
Hybrid Proton-Exchange Membrane Based on Perfluorosulfonated Polymers and Resorcinol–Formaldehyde Hydrogel


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Membranes with RF Resin
2.3. Membrane–Electrode Assembly Preparation
2.4. Analytical Techniques
3. Results and Discussion
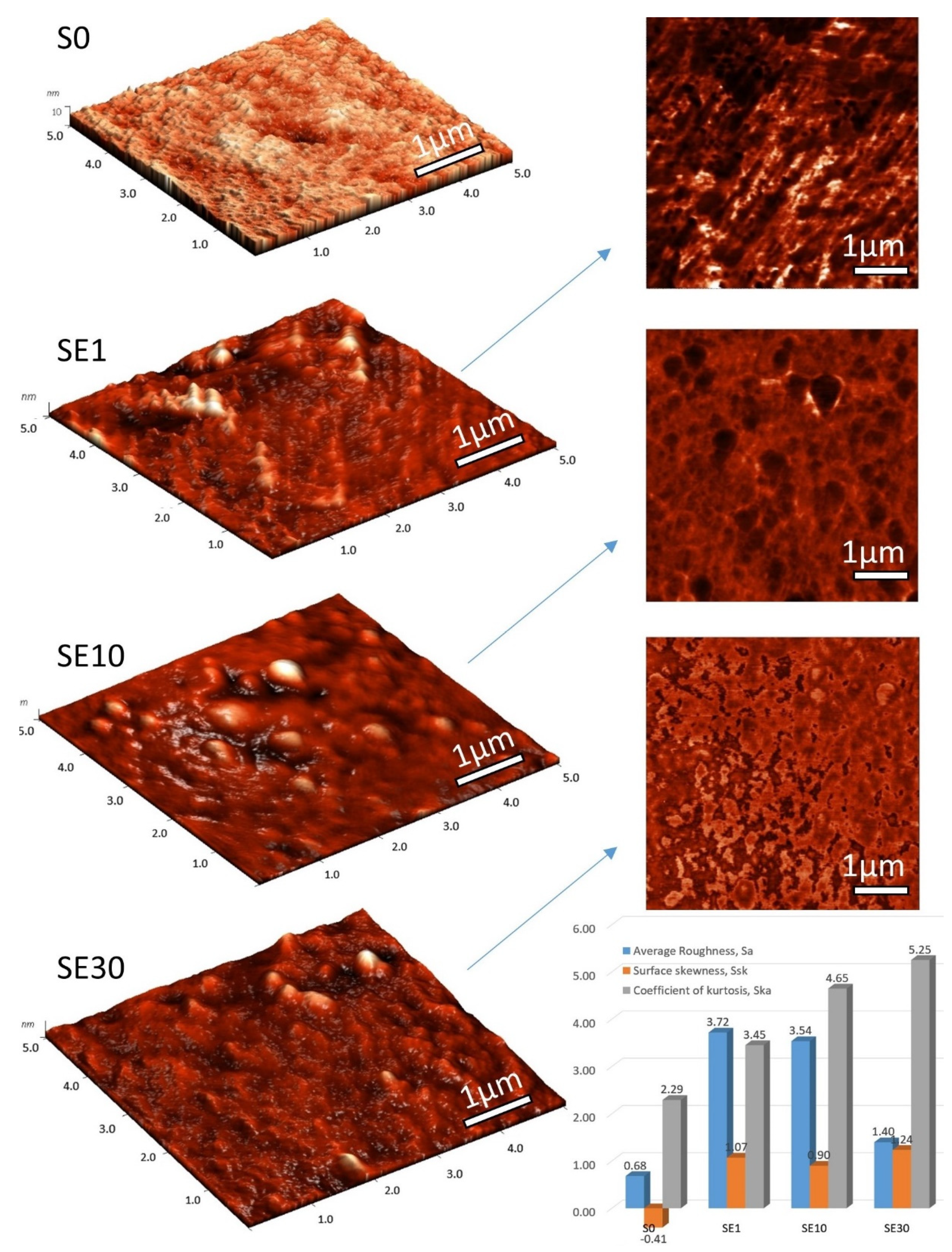
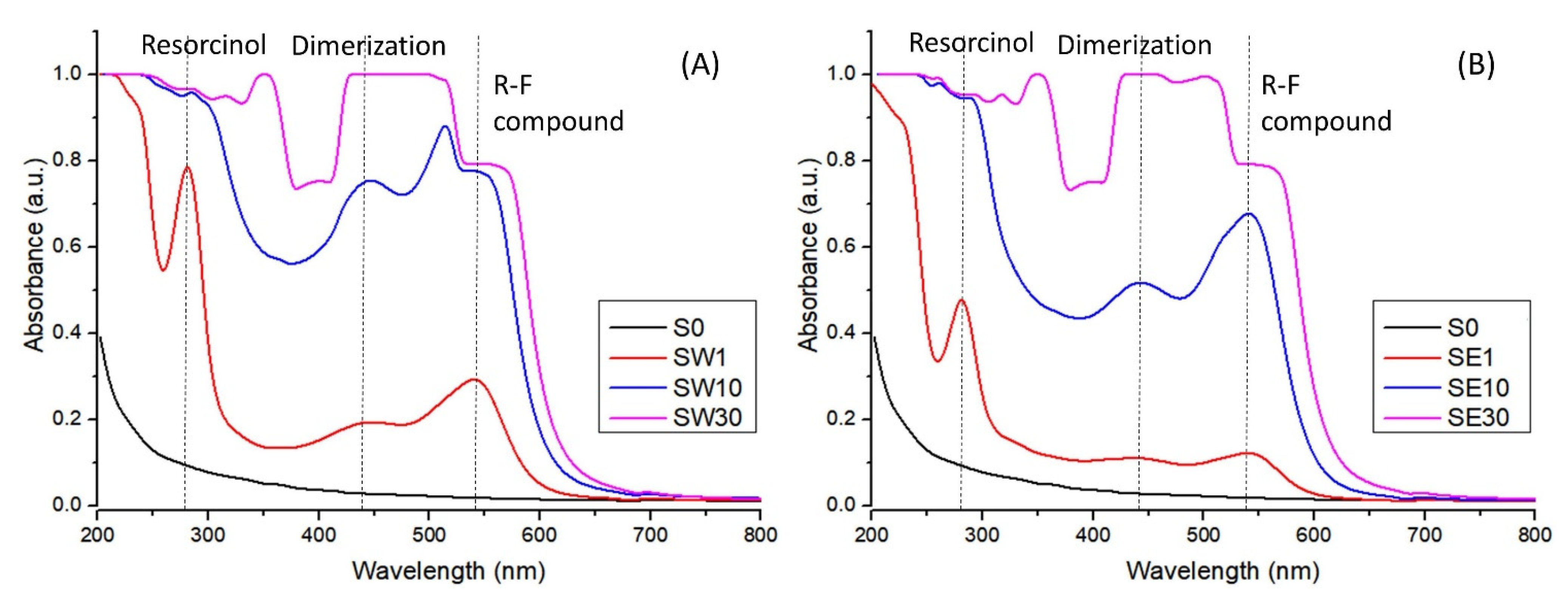
