
Modelling Sorption Thermodynamics and Mass Transport of n-Hexane in a Propylene-Ethylene Elastomer


 , ,
, ,  ,
, 
Abstract
1. Introduction
- -
- Polymerization. The monomer concentration, which can be dissolved in the polymer, determines the concentration at the active site, affecting polymerization rate and molecular weight distribution [3].
- -
- Polymer foaming. The blowing agent concentration, which can be solubilized in the polymer, affects the final expansion ratio, the cellular morphology, and the final foamed shape [4].
- -
- Separation technology: the maximum degree of separation, i.e., solvent concentration at equilibrium that may be obtained for a given system, is the most important parameter of interest to optimize the equipment design, while controlling the strict levels of volatile organic compounds (VOCs) in the final product [5].
2. Theoretical Background
2.1. Modeling Sorption Thermodynamics by NRLF Approach
2.2. Modeling Diffusive Mass Transport of n-Hexane
3. Materials and Methods
3.1. Materials
3.2. Charaterzation of PVT Behavior
3.3. Equilibrium Data for n-Hexane
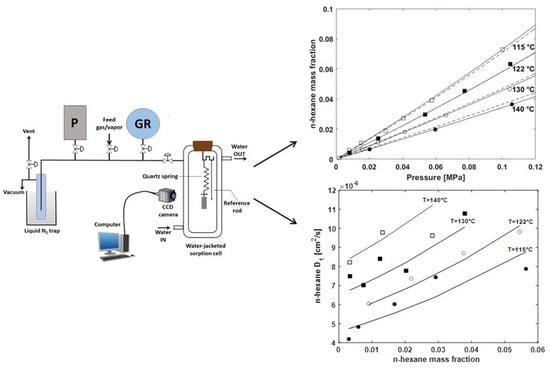
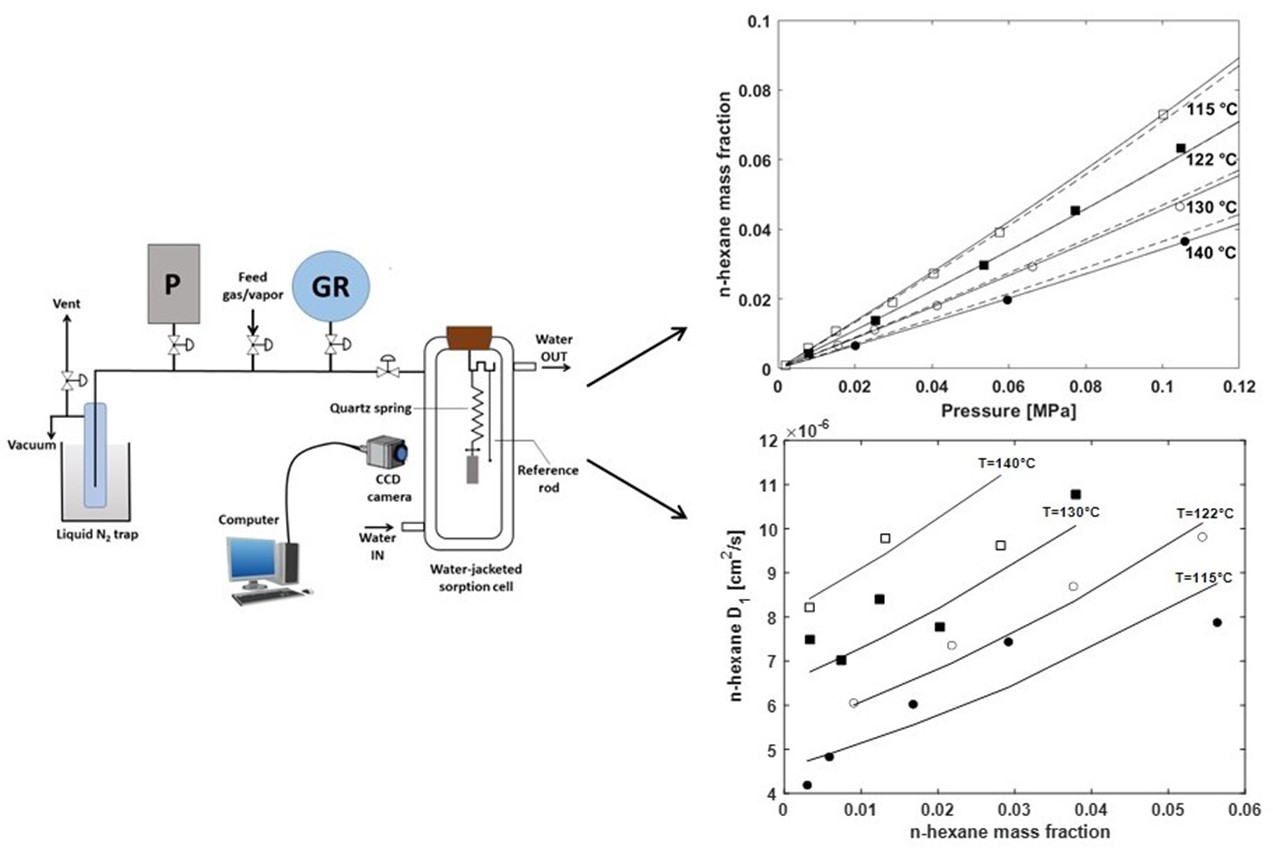
3.4. Gravimetric Sorption Tests
4. Results
5. Discussion
5.1. Determination of NRLF Model Parameters for Pure Components
