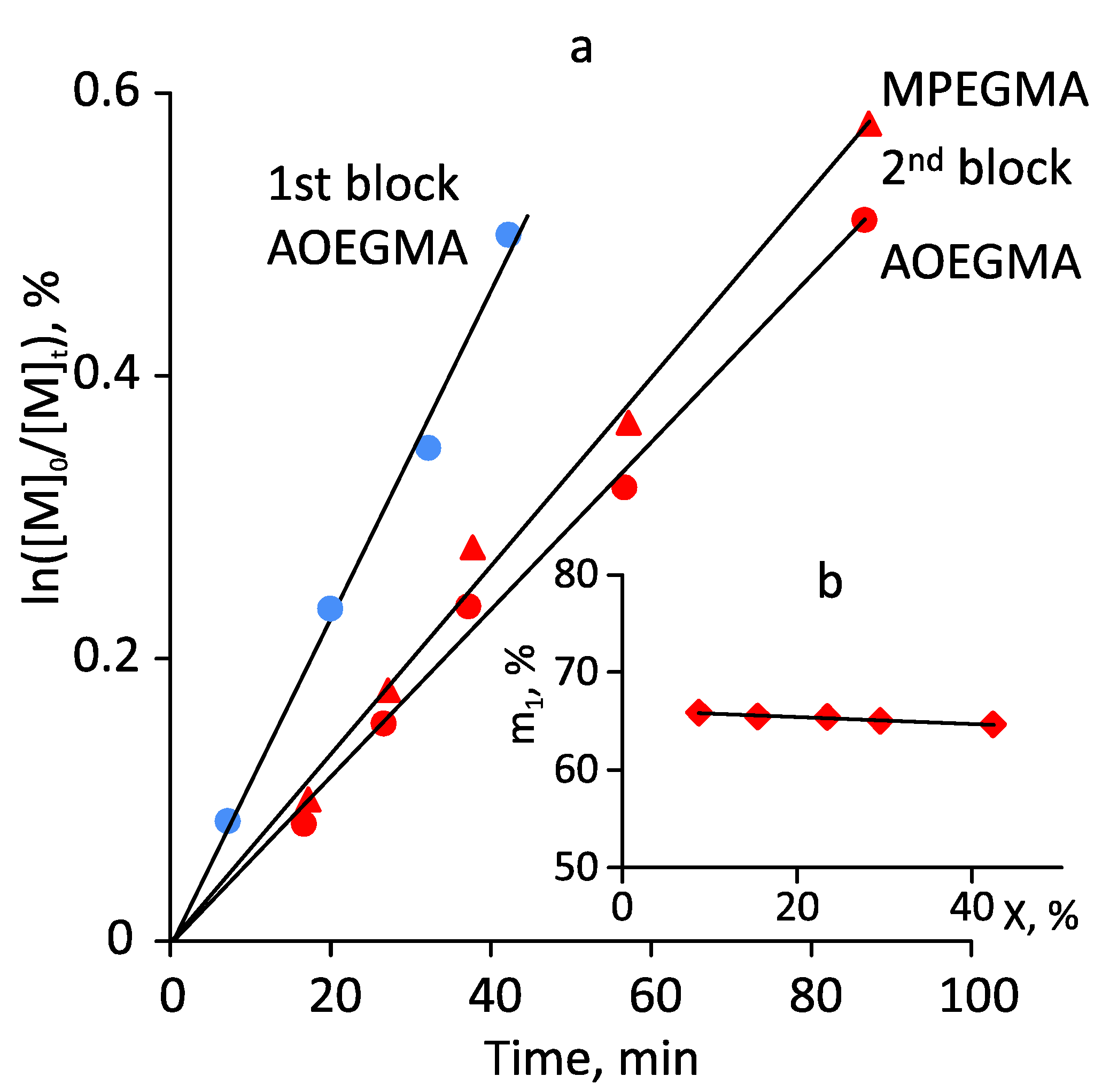
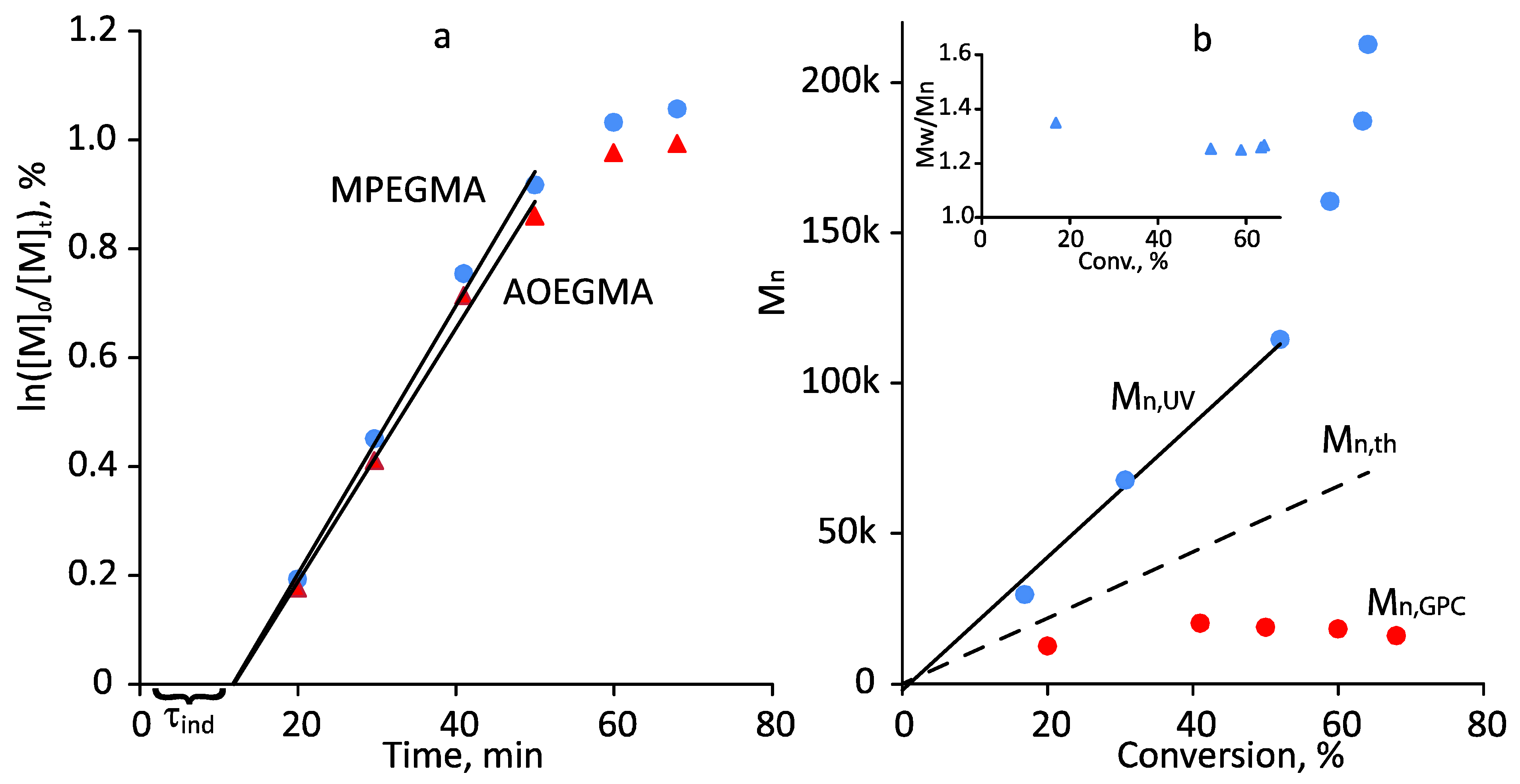
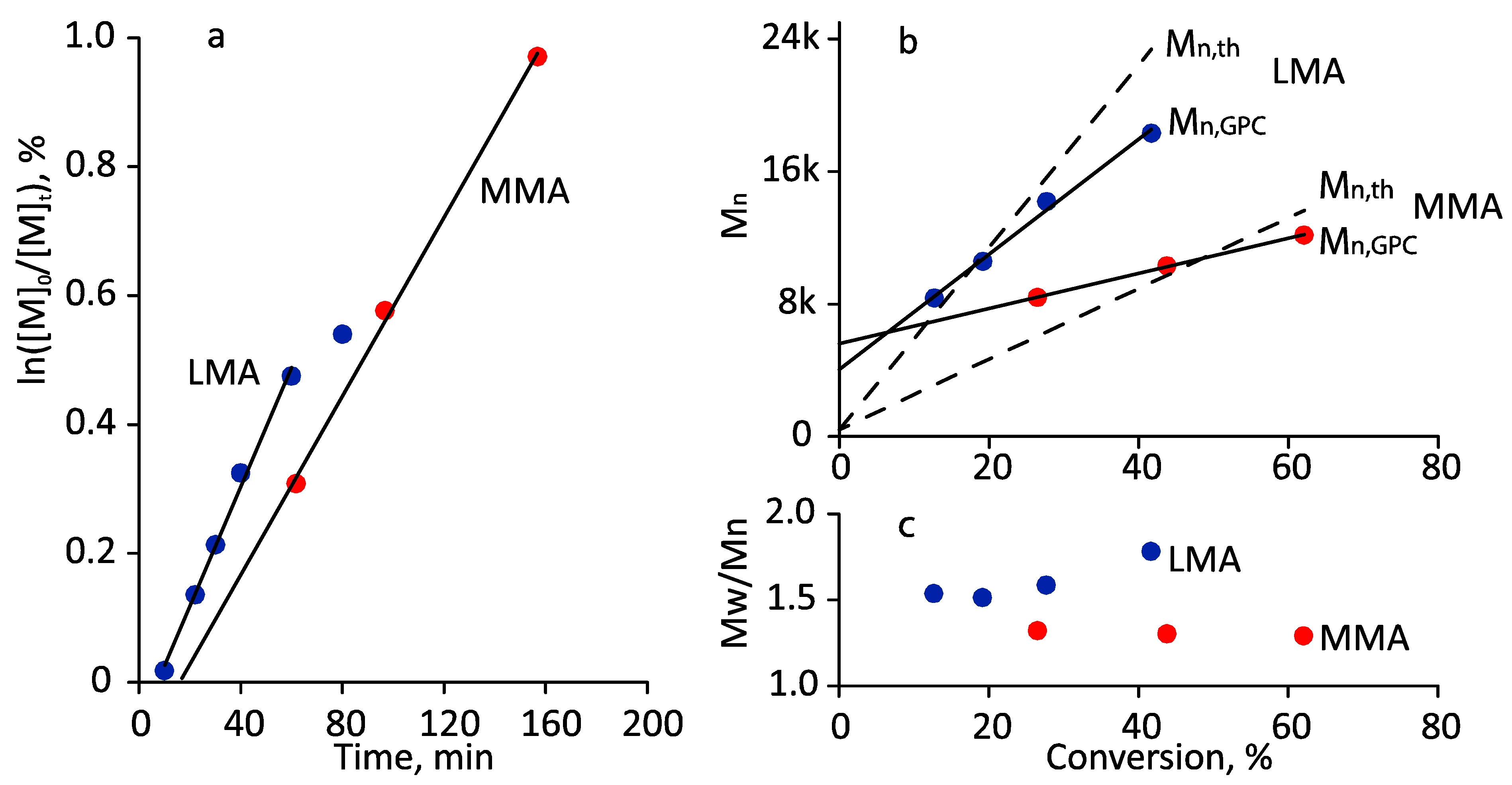
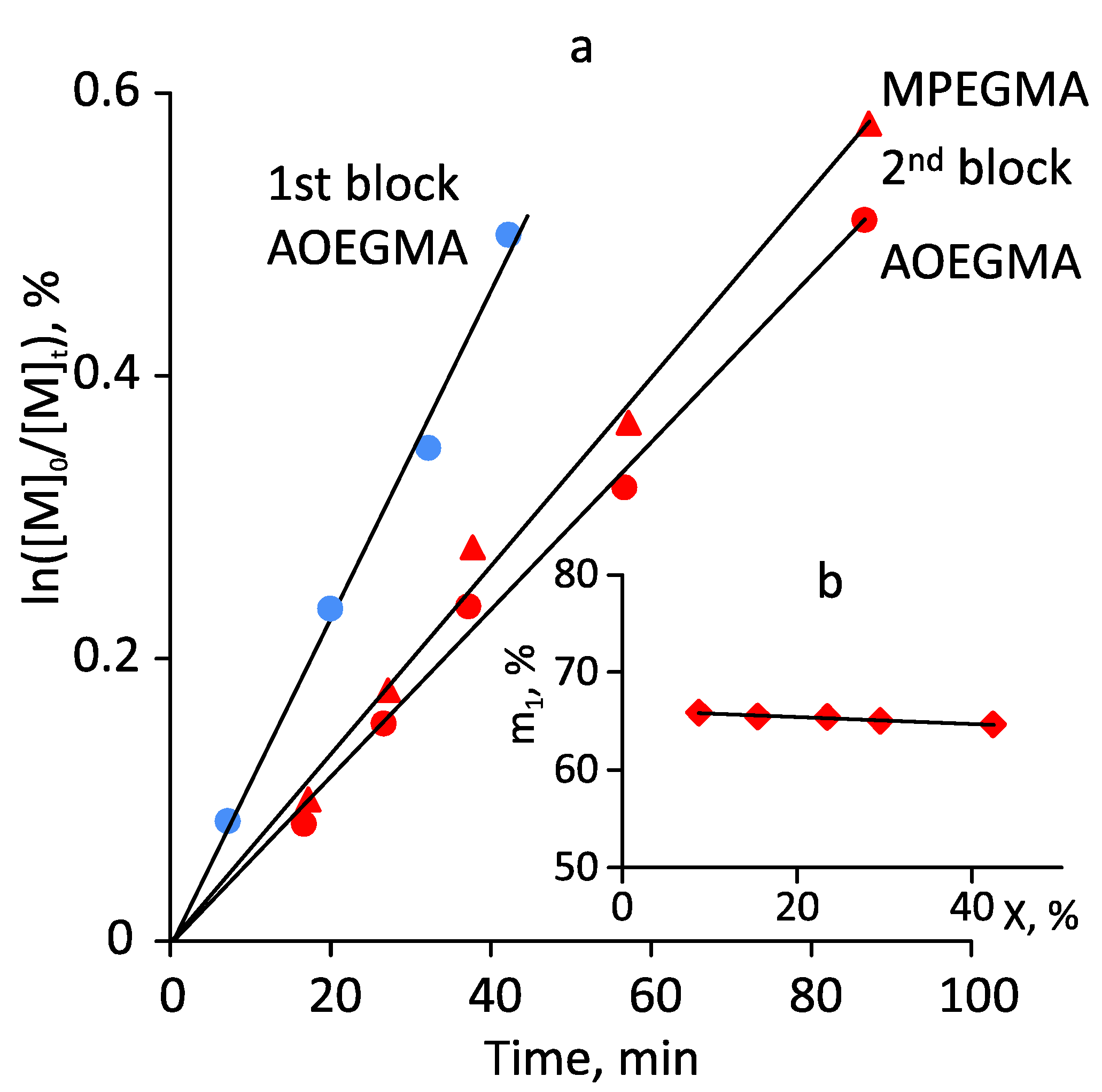
3.2. Photoiniferter RAFT Copolymerization
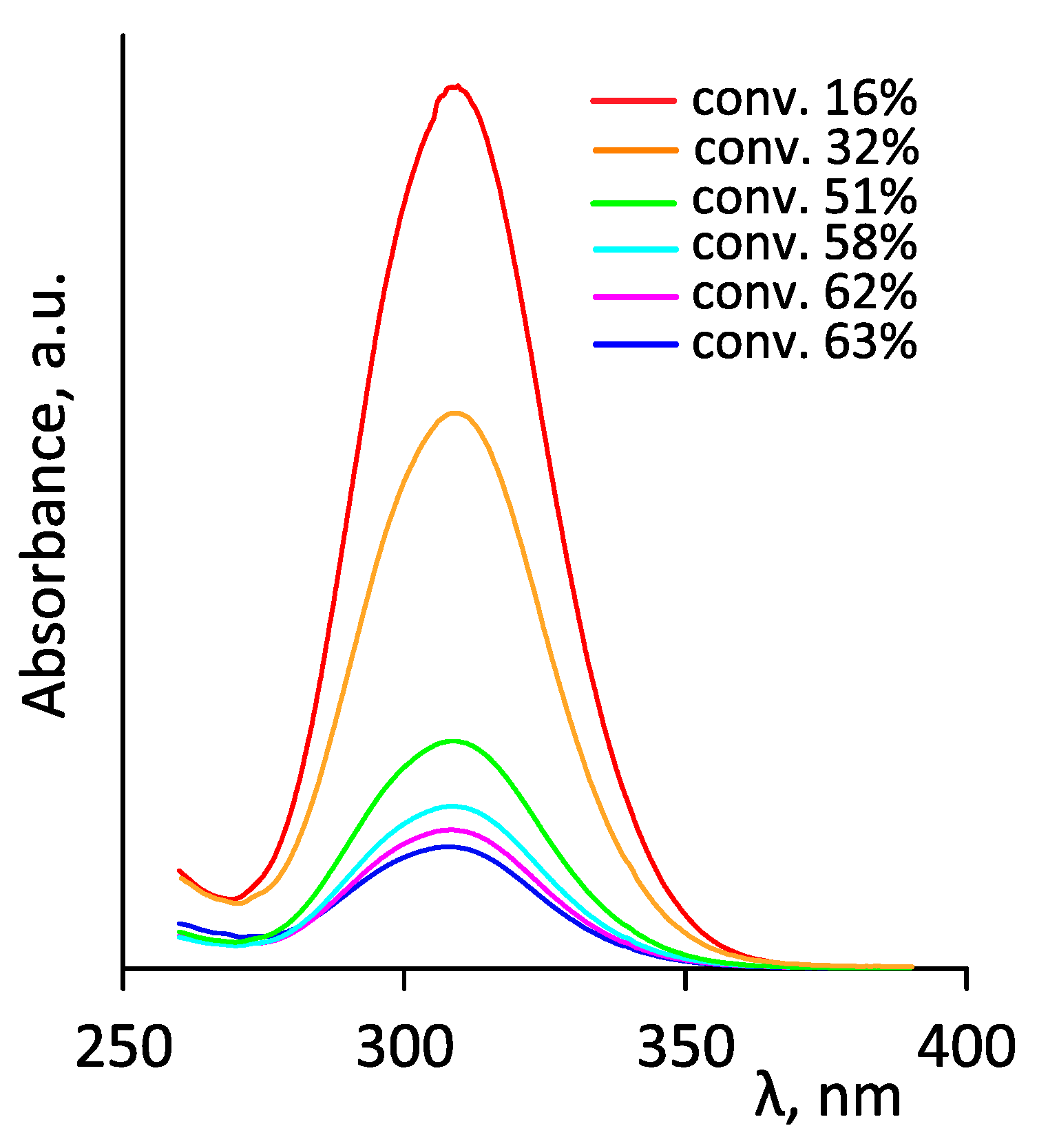
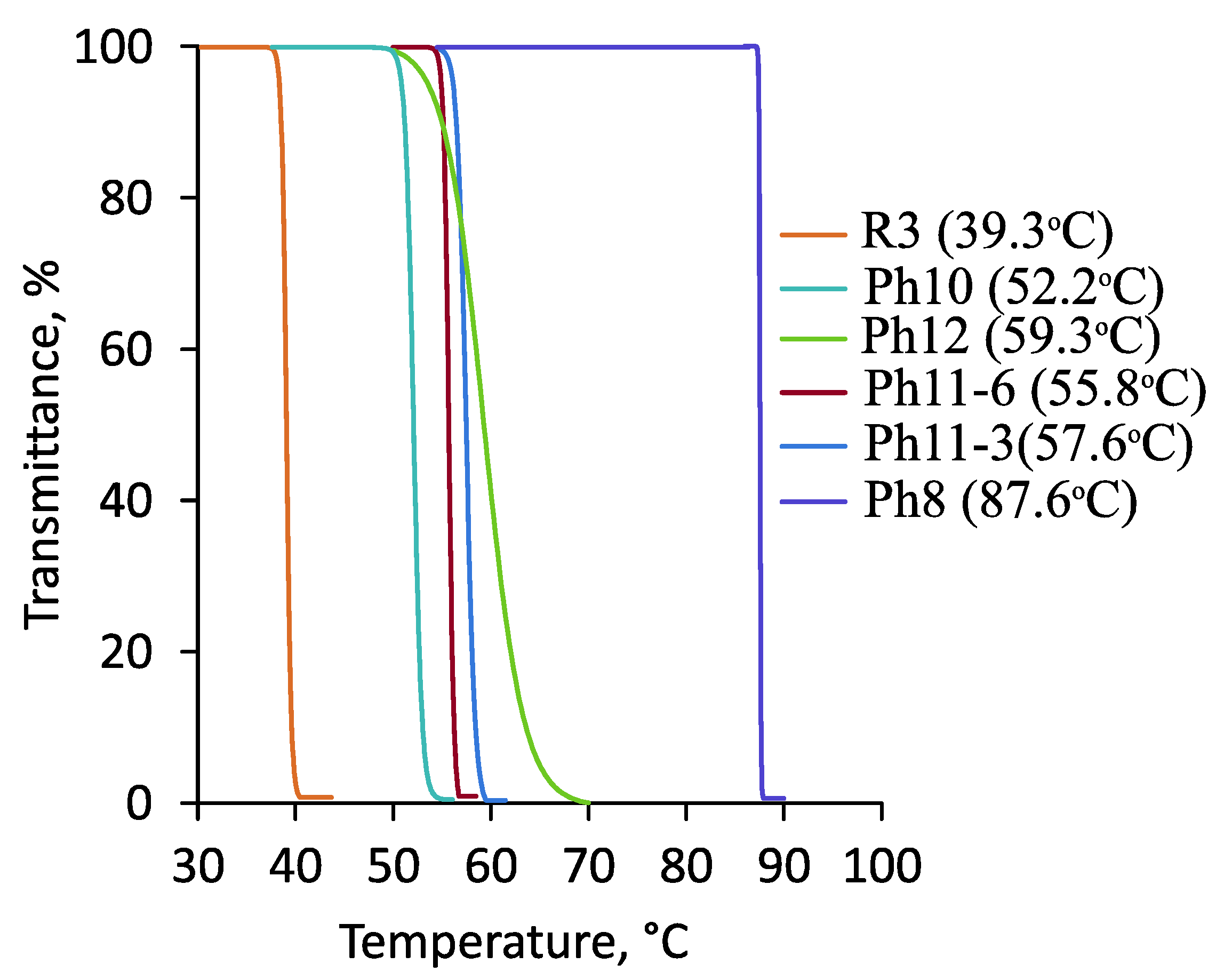
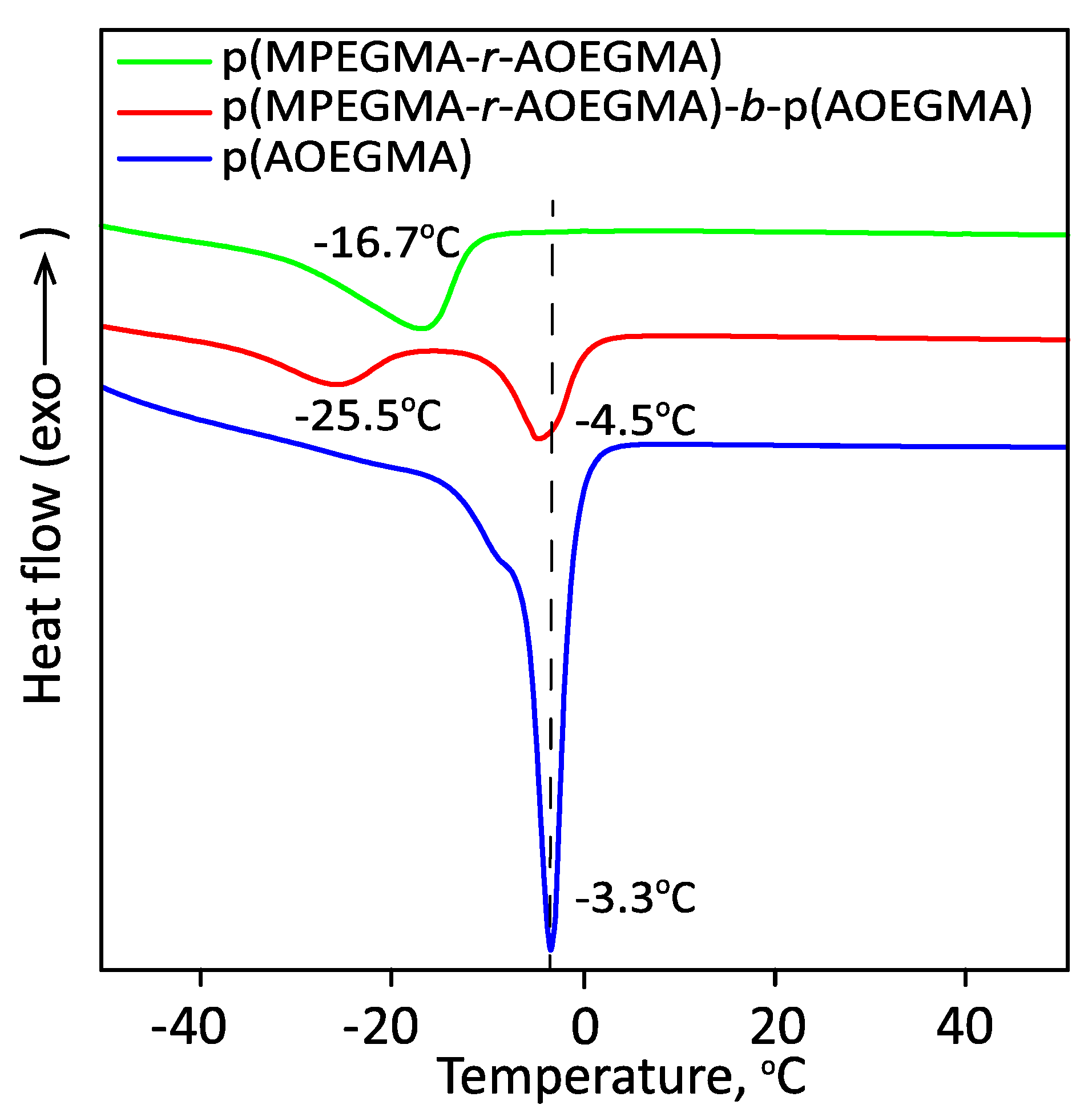
3.3. Thermoresponsive Properties, Hydrodynamic and Molecular Weight Characteristics of the Synthesized (Co)Polymers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AOEGMA | alkoxy oligo(ethylene glycol) methacrylate |
| CDTPA | 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid |
| Cp | cloud point |
| CTA | chain transfer agent |
| DLS | dynamic light scattering |
| DSC | differential scanning calorimetry |
| LCST | lower critical solution temperature |
| LED | light-emitting diode |
| LMA | lauryl methacrylate |
| MA | methacrylic acid |
| MMA | methyl methacrylate |
| Mn,GPC | number average molecular weight determined through gel permeation chromatography |
| Mn,th | theoretical number average molecular weight |
| Mn,UV | Number average molecular weight determined through UV–Vis spectroscopy |
| MPEGMA | methoxy oligo(ethylene glycol) methacrylate |
| OEGMA | oligo(ethylene glycol) methacrylate |
| PEG | polyethylene glycol |
| PEGMA | poly(ethylene glycol) methacrylate |
| PET-RAFT | photo-induced electron/energy transfer RAFT |
| RAFT | reversible addition−fragmentation chain-transfer |
| SCNP | single chain nanoparticle |
| SLS | static light scattering |
References
- Xu, J.; Jung, K.; Atme, A.; Shanmugam, S.; Boyer, C. A Robust and Versatile Photoinduced Living Polymerization of Conjugated and Unconjugated Monomers and Its Oxygen Tolerance. J. Am. Chem. Soc. 2014, 136, 5508–5519. [Google Scholar] [CrossRef]