3.1. Morphological Properties
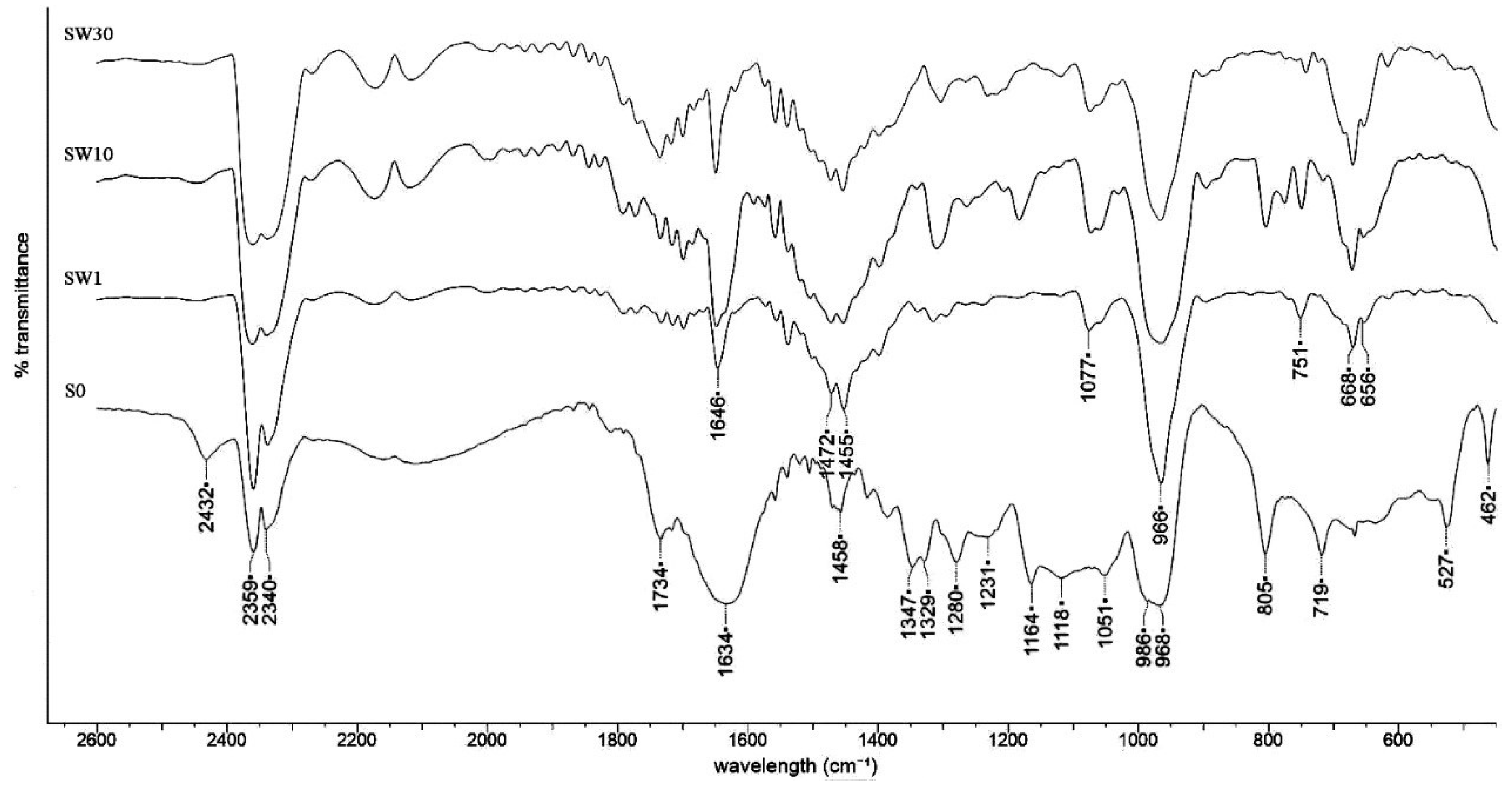
3.2. Structural Characterization
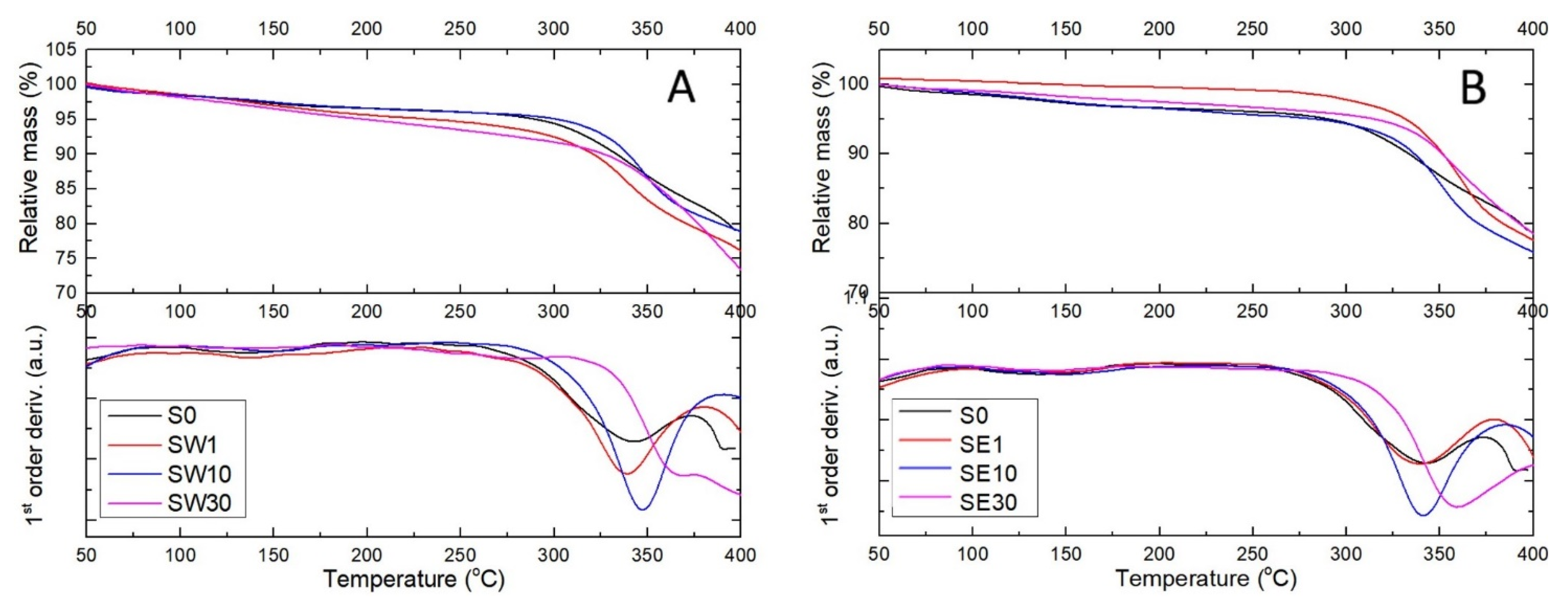
3.3. Thermal Behavior of PFSA–RF Resin Systems
3.4. Water Uptake, Hydration Number, and Ion-Exchange Capacity
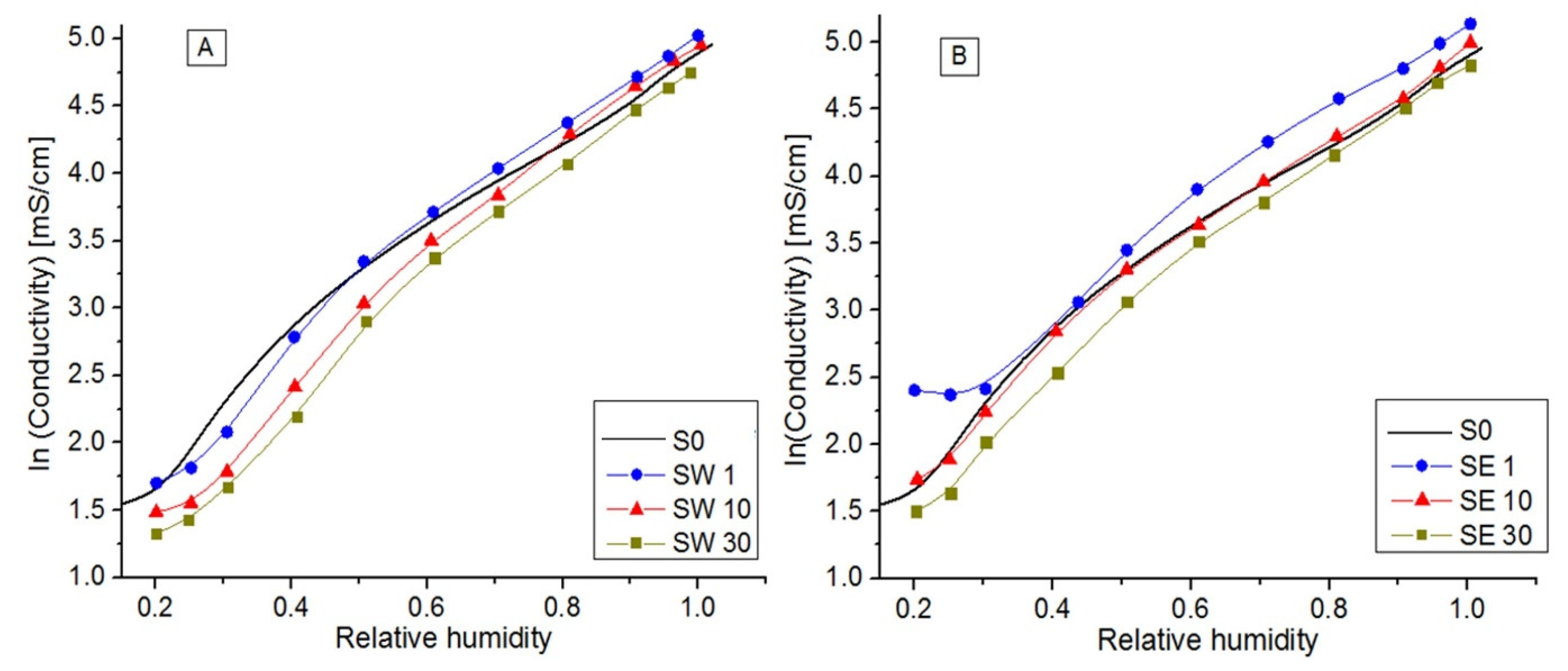
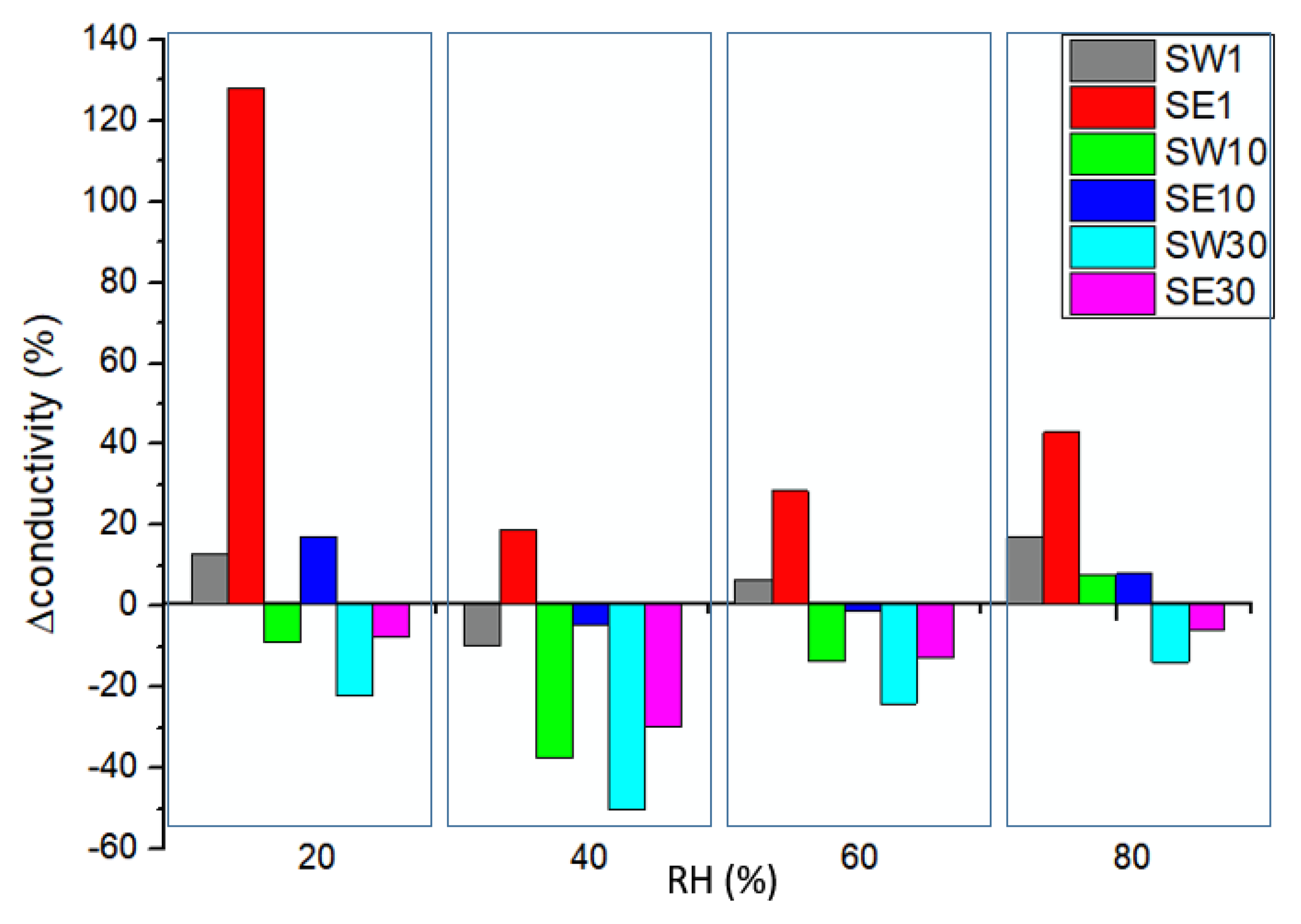
