
Disentangling Self-Atomic Motions in Polyisobutylene by Molecular Dynamics Simulations



Abstract
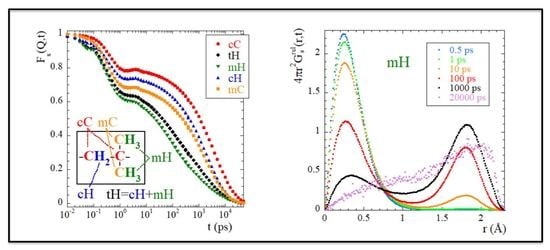
1. Introduction
2. Molecular Dynamics Simulations
Computed Magnitudes
3. Validation
4. Results and Discussion
4.1. Distinguishing the Different Atomic Species: Chemical Dynamic Heterogeneity in PIB
4.2. Methyl Group Dynamics
4.3. Revisiting Neutron Scattering Studies
5. Conclusions
- The motions of the different atomic species are distinct. All atoms manifest Gaussian-like behavior at low Q-values and deviations from it at high Qs.
- The anomalous jump diffusion model captures this behavior by invoking an underlying distribution of elementary jumps at the origin of the sublinear diffusion of atoms in the -relaxation regime. The typical lengths of the jumps deduced are ≈ for main-chain atoms, in accordance with results reported for other polymers.
- Due to rotations, methyl-group hydrogens display an enhanced mobility in the region where the other atoms show decaging processes. These motions are manifested in low values of the shape parameter and shorter characteristic times at small length scales.
- The usually invoked statistically independence of rotation and translational motions does not work in polyisobutylene. The methyl-group rotations could be fully characterized by analyzing the relative motion of these atoms with respect to the carbon at which the are linked. The rotational rate distribution model gives account for these motions, though deviations are found at high Q-values. They can be rationalized as due to non-Gaussian processes taking place during the cageing regime of backbone atoms, which allow a superimposed motion of the rotation axis.
- The methyl-group rotations are coupled with the main-chain dynamics, as deduced from the coincidence of the characteristic times for rotations and non-Gaussian events within the cage of main-chain carbons. This coupling was also concluded from a study combining MD-simulations and NMR experiments [38] and has also been deduced from the analysis of the coherent structure factor of the present simulations [55]. This shall be the origin of the high value observed for the average potential barrier for methyl-group dynamics, leading consequently to a high value of the librational frequency. In fact, a double peak appears in the VDOS of methyl-group hydrogens, where the peak at higher energies occurs where main-chain carbons also present a characteristic peak.
- The information provided by the simulations in real space rules out the occurrence of large amplitude jumps (≈) associated to the -process, as proposed from the analysis of neutron scattering data on protonated samples [34]. The deduction of such a motion is an artifact arising when the hypothesis of statistically independence of -relaxation and localized processes is assumed. The reason is the coupling of methyl-group rotations and main-chain dynamics, possibility that was already suggested by the authors of Ref. [34] as an explanation of the apparently incompatible observations from incoherent and coherent scattering studies.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NS | Neutron scattering |