5.1.1. Fitting of PVT Data of V8880 with NRLF EoS
5.1.2. Fitting of n-Hexane Vapor Pressure Data and Density Data at Vapor–Liquid Equilibrium
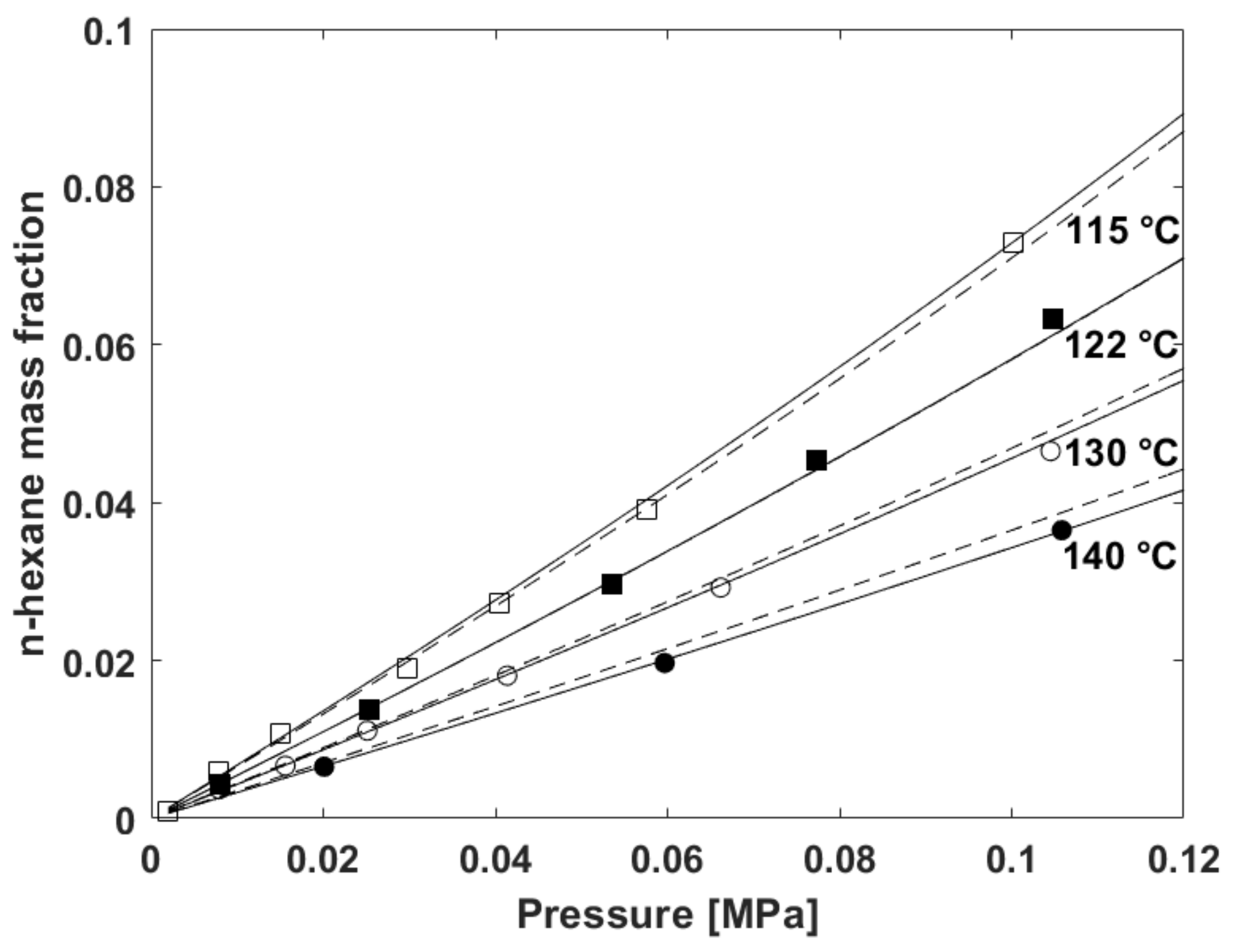
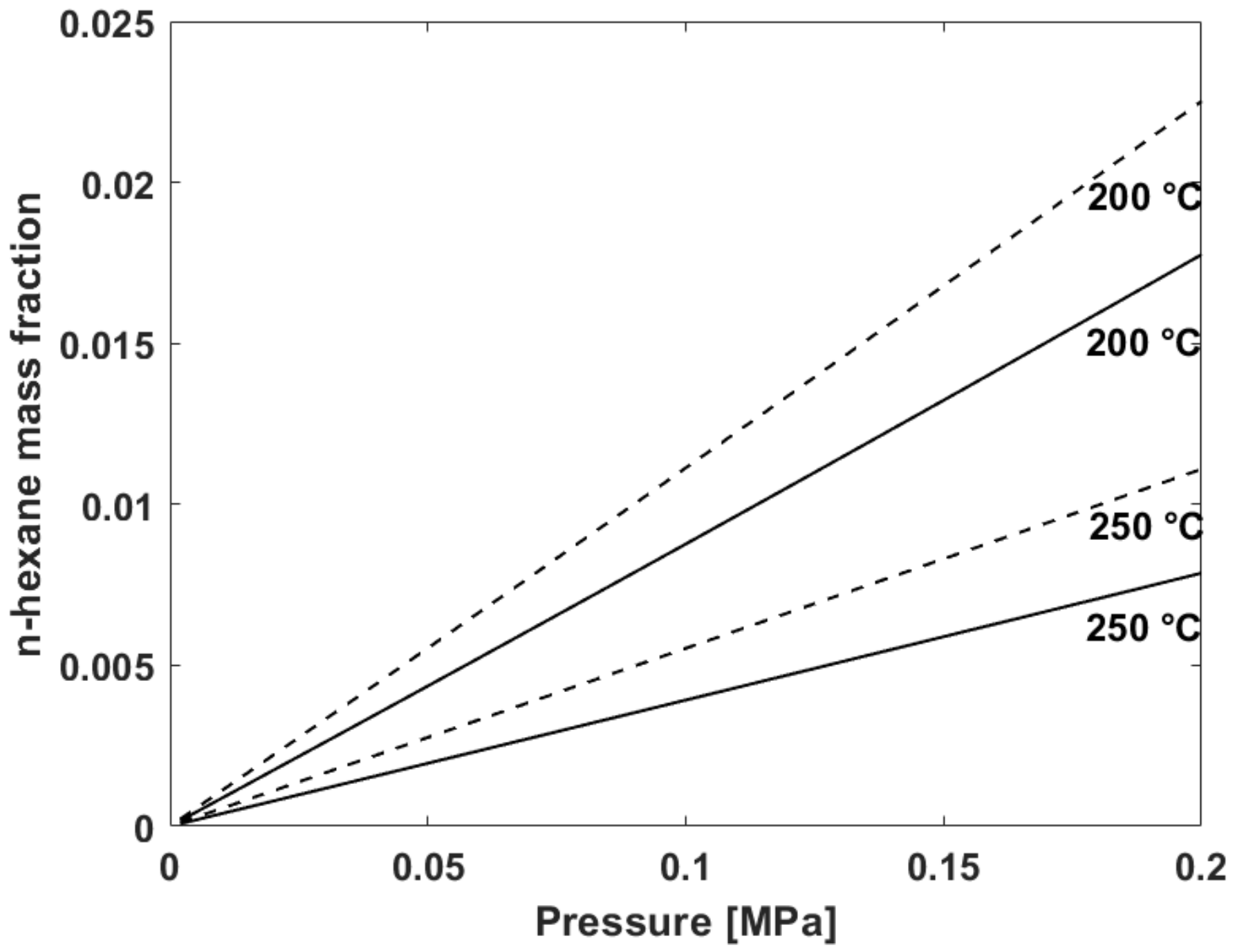
5.2. Fitting of Sorption Isotherms
- Use of a temperature independent binary interaction parameter
- Use of a temperature dependent binary interaction parameter
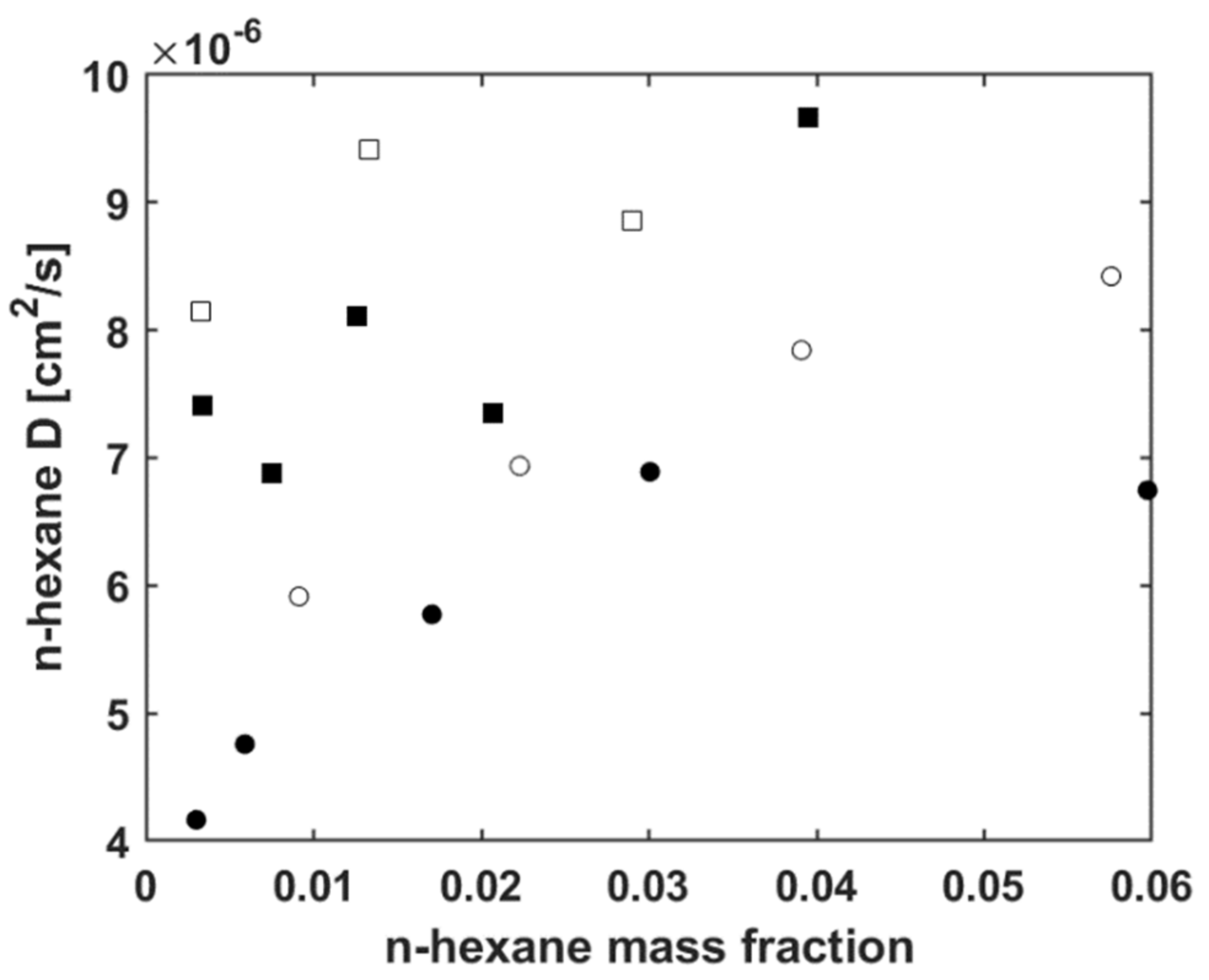
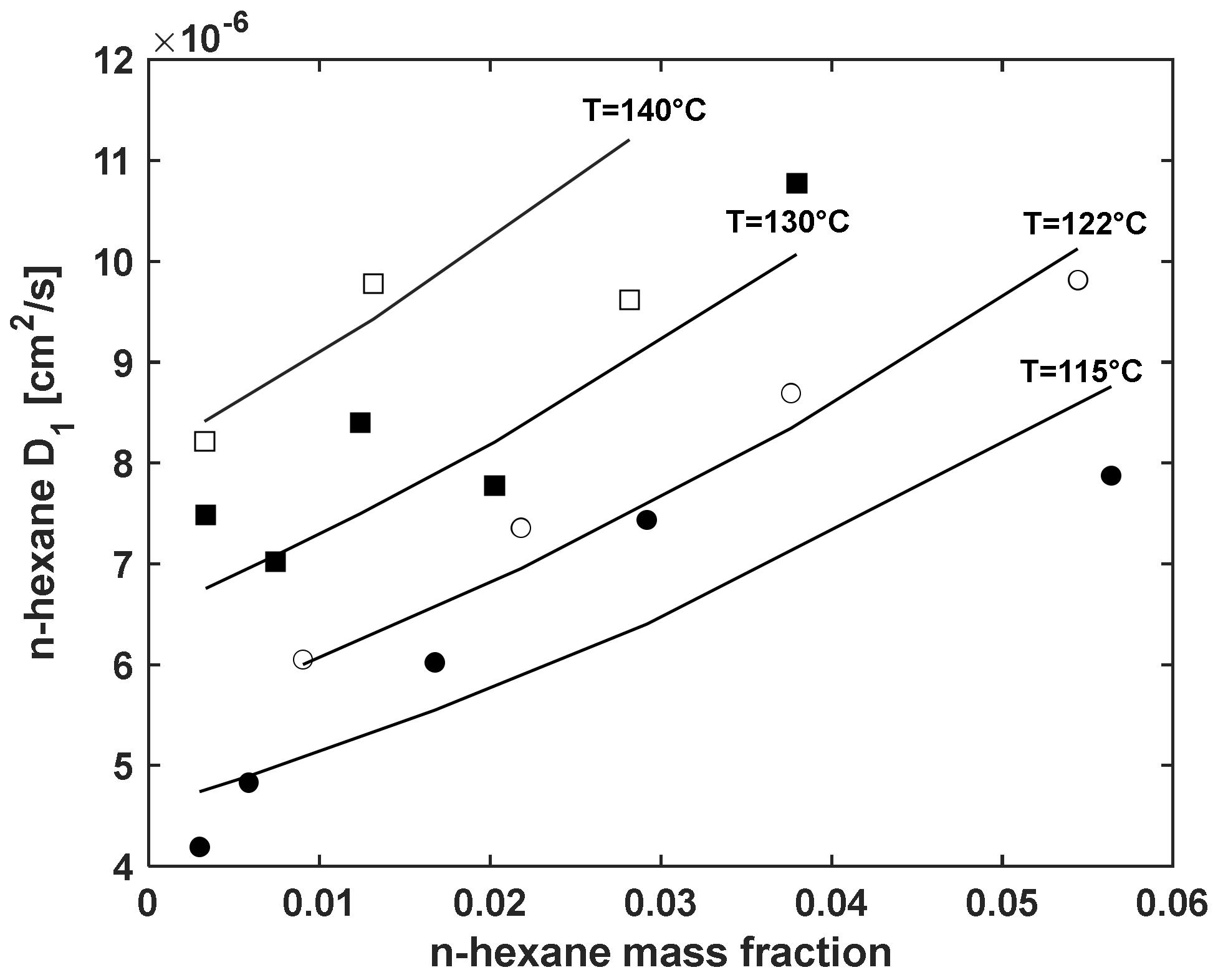
5.3. Modelling n-Hexane Diffusivity
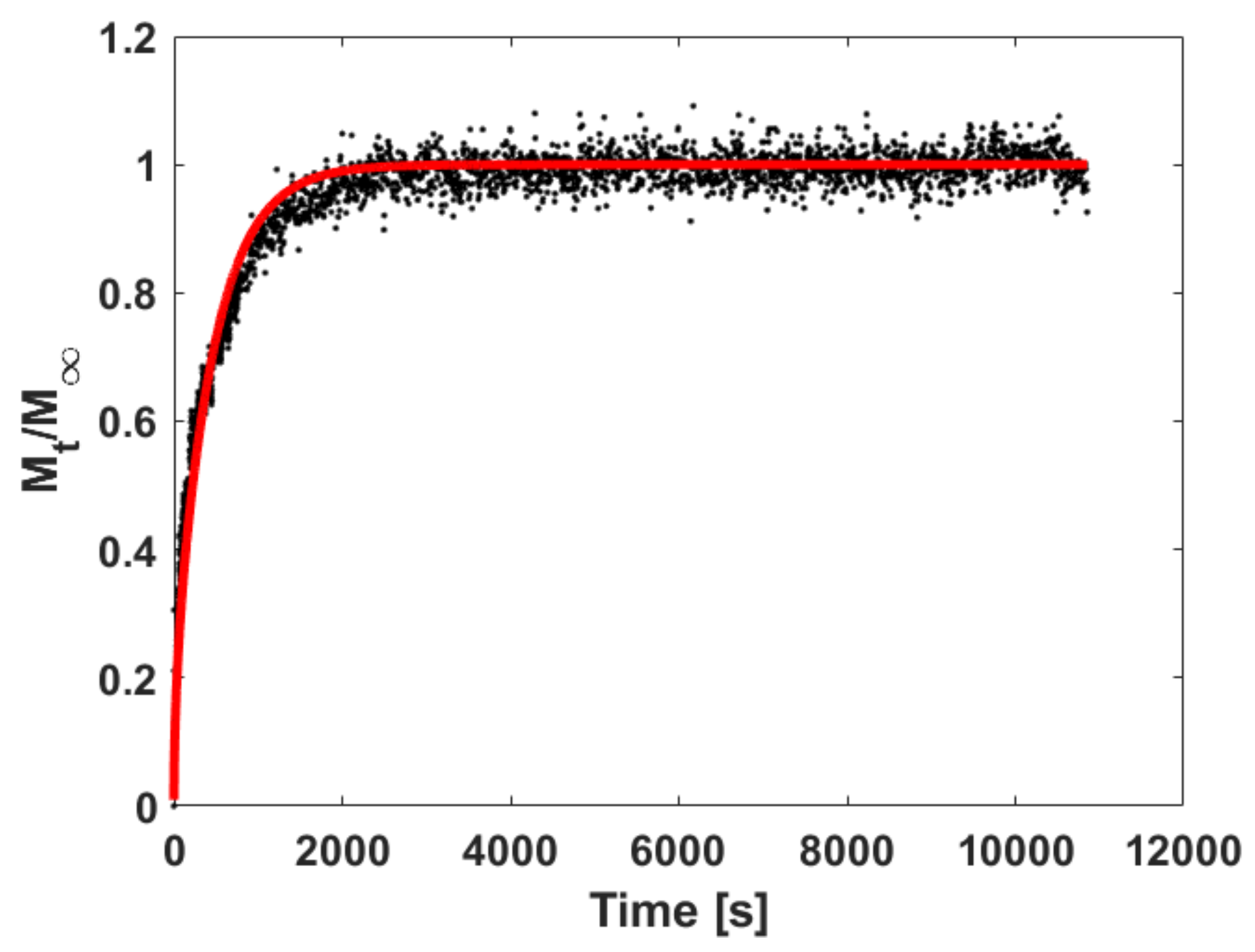
5.3.1. Fitting Sorption Kinetics
5.3.2. Fitting Sorption Kinetics
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shamiri, A.; Chakrabarti, M.H.; Jahan, S.; Hussain, M.A.; Kaminsky, W.; Aravind, P.V.; Yehye, W.A. The influence of Ziegler-Natta and metallocene catalysts on polyolefin structure, properties, and processing ability. Materials 2014, 7, 5069–5108. [Google Scholar] [CrossRef]
- Song, X.; Cao, L.; Tanaka, R.; Shiono, T.; Cai, Z. Optically transparent functional polyolefin elastomer with excellent mechanical and thermal properties. ACS Macro Lett. 2019, 8, 299–303. [Google Scholar] [CrossRef]
- Tammaro, D.; Iannace, S.; Di Maio, E. Insight into bubble nucleation at high-pressure drop rate. J. Cell. Plast. 2017, 53, 551–560. [Google Scholar] [CrossRef]