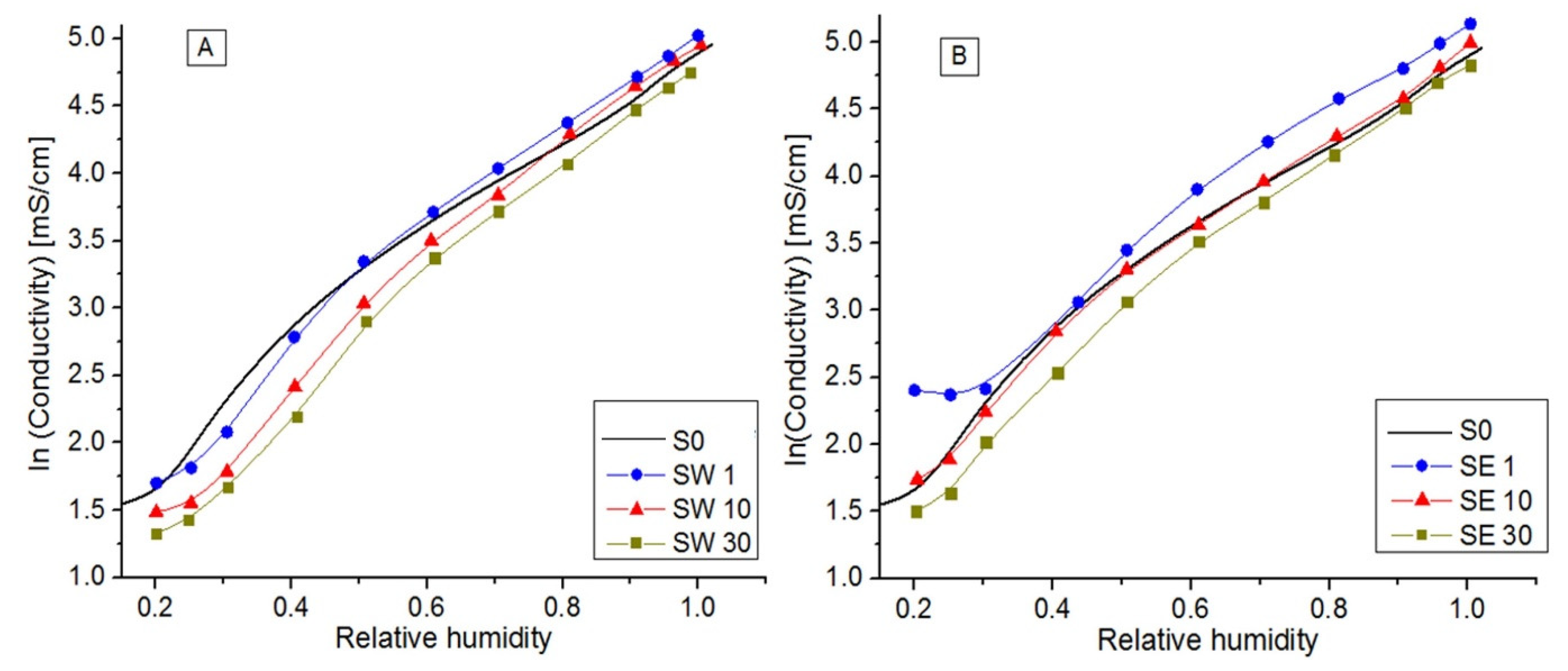
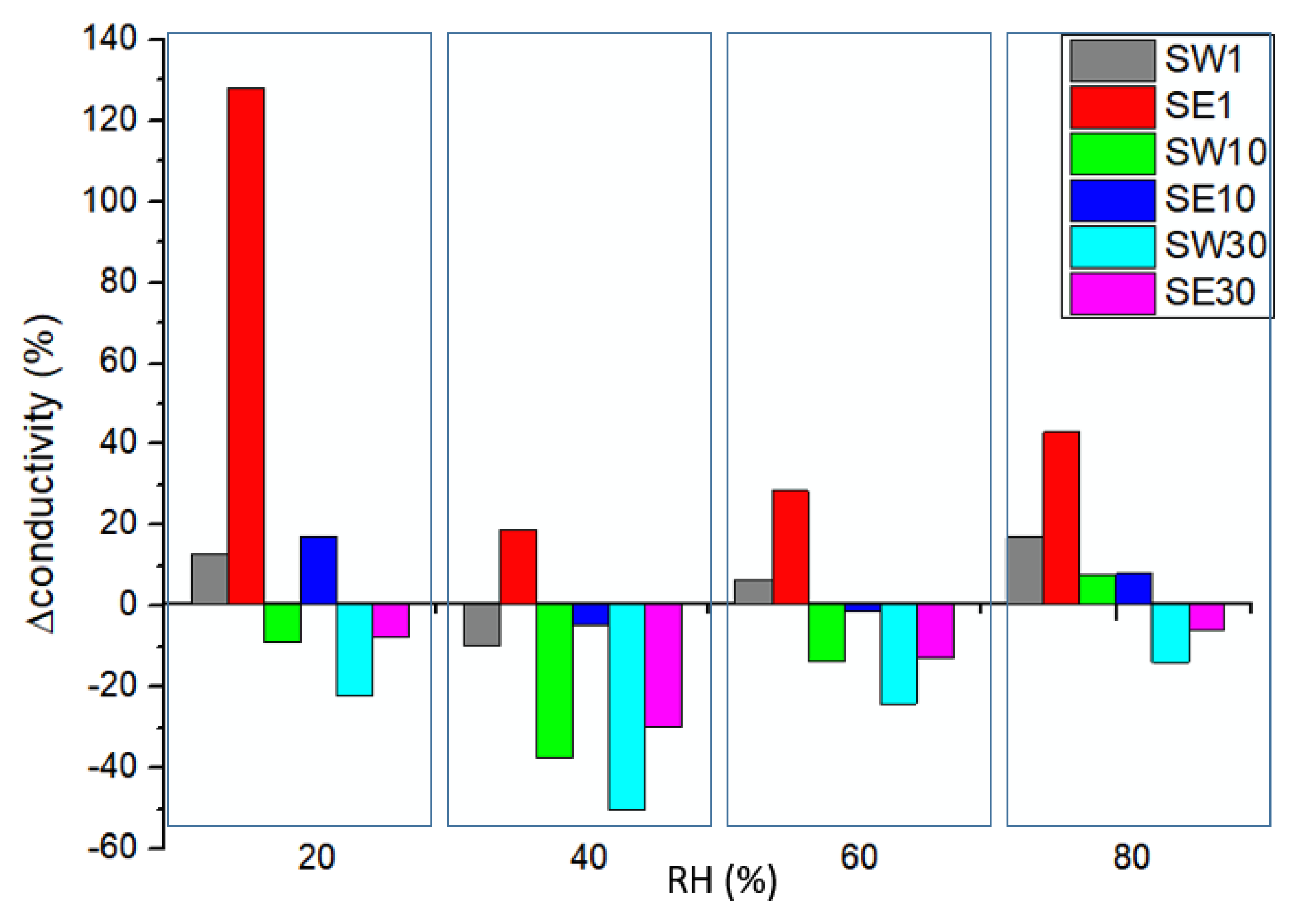
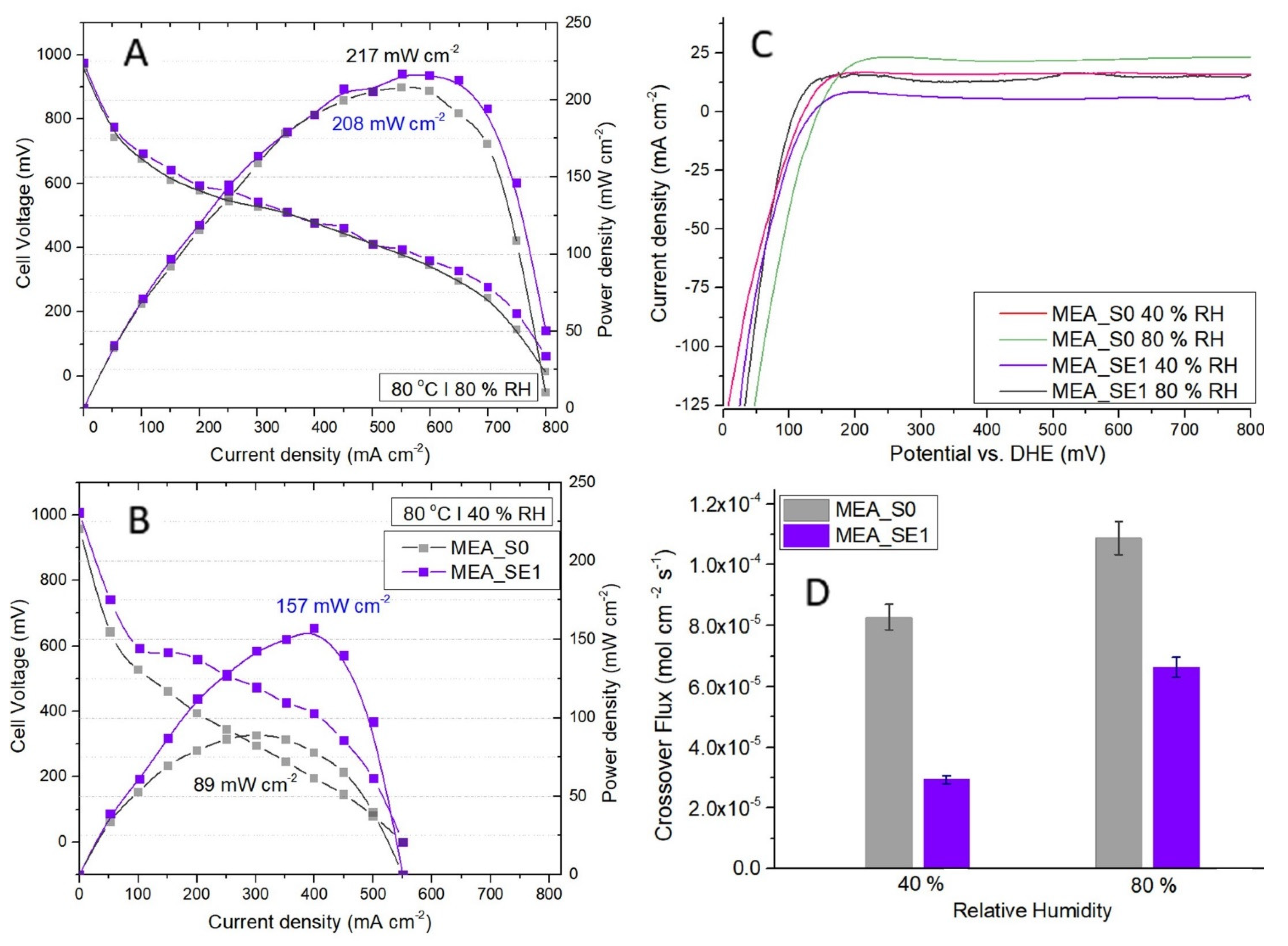
3.5. In-Plane Conductivity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, C.; Negro, E.; Vezzù, K.; Pagot, G.; Cavinato, G.; Nale, A.; Bang, Y.H.; Di Noto, V. Hybrid inorganic-organic proton-conducting membranes based on SPEEK doped with WO3 nanoparticles for application in vanadium redox flow batteries. Electrochim. Acta 2019, 309, 311–325. [Google Scholar] [CrossRef]
- Teng, X.; Wang, M.; Li, G.; Dai, J. Polypyrrole thin film composite membrane prepared via interfacial polymerization with high selectivity for vanadium redox flow battery. React. Funct. Polym. 2020, 157, 104777. [Google Scholar] [CrossRef]
- Ho, W.S.; Sirkar, K.K. Membrane Handbook; Overview Life Science; Springer: New York, NY, USA, 1992; pp. 3–15. ISBN 978-1-4615-3548-5. [Google Scholar]
- EG&G Services, Inc. Science Applications International Corporation, Fuel Cell Handbook; US Department of Energy: Morgantown, WV, USA, 2004.