| PVME | poly(vinyl methyl ether) |
| PH | Phenoxy |
| PVC | Poly(vinyl chloride) |
| PB | 1,4-Polybutadiene |
| PVE | Poly(vinyl ethylene) |
| PI | Polyisoprene |
| PIB | Polyisobutylene |
| NSE | Neutron spin echo |
| MD | Molecular dynamics |
| PMMA | Poly(methyl metacrylate) |
| PEP | Poly(ethylene propylene) |
| hh-PP | head-to-head Polypropylene |
| PVAc | Poly(vinyl acetate) |
| PVP | Poly(vinylpyrrolidone) |
| PEMA | Poly(ethyl methacrylate) |
| PTHF | Poly(tetrahydrofurane) |
| VDOS | vibrational density of states |
| cC | main-chain carbons |
| cH | main-chain hydrogens |
| mC | methyl-group carbons |
| mH | methyl-group hydrogens |
| tH | total hydrogens |
| SBR | styrene-butadiene-rubber |
| PS | polystyrene |
References
- Richter, D.; Monkenbusch, M.; Arbe, A.; Colmenero, J. Neutron Spin Echo in Polymer Systems; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2005; Volume 174. [Google Scholar]
- Colmenero, J.; Arbe, A. Recent progress on polymer dynamics by neutron scattering: From simple polymers to complex materials. J. Polym. Sci. Part B Polym. Phys. 2013, 51, 87–113. [Google Scholar] [CrossRef]
- Arbe, A.; Alvarez, F.; Colmenero, J. Neutron scattering and molecular dynamics simulations: Synergetic tools to unravel structure and dynamics in polymers. Soft Matter 2012, 8, 8257–8270. [Google Scholar] [CrossRef]
- Colmenero, J.; Alegría, A.; Arbe, A.; Frick, B. Correlation between non-Debye behavior and Q behavior of the α relaxation in glass-forming polymeric systems. Phys. Rev. Lett. 1992, 69, 478–481. [Google Scholar] [CrossRef]
- Arbe, A.; Colmenero, J.; Monkenbusch, M.; Richter, D. Dynamics of Glass-Forming Polymers: ‘Homogeneous’ versus ’Heterogeneous’ Scenario. Phys. Rev. Lett. 1998, 81, 590–593. [Google Scholar] [CrossRef]
- Triolo, A.; Russina, O.; Arrighi, V.; Juranyi, F.; Janssen, S.; Gordon, C.M. Quasielastic neutron scattering characterization of the relaxation processes in a room temperature ionic liquid. J. Chem. Phys. 2003, 119, 8549–8557. [Google Scholar] [CrossRef]
- Kofu, M.; Faraone, A.; Tyagi, M.; Nagao, M.; Yamamuro, O. Two inherent crossovers of the diffusion process in glass-forming liquids. Phys. Rev. E 2018, 98, 042601. [Google Scholar] [CrossRef]
- Sobolev, O.; Novikov, A.; Pieper, J. Quasielastic neutron scattering and microscopic dynamics of liquid ethylene glycol. Chem. Phys. 2007, 334, 36–44. [Google Scholar] [CrossRef]
- Roh, J.H.; Tyagi, M.; Hogan, T.E.; Roland, C.M. Space-dependent dynamics in 1,4-polybutadiene nanocomposite. Macromolecules 2013, 46, 6667–6669. [Google Scholar] [CrossRef]
- Jhalaria, M.; Buenning, E.; Huang, Y.; Tyagi, M.; Zorn, R.; Zamponi, M.; García-Sakai, V.; Jestin, J.; Benicewicz, B.C.; Kumar, S.K. Accelerated local dynamics in matrix-free polymer grafted nanoparticles. Phys. Rev. Lett. 2019, 123, 158003. [Google Scholar] [CrossRef]
- Toppozini, L.; Roosen-Runge, F.; Bewley, R.I.; M. Dalgliesh, R.; Perring, T.; Seydel, T.; Glyde, H.R.; García Sakai, V.; Rheinstädter, M.C. Anomalous and anisotropic nanoscale diffusion of hydration water molecules in fluid lipid membranes. Soft Matter 2015, 11, 8354–8371. [Google Scholar] [CrossRef]
