
Penetration of Hydrogen into Polymer Electrolyte Membrane for Fuel Cells by Quantum and Molecular Dynamics Simulations


Abstract
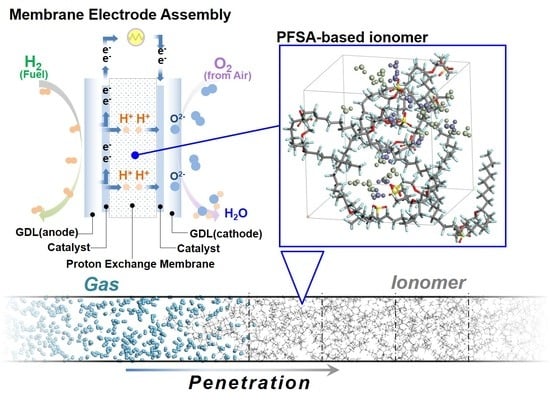
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Simulation Methodology
2.1. Models of Polymer Electrolyte Membranes and Hydrogen
2.2. Simulation Details
3. Results and Discussion
3.1. Quantitative Evaluation Model for the Effect of Side Chain on Penetration
3.2. Hydrogen Adsorption onto the Side Chain
3.3. Physical Obstruction against the Molecular Penetration
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cameron, D.S. Fuel Cell Energy Generators. Platin. Met. Rev. 1978, 22, 38–46. [Google Scholar]
- Wang, Y.; Chen, K.S.; Mishler, J.; Cho, S.C.; Adroher, X.C. A review of polymer electrolyte membrane fuel cells: Technology, applications, and needs on fundamental research. Appl. Energy 2011, 88, 981–1007. [Google Scholar] [CrossRef]
- Mekhilef, S.; Saidur, R.; Safari, A. Comparative study of different fuel cell technologies. Renew. Sustain. Energy Rev. 2012, 16, 981–989. [Google Scholar] [CrossRef]
- Smitha, B.; Sridhar, S.; Khan, A.A. Solid polymer electrolyte membranes for fuel cell applications—A review. J. Memb. Sci. 2005, 259, 10–26. [Google Scholar] [CrossRef]
- Inaba, M.; Kinumoto, T.; Kiriake, M.; Umebayashi, R.; Tasaka, A.; Ogumi, Z. Gas crossover and membrane degradation in polymer electrolyte fuel cells. Electrochim. Acta 2006, 51, 5746–5753. [Google Scholar] [CrossRef]
- Francia, C.; Ijeri, V.S.; Specchia, S.; Spinelli, P. Estimation of hydrogen crossover through Nafion® membranes in PEMFCs. J. Power Sources 2011, 196, 1833–1839. [Google Scholar] [CrossRef]
- Wu, B.; Zhao, M.; Shi, W.; Liu, W.; Liu, J.; Xing, D.; Yao, Y.; Hou, Z.; Ming, P.; Gu, J.; et al. The degradation study of Nafion/PTFE composite membrane in PEM fuel cell under accelerated stress tests. Int. J. Hydrogen Energy 2014, 39, 14381–14390. [Google Scholar] [CrossRef]
- Umeda, M.; Maruta, T.; Inoue, M.; Nakazawa, A. Cathode platinum degradation in membrane electrode assembly studied using a solid-state electrochemical cell. J. Phys. Chem. C 2008, 112, 18098–18103. [Google Scholar] [CrossRef]
- Dubau, L.; Castanheira, L.; Maillard, F.; Chatenet, M.; Lottin, O.; Maranzana, G.; Dillet, J.; Lamibrac, A.; Perrin, J.C.; Moukheiber, E.; et al. A review of PEM fuel cell durability: Materials degradation, local heterogeneities of aging and possible mitigation strategies. Wiley Interdiscip. Rev. Energy Environ. 2014, 3, 540–560. [Google Scholar] [CrossRef]
- Han, M.; Shul, Y.G.; Lee, H.; Shin, D.; Bae, B. Accelerated testing of polymer electrolyte membranes under open-circuit voltage conditions for durable proton exchange membrane fuel cells. Int. J. Hydrogen Energy 2017, 42, 30787–30791. [Google Scholar] [CrossRef]
- Zhang, S.; Yuan, X.; Wang, H.; Mérida, W.; Zhu, H.; Shen, J.; Wu, S.; Zhang, J. A review of accelerated stress tests of MEA durability in PEM fuel cells. Int. J. Hydrogen Energy 2009, 34, 388–404. [Google Scholar] [CrossRef]
- Liang, M.; Fu, C.; Xiao, B.; Luo, L.; Wang, Z. A fractal study for the effective electrolyte diffusion through charged porous media. Int. J. Heat Mass Transf. 2019, 137, 365–371. [Google Scholar] [CrossRef]
- Xiao, B.; Wang, W.E.I.; Zhang, X.; Long, G.; Chen, H.; Cai, H.; Deng, L.I.N. A novel fractal model for relative permeability of gas diffusion layer in proton exchange membrane fuel cell with capillary pressure effect. Fractals 2019, 27, 1950012. [Google Scholar] [CrossRef]
- Baik, K.D.; Hong, B.K.; Kim, M.S. Novel technique for measuring oxygen crossover through the membrane in polymer electrolyte membrane fuel cells. Int. J. Hydrogen Energy 2013, 38, 8927–8933. [Google Scholar] [CrossRef]
