
Polybutadiene Vitrimers with Tunable Epoxy Ratios: Preparation and Properties


Abstract
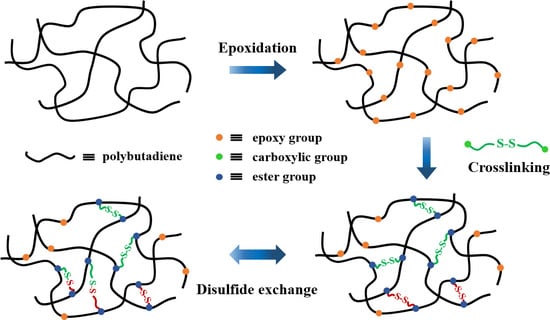
:
1. Introduction
2. Materials and Methods
2.1. Materials
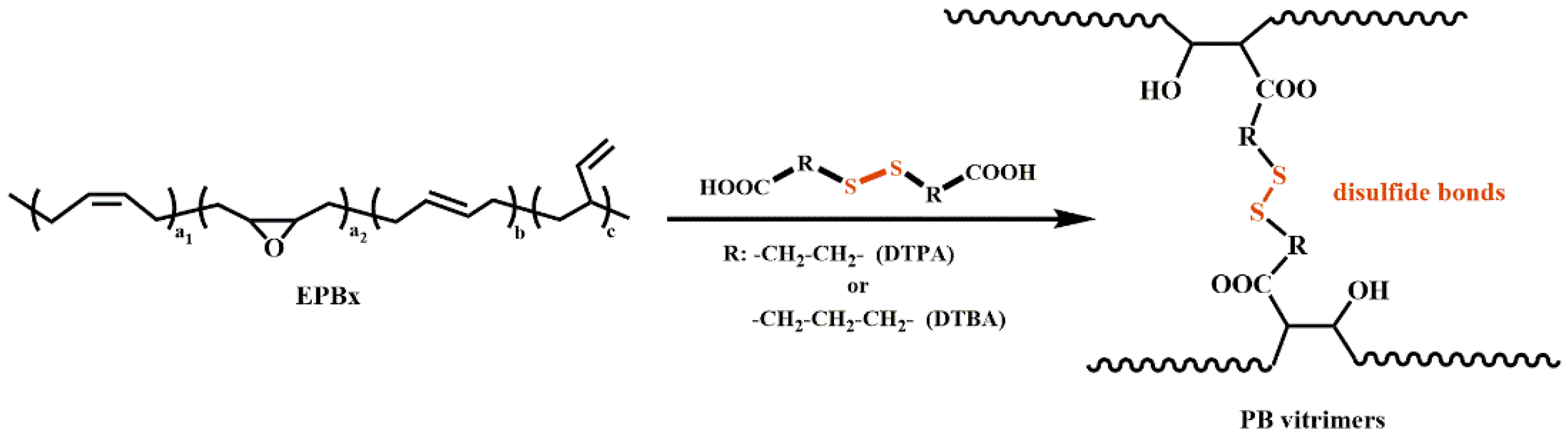
2.2. Preparation of Polybutadiene Vitrimers (PB Vitrimers)
2.3. Characterization
3. Results and Discussion
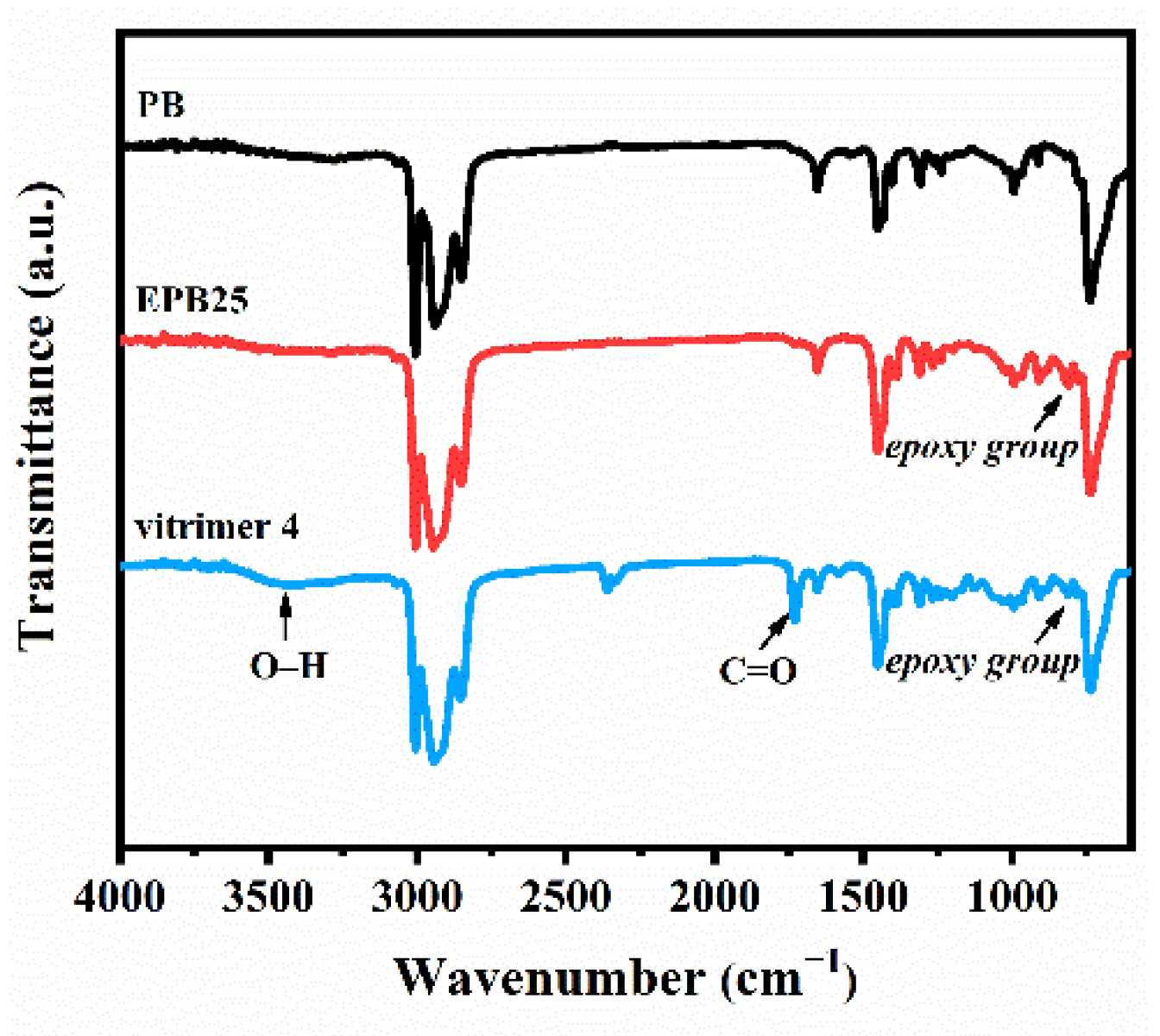
3.1. Design and Characterization of PB Vitrimers
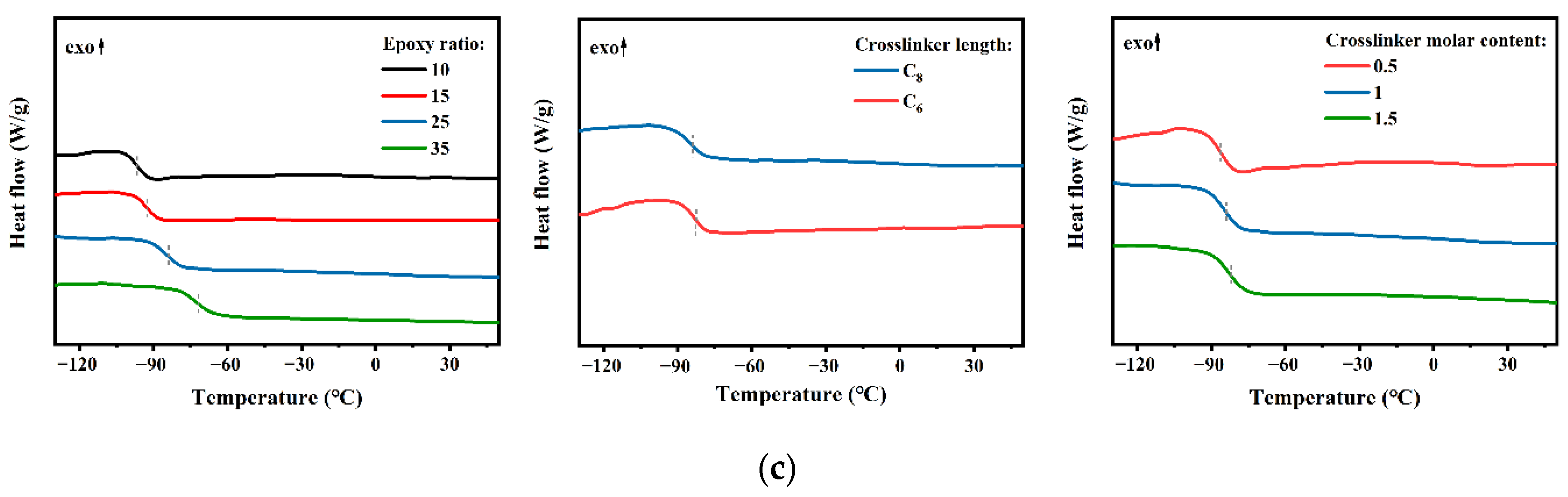
3.2. Thermal Properties of PB Vitrimers
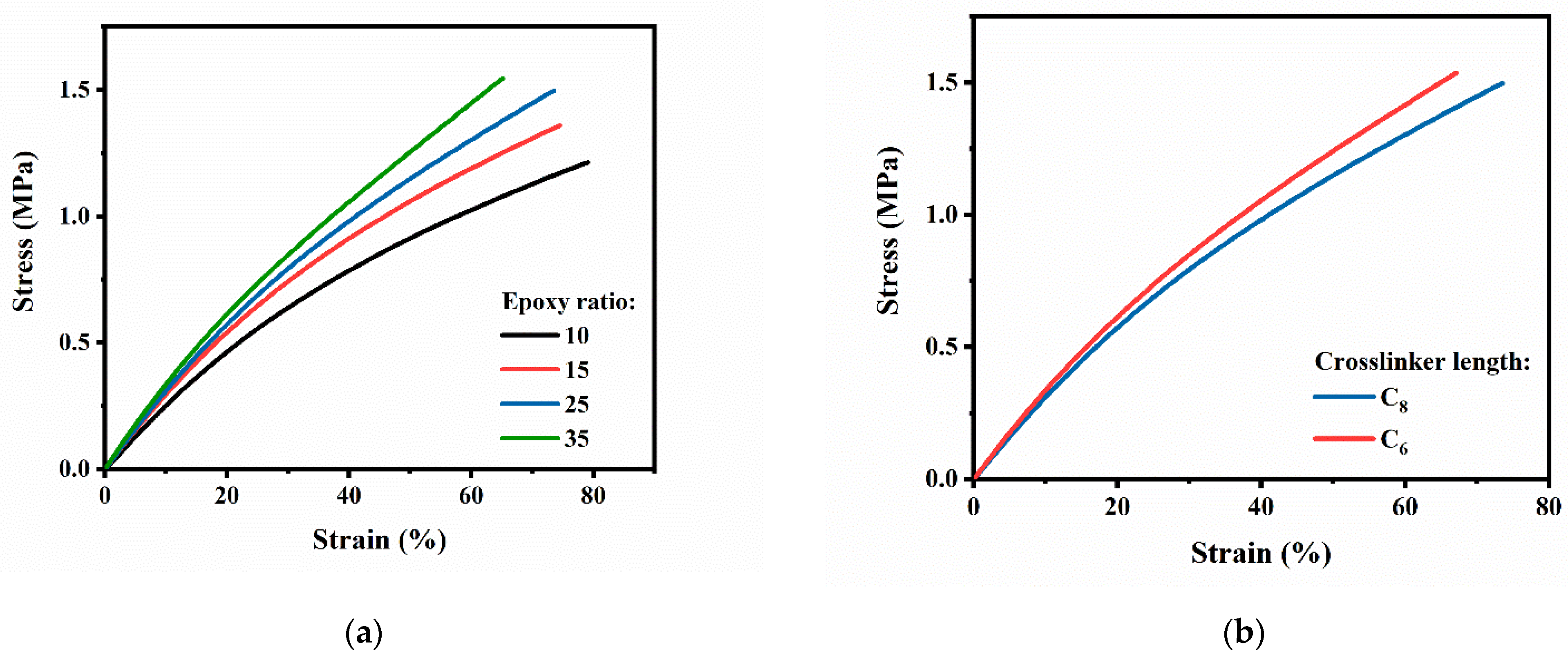
3.3. Mechanical Properties of PB Vitrimers
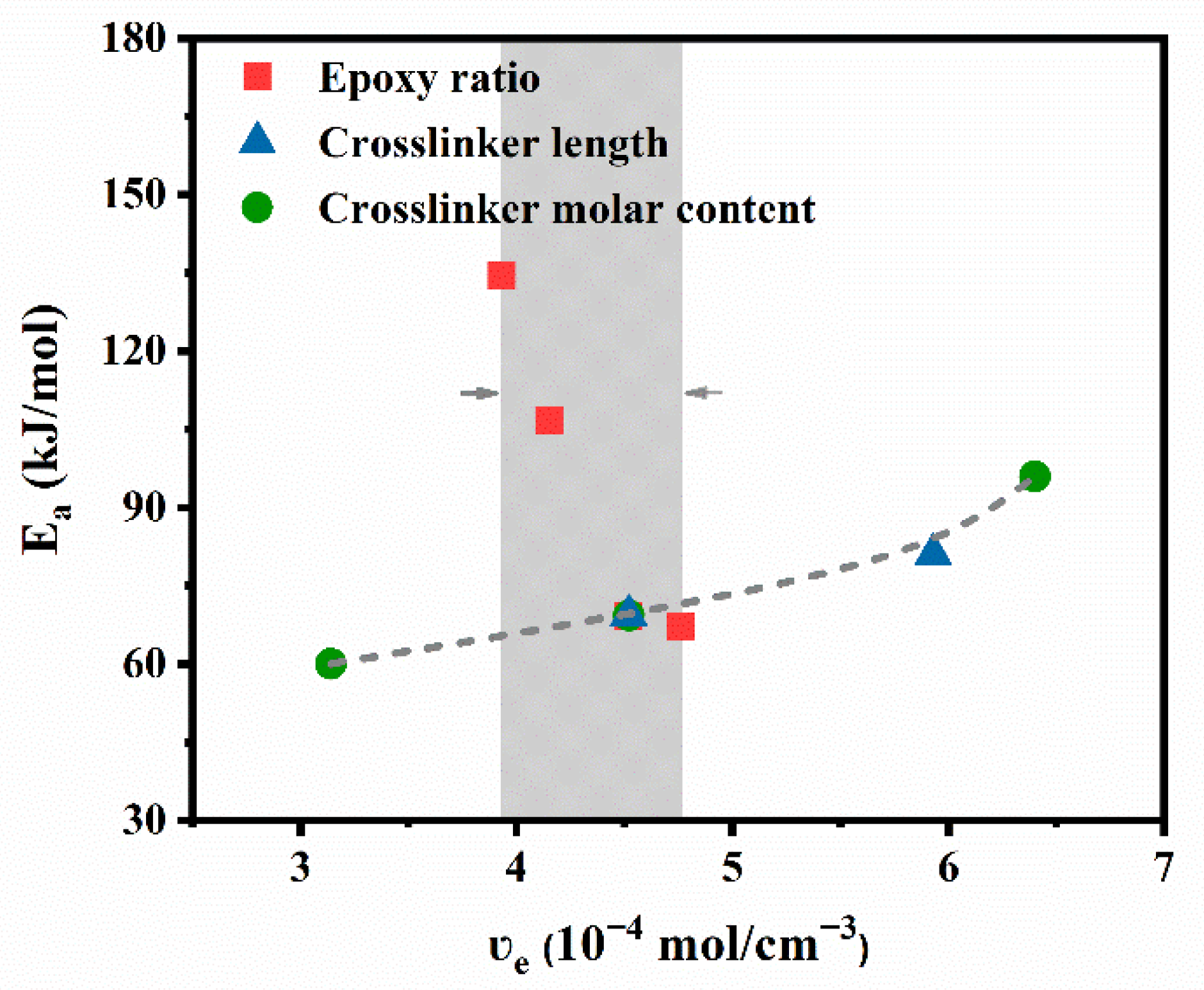
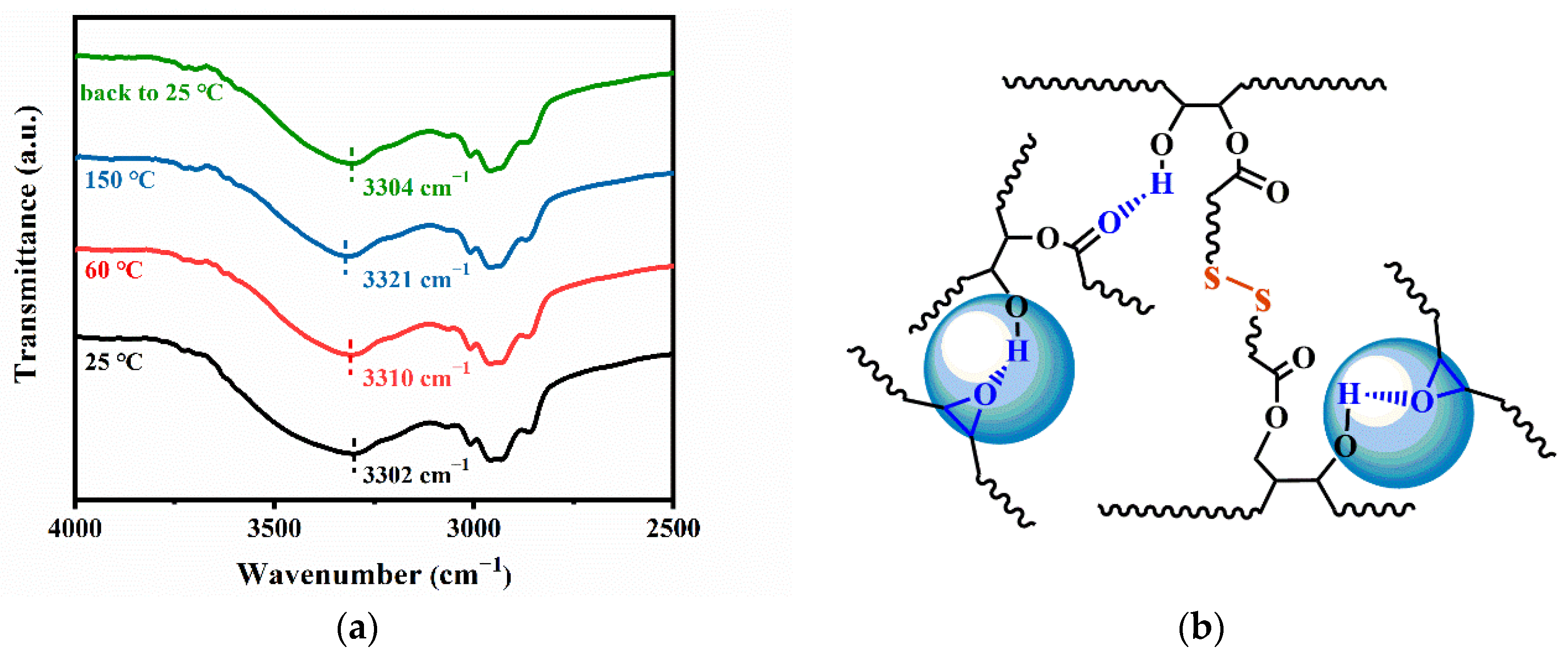
3.4. Topological Network Rearrangements of PB Vitrimers
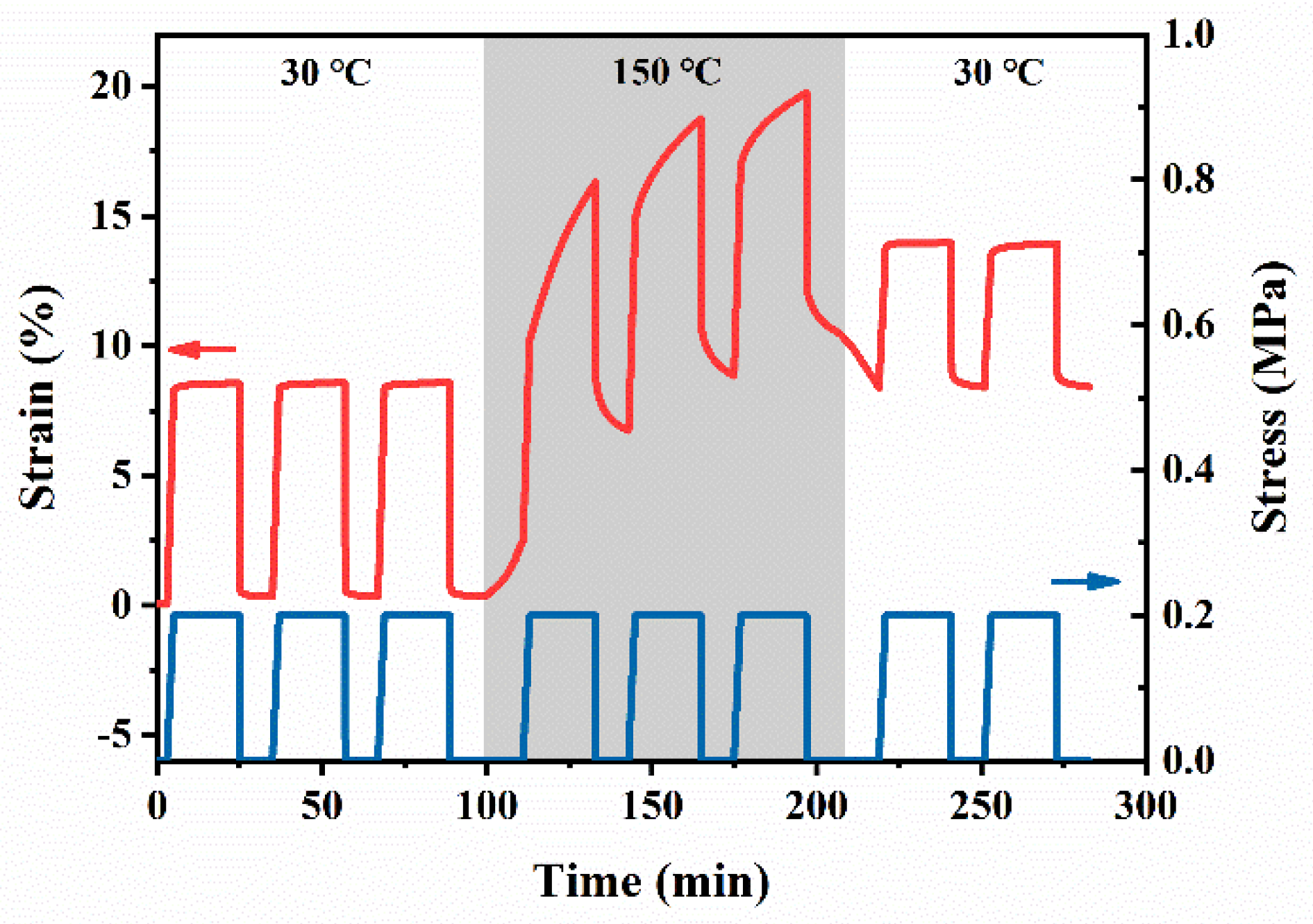
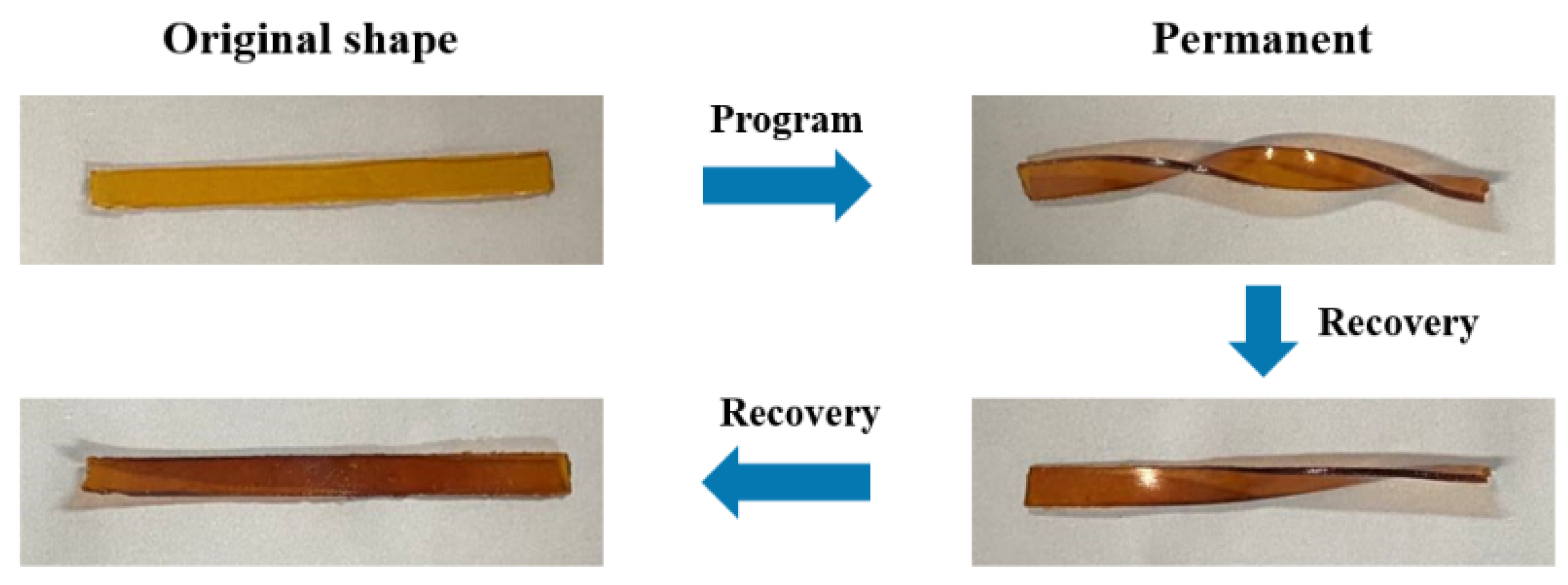
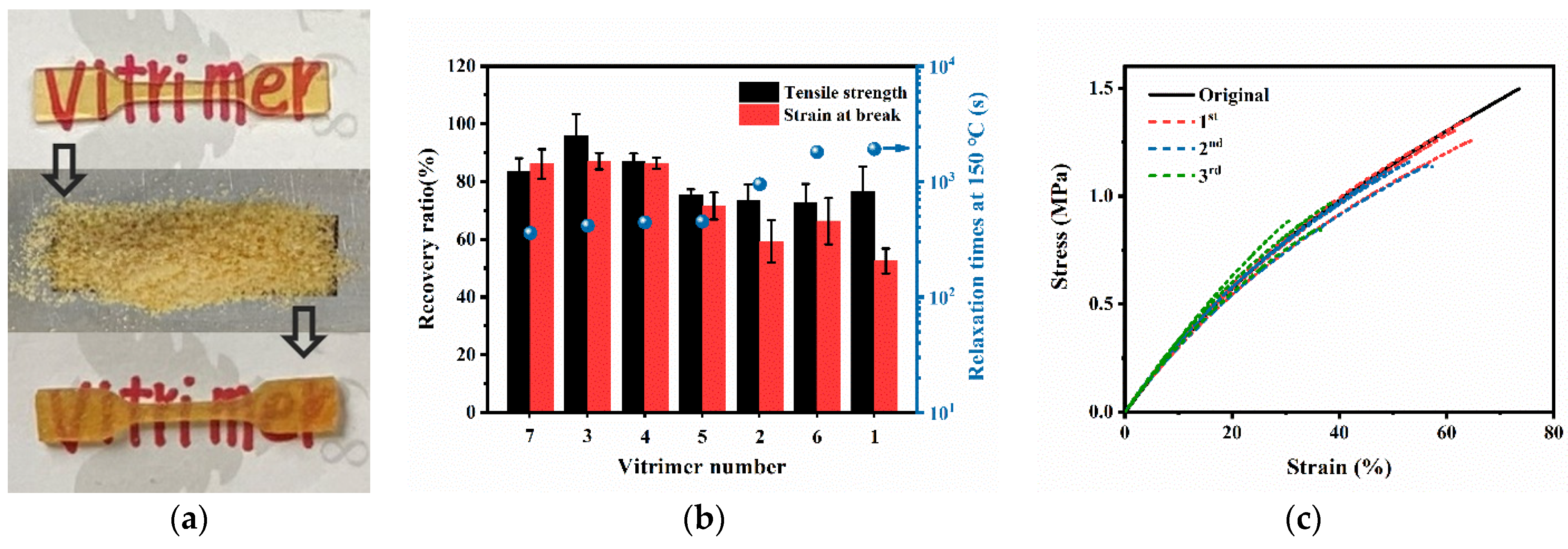
3.5. Recycling Properties of PB Vitrimers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adhikari, B.; De, D.; Maiti, S. Reclamation and recycling of waste rubber. Prog. Polym. Sci. 2000, 25, 909–948. [Google Scholar] [CrossRef]
- Imbernon, L.; Norvez, S. From landfilling to vitrimer chemistry in rubber life cycle. Eur. Polym. J. 2016, 82, 347–376. [Google Scholar] [CrossRef]
- Montarnal, D.; Capelot, M.; Tournilhac, F.; Leibler, L. Silica-Like Malleable Materials from Permanent Organic Networks. Science 2011, 334, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Ruiz de Luzuriaga, A.; Martin, R.; Markaide, N.; Rekondo, A.; Cabañero, G.; Rodríguez, J.; Odriozola, I. Epoxy resin with exchangeable disulfide crosslinks to obtain reprocessable, repairable and recyclable fiber-reinforced thermoset composites. Mater. Horiz. 2016, 3, 241–247. [Google Scholar] [CrossRef]