- Cancelas, A.J.; Plata, M.A.; Bashir, M.A.; Bartke, M.; Monteil, V.; McKenna, T.F. Solubility and diffusivity of propylene, ethylene, and propylene–ethylene mixtures in polypropylene. Macromol. Chem. Phys. 2018, 219, 542–565. [Google Scholar] [CrossRef]
- Merrill, E.W. Thermodynamic Aspects of Devolatilization of Polymers. In Polymer Devolatilization, 1st ed.; Albalak, R.J., Ed.; CRC Press: Boca Raton, FL, USA, 1997; pp. 13–34. [Google Scholar]
- Lee, J.K.; Yao, S.X.; Li, G.; Jun, M.B.G.; Lee, P.C. Measurement Methods for Solubility and Diffusivity of Gases and Supercritical Fluids in Polymers and Its Applications. Polym. Rev. 2017, 57, 695–747. [Google Scholar] [CrossRef]
- Villaluenga, J.P.G.; Khayet, M.; López-Manchado, M.A.; Valentin, J.L.; Seoane, B.; Mengual, J.I. Gas transport properties of polypropylene/clay composite membranes. Eur. Polym. J. 2007, 43, 1132–1143. [Google Scholar] [CrossRef]
- Guo, B.; Tang, Z.; Zhang, L. Transport performance in novel elastomer nanocomposites: Mechanism, design and control. Prog. Polym. Sci. 2016, 61, 29–66. [Google Scholar] [CrossRef]
- Guggenheim, E.A. Mixtures, 1st ed.; Oxford University Press: Oxford, UK, 1952. [Google Scholar]
- Flory, P.J. Principles of Polymer Chemistry, 1st ed.; Cornwell University Press: Ithaca, NY, USA, 1953; Chapters 12 and 13. [Google Scholar]