- Nomeir, B.; Fabre, O.; Ferji, K. Effect of Tertiary Amines on the Photoinduced Electron Transfer-Reversible Addition–Fragmentation Chain Transfer (PET-RAFT) Polymerization. Macromolecules 2019, 52, 6898–6903. [Google Scholar] [CrossRef]
- Peng, J.; Xu, Q.; Ni, Y.; Zhang, L.; Cheng, Z.; Zhu, X. Visible light controlled aqueous RAFT continuous flow polymerization with oxygen tolerance. Polym. Chem. 2019, 10, 2064–2072. [Google Scholar] [CrossRef]
- Lamb, J.R.; Qin, K.P.; Johnson, J.A. Visible-light-mediated, additive-free, and open-to-air controlled radical polymerization of acrylates and acrylamides. Polym. Chem. 2019, 10, 1585–1590. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Abetz, V. Nonionic UCST–LCST Diblock Copolymers with Tunable Thermoresponsiveness Synthesized via PhotoRAFT Polymerization. Macromol. Rapid Commun. 2021, 42, 2000648. [Google Scholar] [CrossRef]
- Lu, Z.; Yang, H.; Fu, X.; Zhao, Y.; Xiao, L.; Zhang, Z.; Hou, L. Visible Light-Regulated Heterogeneous Catalytic PET-RAFT by High Crystallinity Covalent Organic Framework. Macromol. Rapid Commun. 2021, 42, 2100384. [Google Scholar] [CrossRef] [PubMed]
- Rasines Mazo, A.; Tran, T.N.; Zhang, W.; Meng, Y.; Reyhani, A.; Pascual, S.; Fontaine, L.; Qiao, G.G.; Piogé, S. Blue LED light-activated RAFT polymerization of PEG acrylate with high chain-end fidelity for efficient PEGylation. Polym. Chem. 2020, 11, 5238–5248. [Google Scholar] [CrossRef]
- Wang, J.; Hu, X.; Zhu, N.; Guo, K. Continuous flow photo-RAFT and light-PISA. Chem. Eng. J. 2021, 420, 127663. [Google Scholar] [CrossRef]
- Arrington, K.J.; Matson, J.B. Assembly of a visible light photoreactor: An inexpensive tool for bottlebrush polymer synthesis via photoiniferter polymerization. Polym. Chem. 2017, 8, 7452–7456. [Google Scholar] [CrossRef]
- Thum, M.D.; Wolf, S.; Falvey, D.E. State-Dependent Photochemical and Photophysical Behavior of Dithiolate Ester and Trithiocarbonate Reversible Addition–Fragmentation Chain Transfer Polymerization Agents. J. Phys. Chem. A 2020, 124, 4211–4222. [Google Scholar] [CrossRef]