- Kwon, S.H.; Kang, H.; Lee, J.H.; Shim, S.; Lee, J.; Lee, D.S.; Kim, C.M.; Lee, S.G. Investigating the influence of the side-chain pendants of perfluorosulfonic acid membranes in a PEMFC by molecular dynamics simulations. Mater. Today Commun. 2019, 21, 100625. [Google Scholar] [CrossRef]
- Cha, J.H. Morphological effect of side chain on H3O+ transfer inside polymer electrolyte membranes across polymeric chain via molecular dynamics simulation. Sci. Rep. 2020, 10, 22014. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Kuo, A.T.; Hirai, T.; Miyajima, T.; Urata, S.; Terazono, S.; Okazaki, S.; Shinoda, W. Hydrogen Permeation in Hydrated Perfluorosulfonic Acid Polymer Membranes: Effect of Polymer Crystallinity and Equivalent Weight. J. Phys. Chem. C 2019, 123, 20628–20638. [Google Scholar] [CrossRef]
- Jinnouchi, R.; Kudo, K.; Kitano, N.; Morimoto, Y. Molecular Dynamics Simulations on O2 Permeation through Nafion Ionomer on Platinum Surface. Electrochim. Acta 2016, 188, 767–776. [Google Scholar] [CrossRef]
- Lee, S.; Jang, W.; Kim, M.; Shin, J.E.; Park, H.B.; Jung, N.; Whang, D. Rational Design of Ultrathin Gas Barrier Layer via Reconstruction of Hexagonal Boron Nitride Nanoflakes to Enhance the Chemical Stability of Proton Exchange Membrane Fuel Cells. Small 2019, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Grot, W.G. Perfluorinated ion exchange polymers and their use in research and industry. Macromol. Symp. 2011, 82, 161–172. [Google Scholar] [CrossRef]
- Morohoshi, K.; Hayashi, T. Modeling and simulation for fuel cell polymer electrolyte membrane. Polymers 2013, 5, 56–76. [Google Scholar] [CrossRef]
- Sun, H. COMPASS: An ab Initio Force-Field Optimized for Condensed-Phase Applicationss Overview with Details on Alkane and Benzene Compounds. J. Phys. Chem. B 1998, 5647, 7338–7364. [Google Scholar] [CrossRef]
- Sun, H.; Ren, P.; Fried, J.R. The COMPASS and validation. Comput. Theor. Polym. Sci. 1998, 8, 229–246. [Google Scholar] [CrossRef]
- Loup Verlet Computer “Experiments” on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones Molecules. Phys. Rev. 1967, 159, 98–103. [CrossRef]
- Samoletov, A.A.; Dettmann, C.P.; Chaplain, M.A.J. Thermostats for “ Slow ” Configurational Modes. J. Stat. Phys. 2007, 1321–1336. [Google Scholar] [CrossRef]
- Leimkuhler, B.; Noorizadeh, E.; Penrose, O. Comparing the Efficiencies of Stochastic Isothermal Molecular Dynamics Methods. J. Stat. Phys. 2011, 921–942. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Gustavo, S. Assessment of the Perdew-Burke-Ernzerhof exchange-correlation functional. J. Chem. Phys. 1991, 110, 4173–4184. [Google Scholar] [CrossRef]
- Wu, G.; Almquist, C.L.B.; Hwang, S.T. High gas permeability in open-structure membranes. Korean J. Chem. Eng. 2004, 21, 442–453. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cha, J.; Lee, W.; Baek, J. Penetration of Hydrogen into Polymer Electrolyte Membrane for Fuel Cells by Quantum and Molecular Dynamics Simulations. Polymers 2021, 13, 947. https://doi.org/10.3390/polym13060947
Cha J, Lee W, Baek J. Penetration of Hydrogen into Polymer Electrolyte Membrane for Fuel Cells by Quantum and Molecular Dynamics Simulations. Polymers. 2021; 13(6):947. https://doi.org/10.3390/polym13060947
Chicago/Turabian StyleCha, JinHyeok, Wooju Lee, and Jihye Baek. 2021. "Penetration of Hydrogen into Polymer Electrolyte Membrane for Fuel Cells by Quantum and Molecular Dynamics Simulations" Polymers 13, no. 6: 947. https://doi.org/10.3390/polym13060947
APA StyleCha, J., Lee, W., & Baek, J. (2021). Penetration of Hydrogen into Polymer Electrolyte Membrane for Fuel Cells by Quantum and Molecular Dynamics Simulations. Polymers, 13(6), 947. https://doi.org/10.3390/polym13060947