- Denissen, W.; Winne, J.M.; Du Prez, F.E. Vitrimers: Permanent organic networks with glass-like fluidity. Chem. Sci. 2016, 7, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.P.; Rong, M.Z.; Zhang, M.Q. Polymer engineering based on reversible covalent chemistry: A promising innovative pathway towards new materials and new functionalities. Prog. Polym. Sci. 2018, 80, 39–93. [Google Scholar] [CrossRef]
- Zou, W.; Dong, J.; Luo, Y.; Zhao, Q.; Xie, T. Dynamic Covalent Polymer Networks: From Old Chemistry to Modern Day Innovations. Adv. Mater. 2017, 29, 1606100. [Google Scholar] [CrossRef]
- Caffy, F.; Nicolaÿ, R. Transformation of polyethylene into a vitrimer by nitroxide radical coupling of a bis-dioxaborolane. Polym. Chem. 2019, 10, 3107–3115. [Google Scholar] [CrossRef]
- Yang, F.; Pan, L.; Ma, Z.; Lou, Y.; Li, Y.; Li, Y. Highly elastic, strong, and reprocessable cross-linked polyolefin elastomers enabled by boronic ester bonds. Polym. Chem. 2020, 11, 3285–3295. [Google Scholar] [CrossRef]
- Kar, G.P.; Saed, M.O.; Terentjev, E.M. Scalable upcycling of thermoplastic polyolefins into vitrimers through transesterification. J. Mater. Chem. A 2020, 8, 24137–24147. [Google Scholar] [CrossRef]
- Feng, Z.; Hu, J.; Zuo, H.; Ning, N.; Zhang, L.; Yu, B.; Tian, M. Photothermal-Induced Self-Healable and Reconfigurable Shape Memory Bio-Based Elastomer with Recyclable Ability. ACS Appl. Mater. Interfaces 2019, 11, 1469–1479. [Google Scholar] [CrossRef]
