
Improving the Efficiency for the Production of Bis-(2-Hydroxyethyl) Terephtalate (BHET) from the Glycolysis Reaction of Poly(Ethylene Terephtalate) (PET) in a Pressure Reactor

 ,
,  and
and
Abstract

1. Introduction
2. Materials and Methods
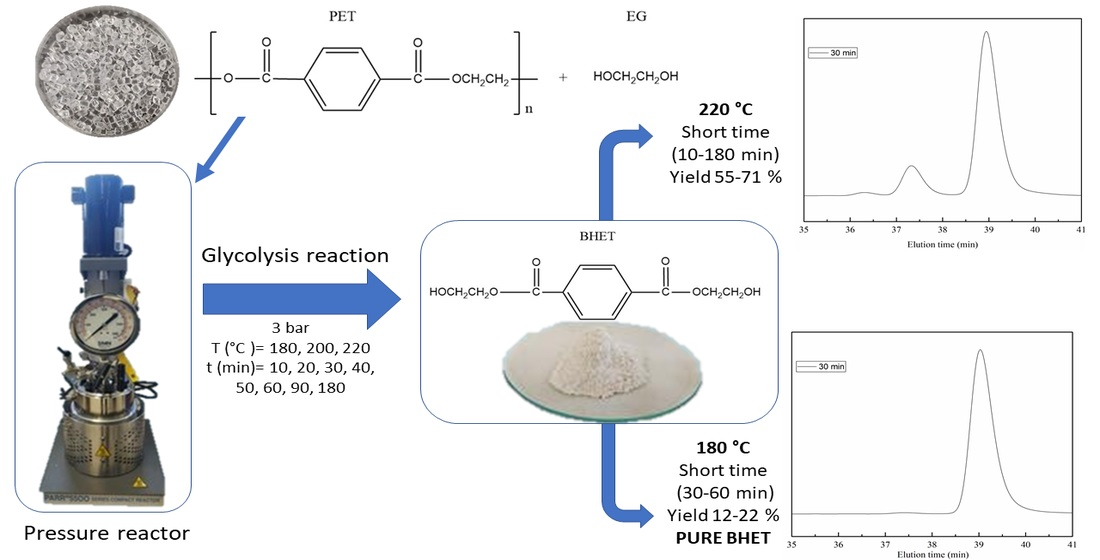
2.1. Glycolysis Reaction
2.2. Characterization Techniques
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wei, H.S.; Liu, K.T.; Chang, Y.C.; Chan, C.H.; Lee, C.C.; Kuo, C.C. Superior mechanical properties of hybrid organic-inorganic superhydrophilic thin film on plastic substrate. Surf. Coat. Technol. 2017, 320, 377–382. [Google Scholar] [CrossRef]
- PlasticsEurope. Plastics—the Facts 2019. Available online: https://www.plasticseurope.org/en (accessed on 18 September 2020).
- Chapa-Martínez, C.A.; Hinojosa-Reyes, L.; Hernández-Ramírez, A.; Ruiz-Ruiz, E.; Maya-Treviño, L.; Guzmán-Mar, J.L. An evaluation of the migration of antimony from polyethylene terephthalate (PET) plastic used for bottled drinking water. Sci. Total Environ. 2016, 565, 511–518. [Google Scholar] [CrossRef]
- Hu, Y.S.; Prattipati, V.; Mehta, S.; Schiraldi, D.A.; Hiltner, A.; Baer, E. Improving gas barrier of PET by blending with aromatic polyamides. Polymer 2005, 46, 2685–2698. [Google Scholar] [CrossRef]
- Petcore Europe. Recycled Products. Available online: https://www.petcore-europe.org/recycled-products.html (accessed on 18 September 2020).