- Vural, D.; Smith, J.C.; Petridis, L. Dynamics of the lignin glass transition. Phys. Chem. Chem. Phys. 2018, 20, 20504–20512. [Google Scholar] [CrossRef]
- Farago, B.; Arbe, A.; Colmenero, J.; Faust, R.; Buchenau, U.; Richter, D. Intermediate length scale dynamics of polyisobutylene. Phys. Rev. E 2002, 65, 051803. [Google Scholar] [CrossRef]
- Arbe, A.; Colmenero, J.; Alvarez, F.; Monkenbusch, M.; Richter, D.; Farago, B.; Frick, B. Non-Gaussian nature of the α relaxation of glass-forming polyisoprene. Phys. Rev. Lett. 2002, 89, 245701. [Google Scholar] [CrossRef]
- Arbe, A.; Colmenero, J.; Alvarez, F.; Monkenbusch, M.; Richter, D.; Farago, B.; Frick, B. Experimental evidence by neutron scattering of a crossover from Gaussian to non-Gaussian behavior in the α relaxation of polyisoprene. Phys. Rev. E 2003, 67, 051802. [Google Scholar] [CrossRef]
- Arbe, A.; Alvarez, F.; Colmenero, J. Insight into the structure and dynamics of polymers by neutron scattering combined with atomistic molecular dynamics simulations. Polymers 2020, 12, 3067. [Google Scholar] [CrossRef]
- Singwi, K.S.; Sjölander, A. Diffusive motions in water and cold neutron scattering. Phys. Rev. 1960, 119, 863–871. [Google Scholar] [CrossRef]
- Colmenero, J.; Arbe, A.; Alvarez, F.; Narros, A.; Richter, D. Hydrogen motions and the α-relaxation in glass- forming polymers: Molecular dynamics simulation and quasi-elastic neutron scattering results. Pramana J. Phys. 2004, 63, 25–32. [Google Scholar] [CrossRef]
- Narros, A.; Alvarez, F.; Arbe, A.; Colmenero, J.; Richter, D.; Farago, B. Hydrogen motions in the α-relaxation regime of poly (vinyl ethylene): A molecular dynamics simulation and neutron scattering study. J. Chem. Phys. 2004, 121, 3282. [Google Scholar] [CrossRef]
- Capponi, S.; Arbe, A.; Cerveny, S.; Busselez, R.; Frick, B.; Embs, J.; Colmenero, J. Quasielastic neutron scattering study of hydrogen motions in an aqueous poly(vinyl methyl ether) solution. J. Chem. Phys. 2011, 134, 204906. [Google Scholar] [CrossRef]
- Genix, A.C.; Arbe, A.; Alvarez, F.; Colmenero, J.; Farago, B.; Wischnewski, A.; Richter, D. Self- and collective dynamics of syndiotactic poly(methyl methacrylate). A combined study by quasielastic neutron scattering and atomistic molecular dynamics simulations. Macromolecules 2006, 39, 6260–6272. [Google Scholar] [CrossRef]
- Aparicio, R.P.; Arbe, A.; Colmenero, J.; Frick, B.; Willner, L.; Richter, D. Quasielastic neutron scattering study on the effect of blending on the dynamics of head-to-head poly(propylene) and poly(ethylene-propylene). Macromolecules 2006, 39, 1060–1072. [Google Scholar] [CrossRef]
- Tyagi, M.; Arbe, A.; Alegría, A.; Colmenero, J.; Frick, B. Dynamic confinement effects in polymer blends. A quasielastic neutron scattering study of the slow component in the blend poly (vinyl acetate)/poly (ethylene oxide). Macromolecules 2007, 40, 4568. [Google Scholar] [CrossRef]
- Busselez, R.; Arbe, A.; Alvarez, F.; Colmenero, J.; Frick, B. Study of the structure and dynamics of poly(vinyl pyrrolidone) by molecular dynamics simulations validated by quasielastic neutron scattering and X-ray diffraction experiments. J. Chem. Phys. 2011, 134, 054904. [Google Scholar] [CrossRef] [PubMed]