- Kaiser, S.; Wurzer, S.; Pilz, G.; Kern, W.; Schlogl, S. Stress relaxation and thermally adaptable properties in vitrimer-like elastomers from HXNBR rubber with covalent bonds. Soft Matter 2019, 15, 6062–6072. [Google Scholar] [CrossRef]
- Wang, J.; Chen, S.; Lin, T.; Ke, J.; Chen, T.; Wu, X.; Lin, C. A catalyst-free and recycle-reinforcing elastomer vitrimer with exchangeable links. RSC Adv. 2020, 10, 39271–39276. [Google Scholar] [CrossRef]
- Xu, C.; Cui, R.; Fu, L.; Lin, B. Recyclable and heat-healable epoxidized natural rubber/bentonite composites. Compos. Sci. Technol. 2018, 167, 421–430. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, C.; Shi, Z.; Yin, J.; Tian, M. Tailoring vinylogous urethane chemistry for the cross-linked polybutadiene: Wide freedom design, multiple recycling methods, good shape memory behavior. Polymer 2018, 148, 202–210. [Google Scholar] [CrossRef]
- Xiang, H.; Yin, J.; Lin, G.; Liu, X.; Rong, M.; Zhang, M. Photo-crosslinkable, self-healable and reprocessable rubbers. Chem. Eng. J. 2019, 358, 878–890. [Google Scholar] [CrossRef]
- Chen, Y.; Tang, Z.; Zhang, X.; Liu, Y.; Wu, S.; Guo, B. Covalently Cross-Linked Elastomers with Self-Healing and Malleable Abilities Enabled by Boronic Ester Bonds. ACS Appl. Mater. Interfaces 2018, 10, 24224–24231. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Tang, Z.; Liu, Y.; Wu, S.; Guo, B. Mechanically Robust, Self-Healable, and Reprocessable Elastomers Enabled by Dynamic Dual Cross-Links. Macromolecules 2019, 52, 3805–3812. [Google Scholar] [CrossRef]
- Cromwell, O.R.; Chung, J.; Guan, Z. Malleable and Self-Healing Covalent Polymer Networks through Tunable Dynamic Boronic Ester Bonds. J. Am. Chem. Soc. 2015, 137, 6492–6495. [Google Scholar] [CrossRef] [PubMed]