- Sanchez, I.C.; Lacombe, R.H. An elementary molecular theory of classical fluids. Pure fluids. J. Phys. Chem. 1976, 80, 2352–2362. [Google Scholar] [CrossRef]
- Lacombe, R.H.; Sanchez, I.C. Statistical thermodynamics of fluid mixtures. J. Phys. Chem. 1976, 80, 2568–2580. [Google Scholar] [CrossRef]
- Sanchez, I.C.; Lacombe, R.H. Statistical Thermodynamics of Polymer Solutions. Macromolecules 1978, 11, 1145–1156. [Google Scholar] [CrossRef]
- Panayiotou, C.G. Thermodynamics of Alkanol-Alkane Mixtures. J. Phys. Chem. 1988, 92, 2960–2969. [Google Scholar] [CrossRef]
- Taimoori, M.; Panayiotou, C. The non-random distribution of free volume in fluids: Polydisperse polymer systems. Fluid Phase Equilibria 2003, 205, 249–265. [Google Scholar] [CrossRef]
- Panayiotou, C. The QCHB model of fluids and their mixtures. J. Chem. Thermodyn. 2003, 35, 349–381. [Google Scholar] [CrossRef]
- Panayiotou, C.; Pantoula, M.; Stefanis, E.; Tsivintzelis, I.; Economou, I.G. Nonrandom Hydrogen-Bonding Model of Fluids and Their Mixtures. 1. Pure Fluids. Ind. Eng. Chem. Res. 2004, 43, 6592–6606. [Google Scholar] [CrossRef]
- Panayiotou, C.; Tsivintzelis, I.; Economou, I.G. Nonrandom Hydrogen-Bonding Model of Fluids and Their Mixtures. 2. Multicomponent Mixtures. Ind. Eng. Chem. Res. 2007, 46, 2628–2636. [Google Scholar] [CrossRef]