- Dubelley, F.; Planes, E.; Bas, C.; Pons, E.; Yrieix, B.; Flandin, L. The hygrothermal degradation of PET in laminated multilayer. Eur. Polym. J. 2017, 87, 1–13. [Google Scholar] [CrossRef]
- Sammon, C.; Yarwood, J.; Everall, N. An FT–IR study of the effect of hydrolytic degradation on the structure of thin PET films. Polym. Degrad. Stab. 2000, 67, 149–158. [Google Scholar] [CrossRef]
- Holland, B.J.; Hay, J.N. The thermal degradation of PET and analogous polyesters measured by thermal analysis—Fourier transform infrared spectroscopy. Polymer 2002, 43, 1835–1847. [Google Scholar] [CrossRef]
- Pirzadeh, E.; Zadhoush, A.; Haghighat, M. Hydrolytic and thermal degradation of PET fibers and PET granule: The effects of crystallization, temperature, and humidity. J. Appl. Polym. Sci. 2007, 106, 1544–1549. [Google Scholar]
- El’Darov, E.G.; Mamedov, F.V.; Gol’Dberg, V.M.; Zaikov, G.E. A kinetic model of polymer degradation during extrusion. Polym. Degrad. Stab. 1996, 51, 271–279. [Google Scholar]
- Petcore Europe. Processing. Available online: https://www.petcore-europe.org/processing.html (accessed on 22 September 2020).
- Ghaderian, A.; Haghighi, A.H.; Taromi, F.A.; Abdeen, Z.; Boroomand, A.; Taheri, S.M.R. Characterization of rigid polyurethane foam prepared from recycling of PET waste. Period. Polytech. Chem. Eng. 2015, 59, 296–305. [Google Scholar]
- George, N.; Kurian, T. Recent developments in the chemical recycling of postconsumer poly (ethylene terephthalate) waste. Ind. Eng. Chem. Res. 2014, 53, 14185–14198. [Google Scholar]
- Abdelaal, M.Y.; Sobahi, T.R.; Makki, M.S.I. Chemical transformation of pet waste through glycolysis. Constr. Build. Mater. 2011, 25, 3267–3271. [Google Scholar] [CrossRef]
- Nikles, D.E.; Farahat, M.S. New motivation for the depolymerization products derived from poly (ethylene terephthalate) (PET) waste: A review. Macromol. Mater. Eng. 2005, 290, 13–30. [Google Scholar] [CrossRef]
- Güçlü, G.; Kas göz, A.; Özbudak, S.; Özgümüs, S.; Orbay, M. Glycolysis of poly (ethylene terephthalate) wastes in xylene. J. Appl. Polym. Sci. 1998, 69, 2311–2319. [Google Scholar] [CrossRef]
- Al-Sabagh, A.M.; Yehia, F.Z.; Eshaq, G.; Rabie, A.M.; ElMetwally, A.E. Greener routes for recycling of polyethylene terephthalate. Egypt. J. Pet. 2016, 25, 53–64. [Google Scholar] [CrossRef]
- Chaudhary, S.; Surekha, P.; Kumar, D.; Rajagopal, C.; Roy, P.K. Microwave assisted glycolysis of poly (ethylene terepthalate) for preparation of polyester polyols. J. Appl. Polym. Sci. 2013, 129, 2779–2788. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.; Li, Z.; Zhang, X.; Zhang, S.; Zhang, Y. Glycolysis of poly (ethylene terephthalate) catalyzed by ionic liquids. Eur. Polym. J. 2009, 45, 1535–1544. [Google Scholar] [CrossRef]
- Chen, J.W.; Chen, L.W.; Cheng, W.H. Kinetics of glycolysis of polyethylene terephthalate with zinc catalyst. Polym. Int. 1999, 48, 885–888. [Google Scholar] [CrossRef]
- Imran, M.; Al-Masry, W.A.; Mahmood, A.; Hassan, A.; Haider, S.; Ramay, S.M. Manganese-, cobalt-, and zinc-based mixed-oxide spinels as novel catalysts for the chemical recycling of poly (ethylene terephthalate) via glycolysis. Polym. Degrad. Stab. 2013, 98, 904–915. [Google Scholar] [CrossRef]