- Breuillac, A.; Kassalias, A.; Nicolaÿ, R. Polybutadiene Vitrimers Based on Dioxaborolane Chemistry and Dual Networks with Static and Dynamic Cross-links. Macromolecules 2019, 52, 7102–7113. [Google Scholar] [CrossRef]
- Lu, Y.X.; Guan, Z. Olefin metathesis for effective polymer healing via dynamic exchange of strong carbon-carbon double bonds. J. Am. Chem. Soc. 2012, 134, 14226–14231. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.X.; Tournilhac, F.; Leibler, L.; Guan, Z. Making insoluble polymer networks malleable via olefin metathesis. J. Am. Chem. Soc. 2012, 134, 8424–8427. [Google Scholar] [CrossRef] [PubMed]
- Neal, J.A.; Mozhdehi, D.; Guan, Z. Enhancing mechanical performance of a covalent self-healing material by sacrificial noncovalent bonds. J. Am. Chem. Soc. 2015, 137, 4846–4850. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tang, Z.; Chen, Y.; Wu, S.; Guo, B. Programming dynamic imine bond into elastomer/graphene composite toward mechanically strong, malleable, and multi-stimuli responsive vitrimer. Compos. Sci. Technol. 2018, 168, 214–223. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, D.; Liu, W.; Li, P.; Liu, J.; Liu, C.; Zhang, J.; Zhao, N.; Xu, J. Recyclable polybutadiene elastomer based on dynamic imine bond. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 2011–2018. [Google Scholar] [CrossRef]
- Hayashi, M.; Yano, R. Fair Investigation of Cross-Link Density Effects on the Bond-Exchange Properties for Trans-Esterification-Based Vitrimers with Identical Concentrations of Reactive Groups. Macromolecules 2019, 53, 182–189. [Google Scholar] [CrossRef]