- Genix, A.C.; Arbe, A.; Colmenero, J.; Wuttke, J.; Richter, D. Neutron scattering and X-ray investigation of the structure and dynamics of poly(ethyl methacrylate). Macromolecules 2012, 45, 2522. [Google Scholar] [CrossRef]
- Arbe, A.; Rubio, J.; Malo de Molina, P.; Maiz, J.; Pomposo, J.A.; Fouquet, P.; Prevost, S.; Juranyi, F.; Khaneft, M.; Colmenero, J. Melts of single-chain nanoparticles: A neutron scattering investigation. J. Appl. Phys. 2020, 127, 044305. [Google Scholar] [CrossRef]
- Arrighi, V.; Tanchawanich, J.; Telling, M.T.F. Molar mass dependence of polyethylene chain dynamics. A quasi-elastic neutron scattering investigation. Macromolecules 2013, 46, 216–225. [Google Scholar] [CrossRef]
- Busselez, R.; Lefort, R.; Ghoufi, A.; Beuneu, B.; Frick, B.; Affouard, F.; Morineau, D. The non-Gaussian dynamics of glycerol. J. Phys. Condens. Matter 2011, 23, 505102. [Google Scholar] [CrossRef]
- Perticaroli, S.; Mostofian, B.; Ehlers, G.; Neuefeind, J.C.; Diallo, S.O.; Stanley, C.B.; Daemen, L.; Egami, T.; Katsaras, J.; Cheng, X.; et al. Structural relaxation, viscosity, and network connectivity in a hydrogen bonding liquid. Phys. Chem. Chem. Phys. 2017, 19, 25859–25869. [Google Scholar] [CrossRef]
- Busselez, R.; Lefort, R.; Guendouz, M.; Frick, B.; Merdrignac-Conanec, O.; Morineau, D. Molecular dynamics of glycerol and glycerol-trehalose bioprotectant solutions nanoconfined in porous silicon. J. Chem. Phys. 2009, 130, 214502. [Google Scholar] [CrossRef]
- Arbe, A.; Buchenau, U.; Willner, L.; Richter, D.; Farago, B.; Colmenero, J. Study of the dynamic structure factor in the β-relaxation regime of polybutadiene. Phys. Rev. Lett. 1996, 76, 1872. [Google Scholar] [CrossRef] [PubMed]
- Arbe, A.; Richter, D.; Colmenero, J.; Farago, B. Merging of the α and β relaxations in polybutadiene: A neutron spin echo and dielectric study. Phys. Rev. E 1996, 54, 3853. [Google Scholar] [CrossRef] [PubMed]
- Narros, A.; Arbe, A.; Alvarez, A.; Colmenero, J.; Richter, D. Atomic motions in the αβ-merging region of 1,4-polybutadiene: A molecular dynamics simulation study. J. Chem. Phys. 2008, 128, 224905. [Google Scholar] [CrossRef]
- Arbe, A.; Colmenero, J.; Frick, B.; Monkenbusch, M.; Richter, D. Investigation of the dielectric β-process in polyisobutylene by incoherent quasielastic neutron scattering. Macromolecules 1998, 31, 4926–4934. [Google Scholar] [CrossRef] [PubMed]
- Richter, D.; Arbe, A.; Colmenero, J.; Monkenbusch, M.; Farago, B.; Faust, R. Molecular motions in polyisobutylene: A neutron spin echo and dielectric investigation. Macromolecules 1998, 31, 1133–1143. [Google Scholar] [CrossRef]
- Slichter, W.P. NMR studies of multiple relaxations in polymers. J. Polym. Sci. Part C 1966, 14, 33. [Google Scholar] [CrossRef]
- Törmälä, P. Spin label and probe studies of polymeric solids and melts. J. Macromol. Sci. Rev. Macromol. Chem. C 1979, 17, 297. [Google Scholar] [CrossRef]
- Karatasos, K.; Ryckaert, J.P.; Ricciardi, R.; Laupretre, F. Methyl dynamics and β-relaxation in polyisobutylene: Comparison between experiment and molecular dynamics simulations. Macromolecules 2002, 35, 1451–1462. [Google Scholar] [CrossRef]