- Shanmugam, S.; Cuthbert, J.; Kowalewski, T.; Boyer, C.; Matyjaszewski, K. Catalyst-Free Selective Photoactivation of RAFT Polymerization: A Facile Route for Preparation of Comblike and Bottlebrush Polymers. Macromolecules 2018, 51, 7776–7784. [Google Scholar] [CrossRef]
- Corrigan, N.; Xu, J.; Boyer, C. A Photoinitiation System for Conventional and Controlled Radical Polymerization at Visible and NIR Wavelengths. Macromolecules 2016, 49, 3274–3285. [Google Scholar] [CrossRef]
- Yagci, Y.; Jockusch, S.; Turro, N.J. Photoinitiated Polymerization: Advances, Challenges, and Opportunities. Macromolecules 2010, 43, 6245–6260. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, R.; Huang, Z.; Xu, J. How does the single unit monomer insertion technique promote kinetic analysis of activation and initiation in photo-RAFT processes? Polym. Chem. 2021, 12, 581–593. [Google Scholar] [CrossRef]
- Eckert, T.; Abetz, V. Polymethacrylamide—An underrated and easily accessible upper critical solution temperature polymer: Green synthesis via photoiniferter reversible addition–fragmentation chain transfer polymerization and analysis of solution behavior in water/ethanol mixtures. J. Polym. Sci. 2020, 58, 3050–3060. [Google Scholar] [CrossRef]
- Yang, Q.; Ladmiral, V.; Améduri, B. PhotoRAFT Polymerization of Vinylidene Fluoride Using a Household White LED as Light Source at Room Temperature. ChemPhotoChem 2019, 3, 1095–1099. [Google Scholar] [CrossRef]
- Bray, C.; Li, G.; Postma, A.; Strover, L.T.; Wang, J.; Moad, G. Initiation of RAFT Polymerization: Electrochemically Initiated RAFT Polymerization in Emulsion (Emulsion eRAFT), and Direct PhotoRAFT Polymerization of Liquid Crystalline Monomers. Aust. J. Chem. 2021, 74, 56–64. [Google Scholar] [CrossRef]