- Hayashi, M.; Yano, R.; Takasu, A. Synthesis of amorphous low Tg polyesters with multiple COOH side groups and their utilization for elastomeric vitrimers based on post-polymerization cross-linking. Polym. Chem. 2019, 10, 2047–2056. [Google Scholar] [CrossRef]
- Hajj, R.; Duval, A.; Dhers, S.; Avérous, L. Network Design to Control Polyimine Vitrimer Properties: Physical Versus Chemical Approach. Macromolecules 2020, 53, 3796–3805. [Google Scholar] [CrossRef]
- Bai, J.; Li, H.; Shi, Z.; Yin, J. An Eco-Friendly Scheme for the Cross-Linked Polybutadiene Elastomer via Thiol–Ene and Diels–Alder Click Chemistry. Macromolecules 2015, 48, 3539–3546. [Google Scholar] [CrossRef]
- Liu, Y.; Tang, Z.; Wu, S.; Guo, B. Integrating Sacrificial Bonds into Dynamic Covalent Networks toward Mechanically Robust and Malleable Elastomers. ACS Macro Lett. 2019, 8, 193–199. [Google Scholar] [CrossRef]
- Qiu, M.; Wu, S.; Fang, S.; Tang, Z.; Guo, B. Sustainable, recyclable and robust elastomers enabled by exchangeable interfacial cross-linking. J. Mater. Chem. A 2018, 6, 13607–13612. [Google Scholar] [CrossRef]
- Tang, Z.; Liu, Y.; Guo, B.; Zhang, L. Malleable, Mechanically Strong, and Adaptive Elastomers Enabled by Interfacial Exchangeable Bonds. Macromolecules 2017, 50, 7584–7592. [Google Scholar] [CrossRef]
- Liu, Y.; Tang, Z.; Chen, J.; Xiong, J.; Wang, D.; Wang, S.; Wu, S.; Guo, B. Tuning the mechanical and dynamic properties of imine bond crosslinked elastomeric vitrimers by manipulating the crosslinking degree. Polym. Chem. 2020, 11, 1348–1355. [Google Scholar] [CrossRef]