- Frick, B.; Richter, D.; Trevino, S. Inelastic fast relaxation in a weakly fragile polymer glass near Tg. Phys. A 1993, 201, 88–94. [Google Scholar] [CrossRef]
- Frick, B.; Richter, D. Change of the vibrational dynamics near the glass transition in polyisobutylene: Inelastic neutron scattering on a nonfragile polymer. Phys. Rev. B 1993, 47, 14795. [Google Scholar] [CrossRef]
- Colmenero, J.; Moreno, A.J.; Alegría, A. Neutron scattering investigations on methyl group dynamics in polymers. Prog. Polym. Sci. 2005, 30, 1147–1184. [Google Scholar] [CrossRef]
- Adams, M.A.; Gabrys, B.J.; Zajac, W.M.; Peiffer, D.G. High-resolution incoherent inelastic neutron scattering spectra of polyisobutylene and polyisoprene. Macromolecules 2005, 38, 160–166. [Google Scholar] [CrossRef]
- Londono, J.D.; Habenschuss, A.; Curro, J.G.; Rajasekaran, J.J. Short-range order in some polymer melts from X-ray diffraction. J. Polym. Sci. Part B Polym. Phys. 1996, 34, 3055–3061. [Google Scholar] [CrossRef]
- Richter, D.; Monkenbusch, M.; Allgeier, J.; Arbe, A.; Colmenero, J.; Farago, B.; Cheol Bae, Y.; Faust, R. From Rouse dynamics to local relaxation: A neutron spin echo study on polyisobutylene melts. J. Chem. Phys. 1999, 111, 6107–6120. [Google Scholar] [CrossRef]
- Kanaya, T.; Kawaguchi, T.; Kaji, K. Local dynamics of some bulk polymers above Tg as seen by quasielastic neutron scattering. Macromolecules 1999, 32, 1672–1678. [Google Scholar] [CrossRef]
- Kisliuk, A.; Mathers, R.T.; Sokolov, A.P. Crossover in dynamics of polymeric liquids: Back to Tℓℓ? J. Polym. Sci. Part B Polym. Phys. 2000, 38, 2785. [Google Scholar] [CrossRef]
- Triolo, A.; Lechner, R.E.; Desmedt, A.; Telling, M.T.F.; Arrighi, V. Complex dynamics in polyisobutylene melts. Macromolecules 2002, 35, 7039–7043. [Google Scholar] [CrossRef]
- Arrighi, V.; Triolo, A.; Qian, H. Temperature dependence of the segmental dynamics in polyisobutylene melts. J. Non-Cryst. Solids 2002, 307–310, 654–657. [Google Scholar] [CrossRef]
- Ganazzoli, F.; Raffaini, G.; Arrighi, V. The stretched-exponential approximation to the dynamic structure factor in non-entangled polymer melts. Phys. Chem. Chem. Phys. 2002, 4, 3734–3742. [Google Scholar] [CrossRef]
- Ding, Y.; Novikov, V.N.; Sokolov, A.P.; Dalle-Ferrier, C.; Alba-Simionesco, C.; Frick, B. Influence of molecular weight on fast dynamics and fragility of polymers. Macromolecules 2004, 37, 9264–9272. [Google Scholar] [CrossRef]
- Begen, B.; Kisliuk, A.; Novikov, V.; Sokolov, A.; Niss, K.; Chauty-Cailliaux, A.; Alba-Simionesco, C.; Frick, B. Influence of pressure on fast dynamics in polyisobutylene. J. Non-Cryst. Solids 2006, 42–49, 4583–4588. [Google Scholar] [CrossRef]
- Niss, K.; Begen, B.; Frick, B.; Ollivier, J.; Beraud, A.; Sokolov, A.; Novikov, V.N.; Alba-Simionesco, C. Influence of Pressure on the Boson Peak: Stronger than Elastic Medium Transformation. Phys. Rev. Lett. 2007, 99, 055502. [Google Scholar] [CrossRef] [PubMed]
- Niss, K.; Dalle-Ferrier, C.; Giordano, V.M.; Monaco, G.; Frick, B.; Alba-Simionesco, C. Glassy properties and viscous slowing down: An analysis of the correlation between nonergodicity factor and fragility. J. Chem. Phys. 2008, 129, 194513. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Ferrier, C.; Niss, K.; Sokolov, A.P.; Frick, B.; Serrano, J.; Alba-Simionesco, C. The role of chain length in nonergodicity factor and fragility of polymers. Macromolecules 2010, 43, 8977–8984. [Google Scholar] [CrossRef]