- Delafresnaye, L.; Jung, K.; Boyer, C.; Barner-Kowollik, C. Two colours of light drive PET–RAFT photoligation. Polym. Chem. 2020, 11, 6453–6462. [Google Scholar] [CrossRef]
- Lauterbach, F.; Rubens, M.; Abetz, V.; Junkers, T. Ultrafast PhotoRAFT Block Copolymerization of Isoprene and Styrene Facilitated through Continuous-Flow Operation. Angew. Chem. Int. Ed. 2018, 57, 14260–14264. [Google Scholar] [CrossRef] [PubMed]
- Wenn, B.; Junkers, T. Continuous Microflow PhotoRAFT Polymerization. Macromolecules 2016, 49, 6888–6895. [Google Scholar] [CrossRef] [Green Version]
- Li, C.-Y.; Yu, S.-S. Efficient Visible-Light-Driven RAFT Polymerization Mediated by Deep Eutectic Solvents under an Open-to-Air Environment. Macromolecules 2021, 54, 9825–9836. [Google Scholar] [CrossRef]
- Carmean, R.N.; Sims, M.B.; Figg, C.A.; Hurst, P.J.; Patterson, J.P.; Sumerlin, B.S. Ultrahigh Molecular Weight Hydrophobic Acrylic and Styrenic Polymers through Organic-Phase Photoiniferter-Mediated Polymerization. ACS Macro Lett. 2020, 9, 613–618. [Google Scholar] [CrossRef]
- Corrigan, N.; Trujillo, F.J.; Xu, J.; Moad, G.; Hawker, C.J.; Boyer, C. Divergent Synthesis of Graft and Branched Copolymers through Spatially Controlled Photopolymerization in Flow Reactors. Macromolecules 2021, 54, 3430–3446. [Google Scholar] [CrossRef]
- Lauterbach, F.; Abetz, V. An eco-friendly pathway to thermosensitive micellar nanoobjects via photoRAFT PISA: The full guide to poly(N-acryloylpyrrolidin)-block-polystyrene diblock copolymers. Soft Matter 2020, 16, 2321–2331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Soto, M.J.; Haupt, K.; Gonzato, C. Synthesis of molecularly imprinted polymers by photo-iniferter polymerization under visible light. Polym. Chem. 2017, 8, 4830–4834. [Google Scholar] [CrossRef]
- Ommura, Y.; Imai, S.; Takenaka, M.; Ouchi, M.; Terashima, T. Selective Coupling and Polymerization of Folded Polymer Micelles to Nanodomain Self-Assemblies. ACS Macro Lett. 2020, 9, 426–430. [Google Scholar] [CrossRef]