- Imbernon, L.; Oikonomou, E.K.; Norvez, S.; Leibler, L. Chemically crosslinked yet reprocessable epoxidized natural rubber via thermo-activated disulfide rearrangements. Polym. Chem. 2015, 6, 4271–4278. [Google Scholar] [CrossRef]
- Xiang, H.; Rong, M.; Zhang, M. Intrinsic Self-healing and Solid-state Recycling of Vulcanized Natural Rubber. Acta Polym. Sin. 2017, 7, 1130–1140. [Google Scholar] [CrossRef]
- Xiang, H.P.; Qian, H.J.; Lu, Z.Y.; Rong, M.Z.; Zhang, M.Q. Crack healing and reclaiming of vulcanized rubber by triggering the rearrangement of inherent sulfur crosslinked networks. Green Chem. 2015, 17, 4315–4325. [Google Scholar] [CrossRef]
- Cheng, B.; Lu, X.; Zhou, J.; Qin, R.; Yang, Y. Dual Cross-Linked Self-Healing and Recyclable Epoxidized Natural Rubber Based on Multiple Reversible Effects. ACS Sustain. Chem. Eng. 2019, 7, 4443–4455. [Google Scholar] [CrossRef]
- Odriozola, I.; Rekondo, A.; Martiín, R.; Ruiz de Luzuriaga, A.; Cabañero, G.; Grande, H. Self-Healing Elastomer and Process for Its Preparation. WO2015127981A1, 28 February 2014. [Google Scholar]
- Odriozola, I.; Ruiz de Luzuriaga, A.; Rekondo, A.; Martiín, R.; Cabañero, G.; Grande, H. A Self-Healing, Reprocessable and Recyclable Crosslinked Polymer and Process for Its Preparation. WO2016046135A1, 21 September 2021. [Google Scholar]
- Xu, L.; Li, B.-G.; Jie, S.; Li, Z.; Bu, Z. 110th Anniversary: The Epoxidation of Polybutadiene via Reaction-Controlled Phase-Transfer Catalysis. Ind. Eng. Chem. Res. 2019, 58, 13085–13092. [Google Scholar] [CrossRef]