- Wenzl, H. Batteries and Fuel Cells, Encyclopedia of Electrochemical Power Sources; Garche, J., Ed.; Elsevier: Amsterdam, The Netherlands, 2009; ISBN 978-0-444-52745-5. [Google Scholar]
- Perry, M.L.; Weber, A.Z. Advanced Redox-Flow Batteries: A Perspective. J. Electrochem. Soc. 2016, 163, A5064–A5067. [Google Scholar] [CrossRef]
- Verstraete, D.; Lehmkuehler, K.; Gong, A.; Harvey, J.R.; Brian, G.; Palmer, J.L. Characterisation of a hybrid, fuel-cell-based propulsion system, for small unmanned aircraft. J. Power Sources 2014, 250, 204–211. [Google Scholar] [CrossRef]
- Hsu, W.Y.; Gierke, T.D. Elastic theory for ionic clustering in perfluorinated ionomers. Macromolecules 1982, 15, 101–105. [Google Scholar] [CrossRef]
- Gebel, G.; Loppinet, B.; Schlick, S. (Eds.) Ionomers: Characterization, Theory and Application; CRC Press: Boca Raton, FL, USA, 1996; p. 83. ISBN 0-8493-7648-3. [Google Scholar]
- Gierke, T.D.; Munn, G.E.; Wilson, F.C. The morphology in Nafion perfluorinated membrane products, as determined by wide- and small-angle x-ray studies. J. Polym. Sci. Polym. Phys. Ed. 1981, 19, 1687–1704. [Google Scholar] [CrossRef]
- Hsu, W.Y.; Gierke, T.D.J. Ion transport and clustering in nafion perfluorinated membranes. J. Membr. Sci. 1983, 13, 307–326. [Google Scholar] [CrossRef]
- Dobrovolskii, Y.A.; Volkov, E.V.; Pisareva, A.V.; Fedotov, Y.A.; Likhachev, D.Y.; Rusanov, A.L. Proton-Exchange Membranes for Hydrogen-Air Fuel Cells. Russ. J. Gen. Chem. 2007, 77, 766–777. [Google Scholar] [CrossRef]
- Xu, T. Ion exchange membranes: State of their development and perspective. J. Membr. Sci. 2005, 263, 1–29. [Google Scholar] [CrossRef]
- Mauritz, K.A.; Moore, R.B. State of Understanding of Nafion. Chem. Rev. 2004, 104, 4535–4585. [Google Scholar] [CrossRef]
- Bondi, A. Van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Askadskii, A.A. Methods for calculating the physical properties of polymers. Ref. J. Chem. 2015, 5, 83–142. [Google Scholar] [CrossRef]
- Thampan, T.; Malhotra, S.; Tang, H.; Datta, R. Modeling of conductive transport in proton-exchange membranes for fuel cells. J. Electrochem. Soc. 2000, 147, 3242–3250. [Google Scholar] [CrossRef]
- Vielstich, W.; Gasteiger, H.; Lamm, A. (Eds.) Handbook of Fuel Cells: Fundamentals, Technology, Applications; Wiley: Hoboken, NJ, USA, 2003; pp. 351–396. ISBN 0471499269. [Google Scholar]
- Choi, Y.; Youn, D.H.; Lee, O.L.; Kim, Y.; Lee, J.S. Sulfonated resorcinol-formaldehyde polymer gels synthesized in Nafion ion clusters as nanoscale reactors for a filler of hybrid proton exchange membranes. Int. J. Hydrogen Energy 2012, 37, 9766–9774. [Google Scholar] [CrossRef]
- Ren, P.; Pei, P.; Li, Y.; Wu, Z.; Chen, D.; Huang, S. Degradation mechanisms of proton exchange membrane fuel cell under typical automotive operating conditions. Prog. Energy Combust. Sci. 2020, 80, 100859. [Google Scholar] [CrossRef]
- Lufrano, F.; Baglio, V.; Staiti, P.; Antonucci, V.; Arico, A.S. Performance analysis of polymer electrolyte membranes for direct methanol fuel cells. J. Power Sources 2013, 243, 519–534. [Google Scholar] [CrossRef]
- Khattra, N.S.; Lu, Z.; Karlsson, A.M.; Santarea, M.H.; Busby, F.C.; Schmiedel, S. Time-dependent mechanical response of a composite PFSA membrane. J. Power Sources 2013, 228, 256–269. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.H.; Yi, B.; Shao, Z.G.; Xing, D.; Zhang, H. Carbon Nanotubes Reinforced Nafion Composite Membrane for Fuel Cell Applications. Electrochem. Solid-State Lett. 2006, 9, A356–A359. [Google Scholar] [CrossRef]
- Satterfield, M.B.; Majsztrik, P.W.; Ota, H.; Benziger, J.B.; Bocarsly, A.B. Mechanical properties of Nafion and titania/Nafion composite membranes for polymer electrolyte membrane fuel cells. J. Polym. Sci. Part B-Polym. Phy. 2006, 44, 2327–2345. [Google Scholar] [CrossRef] [Green Version]
- Park, H.S.; Kim, Y.J.; Choi, Y.S.; Hong, W.H.; Jung, D. Surface chemistry and physical properties of Nafion/polypyrrole/Pt composite membrane prepared by chemical in situ polymerization for DMF. J. Power Sources 2008, 178, 610–619. [Google Scholar] [CrossRef]