- López-Fonseca, R.; Duque-Ingunza, I.; De Rivas, B.; Arnaiz, S.; Gutiérrez-Ortiz, J.I. Chemical recycling of post-consumer PET wastes by glycolysis in the presence of metal salts. Polym. Degrad. Stab. 2010, 95, 1022–1028. [Google Scholar] [CrossRef]
- Baliga, S.; Wong, W.T. Depolymerization of poly (ethylene terephthalate) recycled from post-consumer soft-drink bottles. J. Polym. Sci. Part A Polym. Chem. 1989, 27, 2071–2082. [Google Scholar] [CrossRef]
- Viana, M.E.; Riul, A.; Carvalho, G.M.; Rubira, A.F.; Muniz, E.C. Chemical recycling of PET by catalyzed glycolysis: Kinetics of the heterogeneous reaction. Chem. Eng. J. 2011, 173, 210–219. [Google Scholar] [CrossRef]
- Chen, J.Y.; Ou, C.F.; Hu, Y.C.; Lin, C.C. Depolymerization of poly (ethylene terephthalate) resin under pressure. J. Appl. Polym. Sci. 1991, 42, 1501–1507. [Google Scholar] [CrossRef]
- Chen, J.W.; Chen, L.W. The glycolysis of poly (ethylene terephthalate). J. Appl. Polym. Sci. 1999, 73, 35–40. [Google Scholar] [CrossRef]
- Campanelli, J.R.; Kamal, M.R.; Cooper, D.G. Kinetics of glycolysis of poly (ethylene terephthalate) melts. J. Appl. Polym. Sci. 1994, 54, 1731–1740. [Google Scholar] [CrossRef]
- Aguado, A.; Martínez, L.; Becerra, L.; Arieta-Araunabeña, M.; Arnaiz, S.; Asueta, A.; Robertson, I. Chemical depolymerisation of PET complex waste: Hydrolysis vs. glycolysis. J. Mater. Cycles Waste Manag. 2014, 16, 201–210. [Google Scholar] [CrossRef]
- Ptiček Siročić, A.; Fijačko, A.; Hrnjak-Murgić, Z. Chemical recycling of postconsumer poly (ethylene-terephthalate) bottles–depolymerization study. Chem. Biochem. Eng. Q. 2013, 27, 65–71. [Google Scholar]
- Imran, M.; Kim, B.K.; Han, M.; Cho, B.G. Sub-and supercritical glycolysis of polyethylene terephthalate (PET) into the monomer bis (2-hydroxyethyl) terephthalate (BHET). Polym. Degrad. Stab. 2010, 95, 1686–1693. [Google Scholar] [CrossRef]
- Fang, P.; Liu, B.; Xu, J.; Zhou, Q.; Zhang, S.; Ma, J. High-efficiency glycolysis of poly (ethylene terephthalate) by sandwich-structure polyoxometalate catalyst with two active sites. Polym. Degrad. Stab. 2018, 156, 22–31. [Google Scholar] [CrossRef]
- Donelli, I.; Freddi, G.; Nierstrasz, V.A.; Taddei, P. Surface structure and properties of poly-(ethylene terephthalate) hydrolyzed by alkali and cutinase. Polym. Degrad. Stab. 2010, 95, 1542–1550. [Google Scholar] [CrossRef]
- López-Fonseca, R.; Duque-Ingunza, I.; de Rivas, B.; Flores-Giraldo, L.; Gutiérrez-Ortiz, J.I. Kinetics of catalytic glycolysis of PET wastes with sodium carbonate. Chem. Eng. J. 2011, 168, 312–320. [Google Scholar] [CrossRef]
- Chen, C.H. Study of glycolysis of poly (ethylene terephthalate) recycled from postconsumer soft-drink bottles. III. Further investigation. J. Appl. Polym. Sci. 2003, 87, 2004–2010. [Google Scholar] [CrossRef]
- Geng, Y.; Dong, T.; Fang, P.; Zhou, Q.; Lu, X.; Zhang, S. Fast and effective glycolysis of poly (ethylene terephthalate) catalyzed by polyoxometalate. Polym. Degrad. Stab. 2015, 117, 30–36. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Method | Time (h) | Temperature (°C) | Pressure (at) | Catalyst | Ratio PET:EG | Yield (%) |
|---|---|---|---|---|---|---|---|
| [16] | Glycolysis in xylene | 1–3 | 170–245 | - | - | 1:0.5–1:3 Molar ratio | 20 |
| [18] | Glycolysis by microwave irradiation | 0.5 | 180–190 | - | ZnAc 0.5 % | 1:2,1:4, 1:6 Molar ratio | 40 |