- Bala, P.; Samantaray, B.K.; Srivastava, S.K.; Nando, G.B. Organomodified Montmorillonite as Filler in Natural and Synthetic Rubber. J. Appl. Polym. Sci. 2004, 92, 3583–3592. [Google Scholar] [CrossRef]
- Flory, P.J. Statistical Mechanics of Swelling of Network Structures. J. Chem. Phys. 1950, 18, 108–111. [Google Scholar] [CrossRef]
- Hernández, M.; Grande, A.M.; Dierkes, W.; Bijleveld, J.; van der Zwaag, S.; García, S.J. Turning Vulcanized Natural Rubber into a Self-Healing Polymer: Effect of the Disulfide/Polysulfide Ratio. ACS Sustain. Chem. Eng. 2016, 4, 5776–5784. [Google Scholar] [CrossRef]
- Capelot, M.; Montarnal, D.; Tournilhac, F.; Leibler, L. Metal-catalyzed transesterification for healing and assembling of thermosets. J. Am. Chem. Soc. 2012, 134, 7664–7667. [Google Scholar] [CrossRef] [PubMed]
- Imbernon, L.; Norvez, S.; Leibler, L. Stress Relaxation and Self-Adhesion of Rubbers with Exchangeable Links. Macromolecules 2016, 49, 2172–2178. [Google Scholar] [CrossRef]
- Matxain, J.M.; Asua, J.M.; Ruiperez, F. Design of new disulfide-based organic compounds for the improvement of self-healing materials. Phys. Chem. Chem. Phys. 2016, 18, 1758–1770. [Google Scholar] [CrossRef] [PubMed]
- Capelot, M.; Unterlass, M.M.; Tournilhac, F.; Leibler, L. Catalytic Control of the Vitrimer Glass Transition. ACS Macro Lett. 2012, 1, 789–792. [Google Scholar] [CrossRef]
- Di Mauro, C.; Malburet, S.; Graillot, A.; Mija, A. Recyclable, Repairable, and Reshapable (3R) Thermoset Materials with Shape Memory Properties from Bio-Based Epoxidized Vegetable Oils. ACS Appl. Bio Mater. 2020, 3, 8094–8104. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vitrimer | Epoxy Ratio % a | Crosslinker Length | Crosslinker Molar Content % d |