- Hirai, Y.; Terashima, T.; Takenaka, M.; Sawamoto, M. Precision Self-Assembly of Amphiphilic Random Copolymers into Uniform and Self-Sorting Nanocompartments in Water. Macromolecules 2016, 49, 5084–5091. [Google Scholar] [CrossRef]
- Imai, S.; Hirai, Y.; Nagao, C.; Sawamoto, M.; Terashima, T. Programmed Self-Assembly Systems of Amphiphilic Random Copolymers into Size-Controlled and Thermoresponsive Micelles in Water. Macromolecules 2018, 51, 398–409. [Google Scholar] [CrossRef]
- Shibata, M.; Matsumoto, M.; Hirai, Y.; Takenaka, M.; Sawamoto, M.; Terashima, T. Intramolecular Folding or Intermolecular Self-Assembly of Amphiphilic Random Copolymers: On-Demand Control by Pendant Design. Macromolecules 2018, 51, 3738–3745. [Google Scholar] [CrossRef]
- Terashima, T.; Sugita, T.; Fukae, K.; Sawamoto, M. Synthesis and Single-Chain Folding of Amphiphilic Random Copolymers in Water. Macromolecules 2014, 47, 589–600. [Google Scholar] [CrossRef]
- Sivokhin, A.P.; Orekhov, D.V.; Kazantsev, O.A.; Gubanova, O.V.; Kamorin, D.M.; Zarubina, I.S.; Bolshakova, E.A.; Zaitsev, S.D. Amphiphilic thermoresponsive copolymer bottlebrushes: Synthesis, characterization, and study of their self-assembly into flower-like micelles. Polym. J. 2021, 53, 655–665. [Google Scholar] [CrossRef]
- Moad, G.; Chong, Y.K.; Postma, A.; Rizzardo, E.; Thang, S.H. Advances in RAFT polymerization: The synthesis of polymers with defined end-groups. Polymer 2005, 46, 8458–8468. [Google Scholar] [CrossRef]
- Sivokhin, A.P.; Kazantsev, O.A. Temperature Dependence of the Rayleigh Ratio for Toluene: Thermoresponsive Polymers Characterization. ChemistrySelect 2021, 6, 9499–9502. [Google Scholar] [CrossRef]