- Panayiotou, C. New expressions for non-randomness in equation-of-state models. Fluid Phase Equilibria 2005, 237, 130–139. [Google Scholar] [CrossRef]
- Panayiotou, C.G. Hydrogen Bonding in Solutions: The Equation-of-State Approach. In Handbook of Surface and Colloid Chemistry, 2nd ed.; Birdi, K.S., Ed.; CRC Press LLC Taylor and Francis Group: New York, NY, USA, 2003; pp. 5–66. [Google Scholar]
- Panayiotou, C.G. Hydrogen Bonding and Nonrandomness in Solution Thermodynamics. In Handbook of Surface and Colloid Chemistry, 3rd ed.; Birdi, K.S., Ed.; CRC Press LLC Taylor and Francis Group: New York, NY, USA, 2009; pp. 45–89. [Google Scholar]
- Mensitieri, G.; Scherillo, G.; Panayiotou, C.; Musto, P. Towards a predictive thermodynamic description of sorption processes in polymers: The synergy between theoretical EoS models and vibrational spectroscopy. Mater. Sci. Eng. R 2020, 140C, 100525. [Google Scholar] [CrossRef]
- Grenner, A.; Tsivintzelis, I.; Kontogeorgis, G.M.; Economou, I.G.; Panayiotou, C. Evaluation of the Nonrandom Hydrogen Bonding (NRHB) Theory and the Simplified Perturbed-Chain—Statistical Associating Fluid Theory (sPC-SAFT). 1. Vapor—Liquid Equilibria. Ind. Eng. Chem. Res. 2008, 47, 5636–5650. [Google Scholar] [CrossRef]