- Khairy, Y.; Alvarez, F.; Arbe, A.; Colmenero, J. Collective features in polyisobutylene. A study of the static and dynamic structure factor by molecular dynamics simulations. Macromolecules 2014, 47, 447. [Google Scholar] [CrossRef]
- Tengroth, C.; Engberg, D.; Wahnström, G.; Börjesson, L.; Carlsson, P.; Ahlström, P.; Howells, W.S. The segmental and rotational dynamics of PPO, above the glass-transition, investigated by neutron scattering and molecular dynamics simulations. Soft Mater. 2005, 3, 1–20. [Google Scholar] [CrossRef]
- Zhang, C.; Arrighi, V.; Gagliardi, S.; McEwen, I.J.; Tanchawanich, J.; Telling, M.T.; Zanotti, J.M. Quasielastic neutron scattering measurements of fast process and methyl group dynamics in glassy poly(vinyl acetate). Chem. Phys. 2006, 328, 53–63. [Google Scholar] [CrossRef]
- Schönhals, A.; Goering, H.; Schick, C.; Frick, B.; Zorn, R. Glassy dynamics of polymers confined to nanoporous glasses revealed by relaxational and scattering experiments. Eur. Phys. J. E 2003, 12, 173–178. [Google Scholar] [CrossRef]
- Arrighi, V.; Gagliardi, S.; Zhang, C.; Ganazzoli, F.; Higgins, J.S.; Ocone, R.; Telling, M.T.F. A unified picture of the local dynamics of poly(dimethylsiloxane) across the melting point. Macromolecules 2003, 36, 8738–8748. [Google Scholar] [CrossRef]
- Chen, C.; Maranas, J.K.; García-Sakai, V. Local dynamics of syndiotactic poly(methyl methacrylate) using molecular dynamics simulation. Macromolecules 2006, 39, 9630–9640. [Google Scholar] [CrossRef]
- Zorn, R.; Frick, B.; Fetters, L.J. Quasielastic neutron scattering study of the methyl group dynamics in polyisoprene. J. Chem. Phys. 2002, 116, 845–853. [Google Scholar] [CrossRef]
- Smuda, C.; Busch, S.; Wagner, B.; Unruh, T. Methyl group dynamics in glassy, polycrystalline, and liquid coenzyme Q10 studied by quasielastic neutron scattering. J. Chem. Phys. 2008, 129, 074507. [Google Scholar] [CrossRef]
- Fetters, L.J.; Lohse, D.J.; Richter, D.; Witten, T.A.; Zirkel, A. Connection between polymer molecular weight, density, chain dimensions, and melt viscoelastic properties. Macromolecules 1994, 27, 4639. [Google Scholar] [CrossRef]
- Springer, T. Quasielastic Neutron Scattering for the Investigation of Diffusive Motions in Solids and Liquids; Springer Tracts in Modern Physics; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 1972; Volume 64. [Google Scholar]
- Squires, G.L. Introduction to the Theory of Thermal Neutron Scattering; Dover Publication Inc.: New York, NY, USA, 1996. [Google Scholar]
- Zorn, R. Deviation from Gaussian behavior in the self-correlation function of the proton motion in polybutadiene. Phys. Rev. B 1997, 55, 6249. [Google Scholar] [CrossRef]
- Rahman, A.; Singwi, K.S.; Sjölander, A. Theory of slow neutron scattering by liquids. I. Phys. Rev. 1962, 126, 986. [Google Scholar] [CrossRef]
- Mezei, F. Neutron Spin Echo; Lecture Notes in Physics; Springer: Heidelberg, Germany, 1980; Volume 28. [Google Scholar]