- Kuckling, D.; Doering, A.; Krahl, F.; Arndt, K.F. 8.15—Stimuli-Responsive Polymer Systems A2—Matyjaszewski, Krzysztof. In Polymer Science: A Comprehensive Reference; Möller, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 377–413. [Google Scholar]
- McKenzie, T.G.; Fu, Q.; Wong, E.H.H.; Dunstan, D.E.; Qiao, G.G. Visible Light Mediated Controlled Radical Polymerization in the Absence of Exogenous Radical Sources or Catalysts. Macromolecules 2015, 48, 3864–3872. [Google Scholar] [CrossRef]
- Corrigan, N.; Rosli, D.; Jones, J.W.J.; Xu, J.; Boyer, C. Oxygen Tolerance in Living Radical Polymerization: Investigation of Mechanism and Implementation in Continuous Flow Polymerization. Macromolecules 2016, 49, 6779–6789. [Google Scholar] [CrossRef]
- Matsumoto, M.; Takenaka, M.; Sawamoto, M.; Terashima, T. Self-assembly of amphiphilic block pendant polymers as microphase separation materials and folded flower micelles. Polym. Chem. 2019, 10, 4954–4961. [Google Scholar] [CrossRef]
- Hattori, G.; Hirai, Y.; Sawamoto, M.; Terashima, T. Self-assembly of PEG/dodecyl-graft amphiphilic copolymers in water: Consequences of the monomer sequence and chain flexibility on uniform micelles. Polym. Chem. 2017, 8, 7248–7259. [Google Scholar] [CrossRef]
- Zehm, D.; Laschewsky, A.; Heunemann, P.; Gradzielski, M.; Prévost, S.; Liang, H.; Rabe, J.P.; Lutz, J.-F. Synthesis and self-assembly of amphiphilic semi-brush and dual brush block copolymers in solution and on surfaces. Polym. Chem. 2011, 2, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.Y.; Krys, P.; Szcześniak, K.; Schroeder, H.; Park, S.; Jurga, S.; Buback, M.; Matyjaszewski, K. Synthesis of Poly(OEOMA) Using Macromonomers via “Grafting-Through” ATRP. Macromolecules 2015, 48, 6385–6395. [Google Scholar] [CrossRef]
- Hazer, B.; Subramaniyan, S.; Zhang, B. Synthesis of Biobased Block Copolymers Using A Novel Methacrylated Methyl Salicylate and Poly(3-Hydroxybutyrate). ChemistrySelect 2021, 6, 12255–12265. [Google Scholar] [CrossRef]