- Li, Y.; Shi, Y.; Mehio, N.; Tan, M.; Wang, Z.; Hu, X.; Chen, G.Z.; Dai, S.; Jin, X. More sustainable electricity generation in hot and dry fuel cells with a novel hybrid membrane of Nafion/nano-silica/hydroxyl ionic liquid. Appl. Energy 2006, 175, 451–458. [Google Scholar] [CrossRef] [Green Version]
- Neburchilov, V.; Martin, J.; Wang, H.; Zhang, J. A review of polymer electrolyte membranes for direct methanol fuel cells. J. Power Sources 2007, 169, 221–238. [Google Scholar] [CrossRef]
- Xu, F.; Innocent, C.; Bonnet, B.; Jones, D.J.; Roziere, J. Chemical Modification of Perfluorosulfonated Membranes with Pyrrole for Fuel Cell Application: Preparation, Characterisation and Methanol Transport. Fuel Cells 2005, 5, 398–405. [Google Scholar] [CrossRef]
- Mohamed, H.F.M.; Abdel-Hady, E.E.; Akihiro, O. Per-fluorinated sulfonic acid/PTFE copolymer studied by positron annihilation lifetime and gas permeation techniques. J. Phys. Conf. Series 2015, 618, 012031. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Buenaposada, I.D.; Rey-Raap, N.; Calvo, E.G.; Menéndez, J.A.; Arenillas, A. Acid-based resorcinol-formaldehyde xerogels synthesized by microwave heating. J. Sol-Gel Sci. Technol. 2017, 84, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Fairen-Jimenez, D.; Carrasco-Marın, F.; Moreno-Castill, C. Porosity and surface area of monolithic carbon aerogels prepared using alkaline carbonates and organic acids as polymerization catalysts. Carbon 2006, 44, 2301–2307. [Google Scholar] [CrossRef]
- Alonso-Buenaposada, I.D.; Garrido, L.; Montes-Morán, M.A.; Menéndez, J.A.; Arenillas, A. An underrated variable essential for tailoring the structure of xerogel: The methanol content of commercial formaldehyde solutions. J. Sol-Gel Sci. Technol. 2017, 83, 478–488. [Google Scholar] [CrossRef] [Green Version]
- Civioc, R.; Lattuada, M.; Koebel, M.M.; Galmarini, S. Monolithic resorcinol–formaldehyde alcogels and their corresponding nitrogen-doped activated carbons. J. Sol-Gel Sci. Technol. 2020, 95, 719–732. [Google Scholar] [CrossRef]
- Chopade, S.P.; Sharma, M.M. Reaction of ethanol and formaldehyde: Use of versatile cation-exchange resins as catalyst in batch reactors and reactive distillation columns. React. Funct. Polym. 1997, 32, 53–65. [Google Scholar] [CrossRef]
- Trefilov, A.M.; Tiliakos, A.; Serban, E.C.; Ceaus, C.; Iordache, S.M.; Voinea, S.; Balan, A. Carbon xerogel as gas diffusion layer in PEM fuel cells. Int. J. Hydrogen Energy 2017, 42, 10448–10454. [Google Scholar] [CrossRef]
- Sun, C.; Zlotorowicz, A.; Nawn, G.; Negro, E.; Bertasi, F.; Pagot, G.; Vezzù, K.; Pace, G.; Guarnieri, M.; Di Noto, V. [Nafion/(WO3)x] hybrid membranes for vanadium redox flow batteries. Solid State Ion. 2018, 319, 110–116. [Google Scholar] [CrossRef]
- Syed Abthagir, P.; Saraswathi, R. Thermal stability of polypyrrole prepared from a ternary eutectic melt. Mater. Chem. Phys. 2005, 92, 21–26. [Google Scholar] [CrossRef]
- Tiliakos, A.; Trefilov, A.M.I.; Tanasa, E.; Balan, A.; Stamatin, I. Laser-induced graphene as the microporous layer in proton exchange membrane fuel cell. Appl. Surf. Sci. 2020, 504, 144096. [Google Scholar] [CrossRef]
- Sahu, A.K.; Ketpang, K.; Shanmugam, S.; Kwon, O.; Lee, S.; Kim, S. Sulfonated Graphene–Nafion Composite Membranes for Polymer Electrolyte Fuel Cells Operating under Reduced Relative Humidity. J. Phys. Chem. C 2016, 120, 15855–15866. [Google Scholar] [CrossRef]
- Gadelmawla, E.S.; Koura, M.M.; Maksoud, T.M.A.; Elewa, I.M.; Soliman, H.H. Roughness parameters. J. Mater. Process. Technol. 2002, 123, 133–145. [Google Scholar] [CrossRef]
- Shiraishi, Y.; Hagi, T.; Matsumoto, M.; Tanaka, S.; Ichikawa, S.; Hirai, T. Solar-to-hydrogen peroxide energy conversion on resorcinol–formaldehyde resin photocatalysts prepared by acid-catalysed polycondensation. Commun. Chem. 2020, 3, 169. [Google Scholar] [CrossRef]