- Tsivintzelis, I.; Spyriouni, T.; Economou, I.G. Modeling of fluid phase equilibria with two thermodynamic theories: Non-random hydrogen bonding (NRHB) and statistical associating fluid theory (SAFT). Fluid Phase Equilibria 2007, 253, 19–28. [Google Scholar] [CrossRef]
- Tsivintzelis, I.; Grenner, A.; Economou, I.G.; Kontogeorgis, G.M. Evaluation of the Nonrandom Hydrogen Bonding (NRHB) Theory and the Simplified Perturbed-Chain−Statistical Associating Fluid Theory (sPC-SAFT). 2. Liquid−Liquid Equilibria and Prediction of Monomer Fraction in Hydrogen Bonding Systems. Ind. Eng. Chem. Res. 2008, 47, 5651–5659. [Google Scholar] [CrossRef]
- Tsivintzelis, I.; Economou, I.G.; Kontogeorgis, G.M. Modeling the solid–liquid equilibrium in pharmaceutical-solvent mixtures: Systems with complex hydrogen bonding behavior. AIChE J. 2009, 55, 756–770. [Google Scholar] [CrossRef]
- Tsivintzelis, I.; Economou, I.G.; Kontogeorgis, G.M. Modeling the Phase Behavior in Mixtures of Pharmaceuticals with Liquid or Supercritical Solvents. J. Phys. Chem. B 2009, 113, 6446–6458. [Google Scholar] [CrossRef] [PubMed]
- Tsivintzelis, I.; Kontogeorgis, G.M. Modeling the vapor-liquid equilibria of polymer-solvent mixtures: Systems with complex hydrogen bonding behaviour. Fluid Phase Equilibria 2009, 280, 100–109. [Google Scholar] [CrossRef]
- Tsioptsias, C.; Tsivintzelis, I.; Panayiotou, C. Equation-of-state modeling of mixtures with ionic liquids. Phys. Chem. Chem. Phys. 2010, 12, 4843–4851. [Google Scholar] [CrossRef]