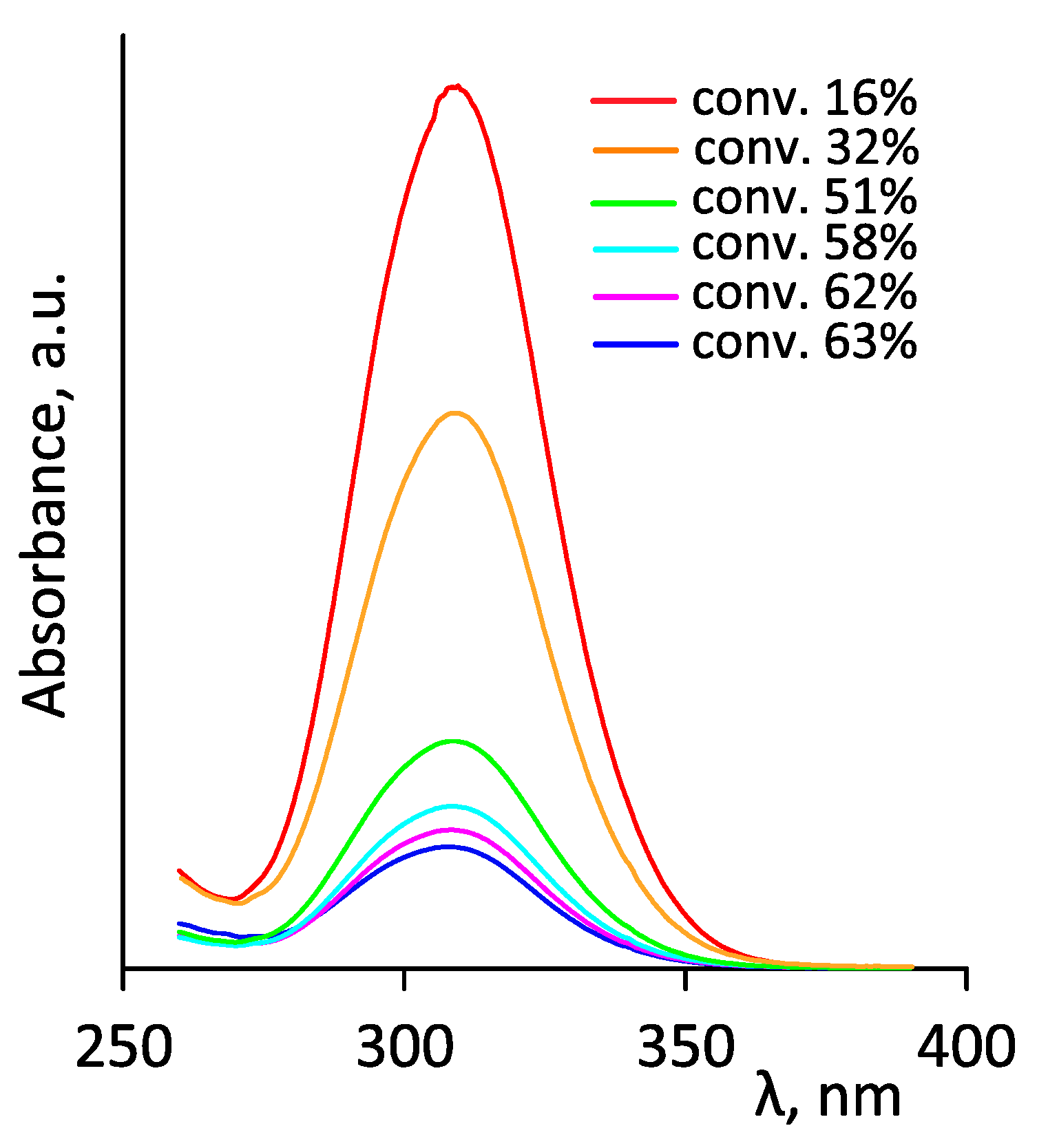
- Skrabania, K.; Miasnikova, A.; Bivigou-Koumba, A.M.; Zehm, D.; Laschewsky, A. Examining the UV-vis absorption of RAFT chain transfer agents and their use for polymer analysis. Polym. Chem. 2011, 2, 2074–2083. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
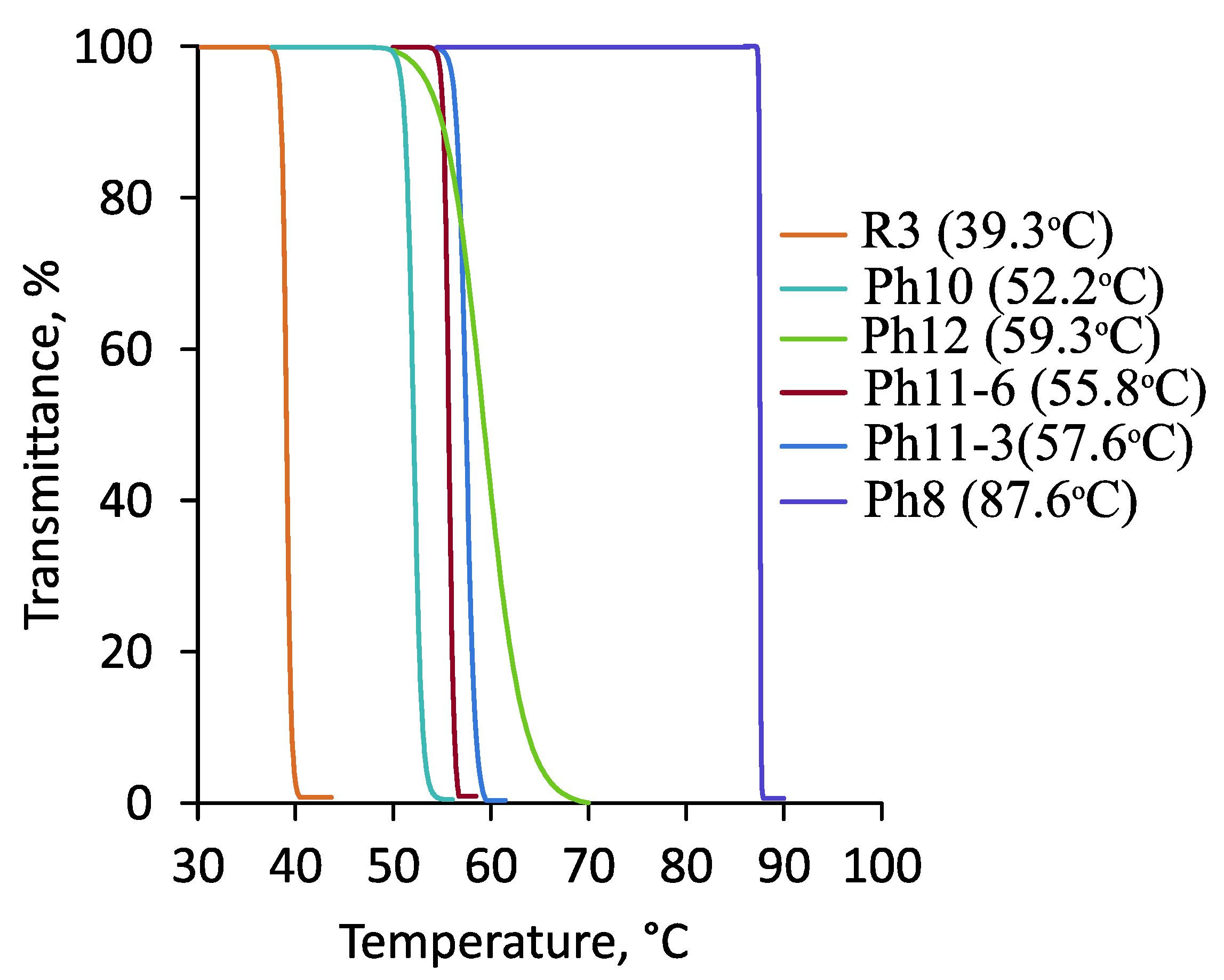{kind=link}
| ID | [MPEGMA]0/[AOEGMA]0/[CDTPA]0 | Solvent | Light (Intensity, mW/cm2) | Time, min | Conversion, % | Composition a, m1:m2 (mol) | Mn,th b | Mn c | MW c | PDI c (MW/Mn) |
|---|---|---|---|---|---|---|---|---|---|---|
| Ph1 | 200:0:1 | Toluene | green (7.3) | 120 | 43 | 100:0 | 41,500 | 16,700 | 23,300 | 1.39 |
| Ph2 | 500:0:1 | Toluene | green (7.3) | 180 | 48 | 100:0 | 121,000 | 15,700 | 26,200 | 1.66 |
| Ph3 | 1000:0:1 | Toluene | green (7.3) | 210 | 47 | 100:0 | 242,400 | 22,200 | 36,700 | 1.64 |
| Ph4 | 0:500:1 | Toluene | green (7.3) | 480 | 45 | 0:100 | 141,600 | 56,000 | 89,700 | 1.60 |
| Ph5 | 200:0:1 | DMSO | blue (7.0) | 75 | 85 | 100:0 | 81,000 | - | - | - |
| Ph6 | 200:0:1 | Toluene | blue (7.0) | 100 | 46 | 100:0 | 44,000 | 3800 | 4800 | 1.25 |
| Ph7 | 200:0:1 | THF | blue (7.0) | 75 | 52 | 100:0 | 50,700 | - | - | - |
| Ph8 | 200:0:1 | Toluene | blue (8.3) | 100 | 72 | 100:0 | 68,400 | 3600 | 4600 | 1.26 |
| Ph9 | 200:0:1 | Toluene | blue (8.3) | 60 | 54 | 100:0 | 51,500 | 3400 | 4300 | 1.26 |
| Ph10 | 100:100:1 | Toluene | green (7.3) | 480 | 65 | 51:49 | 74,000 | 40,800 | 56,200 | 1.27 |
| Ph11-6 d | 100:100:1 | Toluene | blue (8.3) | 68 | 64 | 51:49 | 70,300 | 16,000 | 20,300 | 1.27 |
| Ph12 | 100:100:1 | DMSO | blue (7.0) | 60 | 58 | 56:44 | 59,200 | 11,700 | 15,400 | 1.32 |
| Ph13 | 0:200:1 | THF | blue (8.3) | 60 | 40 | 0:100 | 46,300 | 2900 | 3500 | 1.19 |
| Ph14 e | 800:480:1 | THF | blue (8.3) | 90 | 42 | 42:58 | 390,000 | 40,400 | 49,700 | 1.23 |