| [19] | Ionic liquids as reaction medium | 5–10 | 160–195 | 1 | Ionic liquids 1–4 L | 1:4 (w/w) | Max Yield 78 Conversion 70 |
| [20] | Glycolysis in a pressure reactor | 235–275 | High pressure | ZnAc | 1:1.3 (w/w) | - | |
| [21] | Autoclave reactor | 1 | 260 | 5 | ZnMn2O4 1 % | 5:86 Molar ratio | 92.2 |
| [22] | Glycolysis at atmospheric pressure | 1–2 | 196 | 1 | ZnAc 1 % Or Na2CO3 1% | 1:7.6 Molar ratio | 70 |
| [23] | Glycolysis in potassium nitrate bath | 8 | 190 | - | Metal acetate | 1:4 Molar ratio | - |
| [24] | Glycolysis at atmospheric pressure | 1–4 | 180–190 | 1 | ZnAc 2.35 % | 1:4 (w/w) | 91.6 |
| [25] | Glycolysis under pressure | 0.5–3 | 190–240 | 1–6.12 | - | 1:1–1:4 Molar ratio | - |
| [26] | Glycolysis at atmospheric pressure | 2–10 | 190 | 1 | ZnAc 0.25–0.75 % | 1:4 Molar ratio | - |
| Our work | Glycolysis under pressure | 10–180 | 220 | 3 bar | ZnAc 1 % | 1:3 (w/w) | 55–71 |
| Fraction | Insoluble (%) | ƞ (%) |
|---|---|---|
| G10 | 5.78 | 55.00 ± 10 |
| G20 | 8.03 | 59.67 ± 14 |
| G30 | 7.39 | 67.75 ± 9 |
| G40 | 5.13 | 58.34 ± 4 |
| 650 | 5.47 | 62.95 ± 6 |
| G60 | 5.88 | 63.29 ± 3 |
| 690 | 3.97 | 70.60 ± 9 |
| G180 | 6.41 | 57.71 ± 6 |
| Samples | O1 Mn = 155–167 g/mol Mw = 160–171 g/mol (%) | O2 Mn = 358–379 g/mol Mw = 362–384 g/mol (%) | O3 Mn = 556–595 g/mol Mw = 559–600 g/mol (%) |
|---|---|---|---|
| G10 | 83.83 | 14.61 | 1.46 |
| G20 | 87.40 | 11.71 | 0.89 |
| G30 | 83.97 | 14.58 | 1.45 |
| G40 | 81.56 | 16.67 | 1.67 |
| G50 | 82.97 | 15.51 | 1.47 |
| G60 | 80.05 | 17.66 | 2.11 |
| G90 | 80.25 | 17.65 | 1.93 |
| G180 | 85.43 | 13.31 | 1.26 |
| Samples | 1. Step | 2. Step | ||
|---|---|---|---|---|
| Time (Min) | T1 (°C) | Mass Loss (%) | T2 (°C) | Mass Loss (%) |
| 30 | 183 | 22.62 | 432 | 70.27 |
| 60 | 271 | 27.31 | 434 | 64.4 |
| 90 | 267 | 26.77 | 433 | 64.68 |
| 180 | 198 | 24.41 | 433 | 67.29 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mendiburu-Valor, E.; Mondragon, G.; González, N.; Kortaberria, G.; Eceiza, A.; Peña-Rodriguez, C. Improving the Efficiency for the Production of Bis-(2-Hydroxyethyl) Terephtalate (BHET) from the Glycolysis Reaction of Poly(Ethylene Terephtalate) (PET) in a Pressure Reactor. Polymers 2021, 13, 1461. https://doi.org/10.3390/polym13091461
Mendiburu-Valor E, Mondragon G, González N, Kortaberria G, Eceiza A, Peña-Rodriguez C. Improving the Efficiency for the Production of Bis-(2-Hydroxyethyl) Terephtalate (BHET) from the Glycolysis Reaction of Poly(Ethylene Terephtalate) (PET) in a Pressure Reactor. Polymers. 2021; 13(9):1461. https://doi.org/10.3390/polym13091461
Chicago/Turabian StyleMendiburu-Valor, Eider, Gurutz Mondragon, Nekane González, Galder Kortaberria, Arantxa Eceiza, and Cristina Peña-Rodriguez. 2021. "Improving the Efficiency for the Production of Bis-(2-Hydroxyethyl) Terephtalate (BHET) from the Glycolysis Reaction of Poly(Ethylene Terephtalate) (PET) in a Pressure Reactor" Polymers 13, no. 9: 1461. https://doi.org/10.3390/polym13091461
APA StyleMendiburu-Valor, E., Mondragon, G., González, N., Kortaberria, G., Eceiza, A., & Peña-Rodriguez, C. (2021). Improving the Efficiency for the Production of Bis-(2-Hydroxyethyl) Terephtalate (BHET) from the Glycolysis Reaction of Poly(Ethylene Terephtalate) (PET) in a Pressure Reactor. Polymers, 13(9), 1461. https://doi.org/10.3390/polym13091461