- Fredenslund, A.; Sorensen, M.J. Group Contribution Estimation Methods. In Models for Thermodynamic and Phase Equilibria Calculations, 1st ed.; Sandler, S., Ed.; Marcel Dekker: New York, NY, USA, 1994; pp. 287–362. [Google Scholar]
- Crank, J.; Park, G.S. Diffusion in Polymers, 1st ed.; Academic Press: London, UK; New York, NY, USA, 1968. [Google Scholar]
- Crank, J. The Mathematics of Diffusion, 2nd ed.; Clarendon Press: Oxford, UK, 1975. [Google Scholar]
- Duda, J.L.; Zielinski, J.M. Free-volume theory. In Diffusion in Polymers, 1st ed.; Neogi, K., Ed.; Marcel Dekker Inc.: New York, NY, USA, 1996; pp. 143–172. [Google Scholar]
- Vrentas, J.S.; Duda, J.L. Diffusion in Polymer-Solvent Systems: I. Re-examination of the Free-Volume Theory. J. Polym. Sci. Polym. Phys. Ed. 1977, 15, 403–416. [Google Scholar] [CrossRef]
- Vrentas, J.S.; Duda, J.L. Diffusion in Polymer-Solvent Systems: II. A Predictive Theory for the Dependence of the Diffusion Coefficient on Temperature, Concentration and Molecular Weight. J. Polym. Sci. Polym. Phys. Ed. 1977, 15, 417–439. [Google Scholar] [CrossRef]
- Product Datasheet of VistamaxxTM Performance Polymer 8880 (Propylene Elastomer). Available online: http://exxonmobilchemical.com/ (accessed on 14 July 2020).