| R3 f | 80:80:1:0.2 | Toluene | AIBN | 360 | 83 | 52:48 | 22,800 | 16,300 | 19,800 | 1.22 |
| ID | Hydrodynamic Radius, Rh, nm | Mn,UV a | DPn b | mole cm3 g−2 c | mole cm3 g−2 c | Nagg d | |||
|---|---|---|---|---|---|---|---|---|---|
| Acetonitrile | Water | ||||||||
| Ph1 | 6.4 | 6.8 | 122,800 | 261 | 126,500 | 2.18 | 152,900 | 1.66 | 1.21 |
| Ph10 | 8.4 | 5.9 | 199,400 | 194 | 221,900 | 0.54 | 221,500 | 0.43 | ~1.0 |
| Ph12 | 7.3 | 5.8 | 126,100 | 136 | 214,300 | 0.41 | 225,700 | −0.29 | 1.05 |
| Ph14 | - | 162 | 480,900 | 378 | - | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sivokhin, A.; Orekhov, D.; Kazantsev, O.; Sivokhina, O.; Orekhov, S.; Kamorin, D.; Otopkova, K.; Smirnov, M.; Karpov, R. Random and Diblock Thermoresponsive Oligo(ethylene glycol)-Based Copolymers Synthesized via Photo-Induced RAFT Polymerization. Polymers 2022, 14, 137. https://doi.org/10.3390/polym14010137
Sivokhin A, Orekhov D, Kazantsev O, Sivokhina O, Orekhov S, Kamorin D, Otopkova K, Smirnov M, Karpov R. Random and Diblock Thermoresponsive Oligo(ethylene glycol)-Based Copolymers Synthesized via Photo-Induced RAFT Polymerization. Polymers. 2022; 14(1):137. https://doi.org/10.3390/polym14010137
Chicago/Turabian StyleSivokhin, Alexey, Dmitry Orekhov, Oleg Kazantsev, Olga Sivokhina, Sergey Orekhov, Denis Kamorin, Ksenia Otopkova, Michael Smirnov, and Rostislav Karpov. 2022. "Random and Diblock Thermoresponsive Oligo(ethylene glycol)-Based Copolymers Synthesized via Photo-Induced RAFT Polymerization" Polymers 14, no. 1: 137. https://doi.org/10.3390/polym14010137
APA StyleSivokhin, A., Orekhov, D., Kazantsev, O., Sivokhina, O., Orekhov, S., Kamorin, D., Otopkova, K., Smirnov, M., & Karpov, R. (2022). Random and Diblock Thermoresponsive Oligo(ethylene glycol)-Based Copolymers Synthesized via Photo-Induced RAFT Polymerization. Polymers, 14(1), 137. https://doi.org/10.3390/polym14010137