|---|---|---|---|
| 1 | 10 | C8 b | 1 |
| 2 | 15 | C8 | 1 |
| 3 | 25 | C8 | 0.5 |
| 4 | 25 | C8 | 1 |
| 5 | 25 | C8 | 1.5 |
| 6 | 25 | C6 c | 1 |
| 7 | 35 | C8 | 1 |
| Vitrimer | Parameters | Td (°C) | Tg (°C) a | Tδ (°C) b | G’ at 30 °C (MPa) | ve (10−4 mol·cm−3) |
|---|---|---|---|---|---|---|
| Epoxy ratio | ||||||
| 1 | 10 | 340 | −96.7 | −85.5 | 2.72 | 3.93 |
| 2 | 15 | 341 | −92.7 | −79.9 | 2.92 | 4.15 |
| 4 | 25 | 345 | −84.1 | −69.6 | 2.95 | 4.52 |
| 7 | 35 | 348 | −71.7 | −45.6 | 3.09 | 4.76 |
| Crosslinker length | ||||||
| 4 | C8 | 345 | −84.1 | −69.6 | 2.95 | 4.52 |
| 6 | C6 | 362 | −82.6 | −66.2 | 3.34 | 5.93 |
| Crosslinker molar content | ||||||
| 3 | 0.5 | 341 | −86.2 | −70.1 | 2.47 | 3.14 |
| 4 | 1.0 | 345 | −84.1 | −69.6 | 2.95 | 4.52 |
| 5 | 1.5 | 337 | −82.1 | −69.3 | 4.36 | 6.40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, L.; Xu, L.; Jie, S.; Li, B. Polybutadiene Vitrimers with Tunable Epoxy Ratios: Preparation and Properties. Polymers 2021, 13, 4157. https://doi.org/10.3390/polym13234157
Zhu L, Xu L, Jie S, Li B. Polybutadiene Vitrimers with Tunable Epoxy Ratios: Preparation and Properties. Polymers. 2021; 13(23):4157. https://doi.org/10.3390/polym13234157
Chicago/Turabian StyleZhu, Liqian, Li Xu, Suyun Jie, and Bogeng Li. 2021. "Polybutadiene Vitrimers with Tunable Epoxy Ratios: Preparation and Properties" Polymers 13, no. 23: 4157. https://doi.org/10.3390/polym13234157
APA StyleZhu, L., Xu, L., Jie, S., & Li, B. (2021). Polybutadiene Vitrimers with Tunable Epoxy Ratios: Preparation and Properties. Polymers, 13(23), 4157. https://doi.org/10.3390/polym13234157