- Farago, B. IN11C, medium-resolution multidetector extension of the IN11 NSE spectrometer at the ILL. Phys. B 1997, 241–243, 113. [Google Scholar] [CrossRef]
- Gambino, T.; Alegría, A.; Arbe, A.; Colmenero, J.; Malicki, N.; Dronet, S.; Schnell, B.; Lohstroh, W.; Nemkovski, K. Applying polymer blend dynamics concepts to a simplified industrial system. A combined effort by dielectric spectroscopy and neutron scattering. Macromolecules 2018, 51, 6692–6706. [Google Scholar] [CrossRef]
- Lefort, R.; Morineau, D.; Guégan, R.; Guendouz, M.; Zanotti, J.M.; Frick, B. Relation between static short-range order and dynamic heterogeneities in a nanoconfined liquid crystal. Phys. Rev. E 2008, 78, 040701. [Google Scholar] [CrossRef]
- Hopfenmüller, B.; Zorn, R.; Holderer, O.; Ivanova, O.; Lehnert, W.; Lüke, W.; Ehlers, G.; Jalarvo, N.; Schneider, G.J.; Monkenbusch, M.; et al. Fractal diffusion in high temperature polymer electrolyte fuel cell membranes. J. Chem. Phys. 2018, 148, 204906. [Google Scholar] [CrossRef] [PubMed]
- Götze, W. Liquids, Freezing, Glass Transition; North-Holland: Amsterdam, The Netherlands, 1991; p. 287. [Google Scholar]
- Götze, W. Complex Dynamics of Glass-Forming Liquids. A Mode Coupling Theory; Oxford University Press: New York, NY, USA, 2009. [Google Scholar]
- Khairy, Y.; Alvarez, F.; Arbe, A.; Colmenero, J. Applicability of mode-coupling theory to polyisobutylene: A molecular dynamics simulation study. Phys. Rev. E 2013, 88, 042302. [Google Scholar] [CrossRef]
- Alvarez, F.; Arbe, A.; Colmenero, J. Methyl group dynamics above the glass transition temperature: A molecular dynamics simulation in polyisoprene. Chem. Phys. 2000, 261, 47. [Google Scholar] [CrossRef]
- Chahid, A.; Alegría, A.; Colmenero, J. Methyl group dynamics in poly(vinyl methyl ether). A rotation rate distribution model. Macromolecules 1994, 27, 3282–3288. [Google Scholar] [CrossRef]
- Colmenero, J.; Mukhopadhyay, R.; Alegría, A.; Frick, B. Quantum rotational tunneling of methyl groups in polymers. Phys. Rev. Lett. 1998, 80, 2350–2353. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atomic Species | (Å) | (eV) |
|---|---|---|
| cC | 0.32 ± 0.06 | 0.44 ± 0.13 |
| mC | 0.54 ± 0.03 | 0.47 ± 0.11 |
| cH | 0.41 ± 0.03 | 0.47 ± 0.13 |
| mH | 0.50 ± 0.03 | 0.32 ± 0.10 |
| tH | 0.57 ± 0.03 | 0.42 ± 0.13 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khairy, Y.; Alvarez, F.; Arbe, A.; Colmenero, J. Disentangling Self-Atomic Motions in Polyisobutylene by Molecular Dynamics Simulations. Polymers 2021, 13, 670. https://doi.org/10.3390/polym13040670
Khairy Y, Alvarez F, Arbe A, Colmenero J. Disentangling Self-Atomic Motions in Polyisobutylene by Molecular Dynamics Simulations. Polymers. 2021; 13(4):670. https://doi.org/10.3390/polym13040670
Chicago/Turabian StyleKhairy, Yasmin, Fernando Alvarez, Arantxa Arbe, and Juan Colmenero. 2021. "Disentangling Self-Atomic Motions in Polyisobutylene by Molecular Dynamics Simulations" Polymers 13, no. 4: 670. https://doi.org/10.3390/polym13040670
APA StyleKhairy, Y., Alvarez, F., Arbe, A., & Colmenero, J. (2021). Disentangling Self-Atomic Motions in Polyisobutylene by Molecular Dynamics Simulations. Polymers, 13(4), 670. https://doi.org/10.3390/polym13040670