- Wiederhorn, S.M.; Fields, R.J.; Low, S.; Bahng, G.-W.; Wehrstedt, A.; Hahn, J.; Tomota, Y.; Miyata, T.; Lin, H.; Freeman, B.D.; et al. Section 7.6.5 ‘Experimental Measurement of Gas and Vapor Sorption’. In Springer Handbook of Metrology and Testing, 2nd ed.; Czichos, H., Saito, T., Smith, L., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 438–439, Chapter 7. [Google Scholar]
- Francouer, A.J.B. A mathematical model for the devolatilization of EPDM rubber in a series of steam stripping vessels. Master’s Thesis, Queen’s University, Kingston, ON, Canada, 2012. [Google Scholar]
- Kontogeorgis, G.M.; Folas, G.K. Thermodynamic Models for Industrial Applications, 2nd ed.; John Wiley & Sons: Chichester, UK, 2010. [Google Scholar]
- Krenz, R.A.; Laursen, T.; Heidemann, R.A. The Modified Sanchez-Lacombe Equation of State Applied to Polydisperse Polyethylene Solutions. Ind. Eng. Chem. Res. 2009, 48, 10664–10681. [Google Scholar] [CrossRef]
- Panayiotou, C.; Vera, J.H. Thermodynamics of polymer-polymer solvent and block copolymer solvent systems. Theoretical treatment of data with nonrandom new Flory theory. Polym. J. 1984, 16, 103–112. [Google Scholar] [CrossRef][Green Version]
- Panayiotou, C.; Oehmke, F. Volumetric properties of random copolymers. An experimental and theoretical study. Fluid Phase Equilibria 1996, 126, 289–298. [Google Scholar] [CrossRef]
- Gross, J.; Sadowski, G. Perturbed-Chain SAFT: An equation of state based on a perturbation theory for chain molecules. Ind. Eng. Chem. Res. 2001, 40, 1244–1260. [Google Scholar] [CrossRef]
- Gross, J.; Spuhl, O.; Tumakaka, F.; Sadowski, G. Modeling copolymer systems using the perturbed-chain SAFT equation of state. Ind. Eng. Chem. Res. 2003, 42, 1266–1274. [Google Scholar] [CrossRef]
- Matthews, F.J.; Fair, J.R.; Barlow, J.W.; Paul, D.R.; Cozewith, C. Solvent Removal from Ethylene-Propylene Elastomers. 1. Determination of Diffusion Mechanism. Ind. Eng. Chem. Prod. Res. Dev. 1986, 25, 58–64. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fluid | (J × mol−1) | (J × mol−1 × K−1) | (cm3 × g−1) | s |
|---|---|---|---|---|
| n-hexane | 3986.0 | 1.5009 | 1.2771 | 0.857 1 |
| V8880 | 4292.4 | 2.7679 | 1.1043 | 0.631 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tammaro, D.; Lombardi, L.; Scherillo, G.; Di Maio, E.; Ahuja, N.; Mensitieri, G. Modelling Sorption Thermodynamics and Mass Transport of n-Hexane in a Propylene-Ethylene Elastomer. Polymers 2021, 13, 1157. https://doi.org/10.3390/polym13071157
Tammaro D, Lombardi L, Scherillo G, Di Maio E, Ahuja N, Mensitieri G. Modelling Sorption Thermodynamics and Mass Transport of n-Hexane in a Propylene-Ethylene Elastomer. Polymers. 2021; 13(7):1157. https://doi.org/10.3390/polym13071157
Chicago/Turabian StyleTammaro, Daniele, Lorenzo Lombardi, Giuseppe Scherillo, Ernesto Di Maio, Navanshu Ahuja, and Giuseppe Mensitieri. 2021. "Modelling Sorption Thermodynamics and Mass Transport of n-Hexane in a Propylene-Ethylene Elastomer" Polymers 13, no. 7: 1157. https://doi.org/10.3390/polym13071157
APA StyleTammaro, D., Lombardi, L., Scherillo, G., Di Maio, E., Ahuja, N., & Mensitieri, G. (2021). Modelling Sorption Thermodynamics and Mass Transport of n-Hexane in a Propylene-Ethylene Elastomer. Polymers, 13(7), 1157. https://doi.org/10.3390/polym13071157