- Attia, S.M.; Abdelfatah, M.S.; Mossad, M.M. Characterization of pure and composite resorcinol formaldehyde aerogels doped with silver. IOP Conf. Ser. J. Phys. Conf. Ser. 2017, 869, 012036. [Google Scholar] [CrossRef]
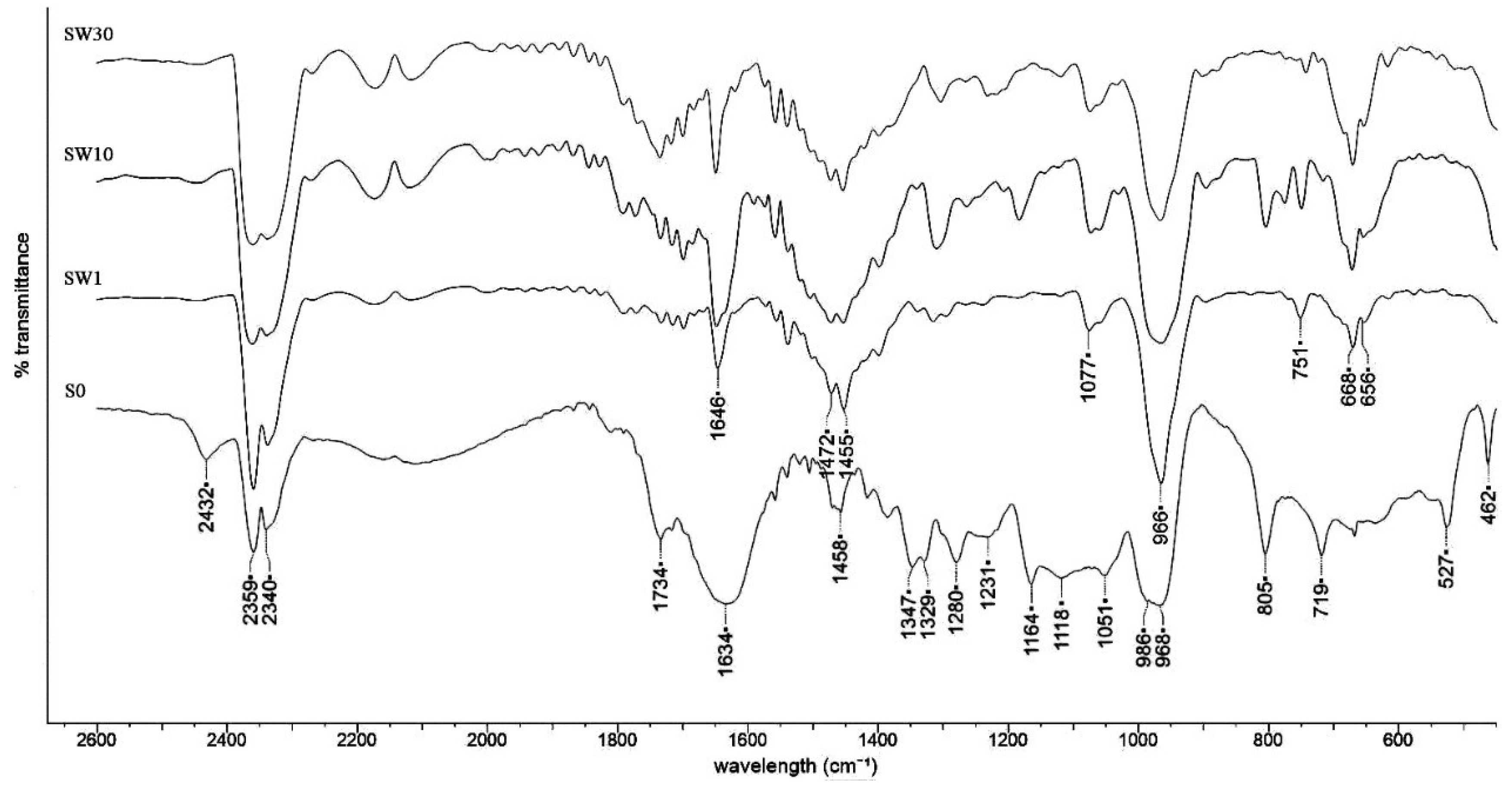
- Liang, Z.; Chen, W.; Liu, J.; Wang, S.; Zhou, Z.; Li, W.; Sun, G.; Xin, Q. FT-IR study of the microstructure of Nafion® membrane. J. Membr. Sci. 2004, 233, 39–44. [Google Scholar] [CrossRef]
- Ludvigsson, M.; Lindgren, J.; Tegenfeldt, J. FTIR study of water in cast Nafion films. Electrochim. Acta 2000, 45, 2267. [Google Scholar] [CrossRef]
- Andronie, A.; Antohe, S.; Iordache, S.M.; Stamatin, S.; Cucu, A.; Ciocanea, A.; Emandi, A.; Ur, E.; Stamatin, I. Proton exchange membrane based on polyperfluorosulfonic acid modified with sulfonamide groups. Optoelectron. Adv. Mater. Rapid Commun. 2010, 4, 1807–1810. [Google Scholar]
- Szczurek, A.; Amaral-Labat, G.; Fierro, V.; Pizzi, A.; Masson, E.; Celzard, A. Porosity of resorcinol-formaldehyde organic and carbon aerogels exchanged and dried with supercritical organic solvents. Mater. Chem. Phys. 2011, 129, 1221–1232. [Google Scholar] [CrossRef]
- Kamal Mohamed, S.M.; Heinrich, C.; Milow, B. Effect of Process Conditions on the Properties of Resorcinol-Formaldehyde Aerogel Microparticles Produced via Emulsion-Gelation Method. Polymers 2021, 13, 2409. [Google Scholar] [CrossRef]
- Principe, I.A.; Fletcher, A.J. Parametric study of factors affecting melamine-resorcinol-formaldehyde xerogels properties. Mater. Today Chem. 2018, 7, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Sha, G.; Guo, S. Resorcinol-formaldehyde aerogels prepared by supercritical acetone drying. J. Non-Crystalline Solids 2000, 271, 167–170. [Google Scholar] [CrossRef]
- Stefanithis, I.D.; Mauritz, K.A. Microstructural evolution of a silicon oxide phase in a perfluorosulfonic acid ionomer by an in situ sol-gel reaction. 3. Thermal analysis studies. Macromolecules 1990, 23, 2397. [Google Scholar] [CrossRef]
- Paddison, S.J. Proton conduction mechanisms at low degrees of hydration in sulfonic acid–based polymer electrolyte membranes. Annu. Rev. Mater. Res. 2003, 33, 289–319. [Google Scholar] [CrossRef]
- Zawodzinski, T.A.; Derouin, C.; Radzinski, S.; Sherman, R.J.; Smith, V.T.; Springer, T.E.; Gottesfeld, S. Water uptake by and transport through Nafion 117 membranes. J. Electrochem. Soc. 1993, 140, 1041–1047. [Google Scholar] [CrossRef]
- Gruger, A.; Regis, A.; Schmatko, T.; Colomban, P. Nanostructure of Nafion® membranes at different states of hydration: An IR and Raman study. Vibr. Spectrosc. 2001, 26, 215–225. [Google Scholar] [CrossRef]
- Kim, Y.S.; Dong, L.; Hickner, M.A.; Glass, T.E.; Webb, V.; McGrath, J.E. State of Water in Disulfonated Poly(arylene ether sulfone) Copolymers and a Perfluorosulfonic Acid Copolymer (Nafion) and Its Effect on Physical and Electrochemical Properties. Macromolecules 2003, 36, 6281. [Google Scholar] [CrossRef]
- Shia, S.; Weber, A.Z.; Kusoglu, A. Structure-transport relationship of perfluorosulfonic-acid membranes in different cationic forms. Electrochim. Acta 2016, 220, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Selvanathan, V.; Ruslan, M.H.; Aminuzzaman, M.; Muhammad, G.; Amin, N.; Sopian, K.; Akhtaruzzaman, M. Resorcinol-Formaldehyde (RF) as a Novel Plasticizer for Starch-Based Solid Biopolymer Electrolyte. Polymers 2020, 12, 2170. [Google Scholar] [CrossRef] [PubMed]
- Schoemaker, M.; Misz, U.; Beckhaus, P.; Heinzel, A. Evaluation of hydrogen crossover through fuel cell membranes. Fuel Cells 2014, 14, 412–415. [Google Scholar] [CrossRef]
- Cheng, X.; Zhang, J.; Tang, Y.; Song, C.; Shen, J.; Song, D.; Zhang, J. Hydrogen cross over in high-temperature PEM fuel cells. J. Power Sources 2007, 167, 25–31. [Google Scholar] [CrossRef]
- Francia, C.; Ijeri, V.S.; Specchia, S.; Spinelli, P. Estimation of hydrogen crossover through Nafion® membranes in PEMFCs. J. Power Sources 2011, 196, 1833–1839. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
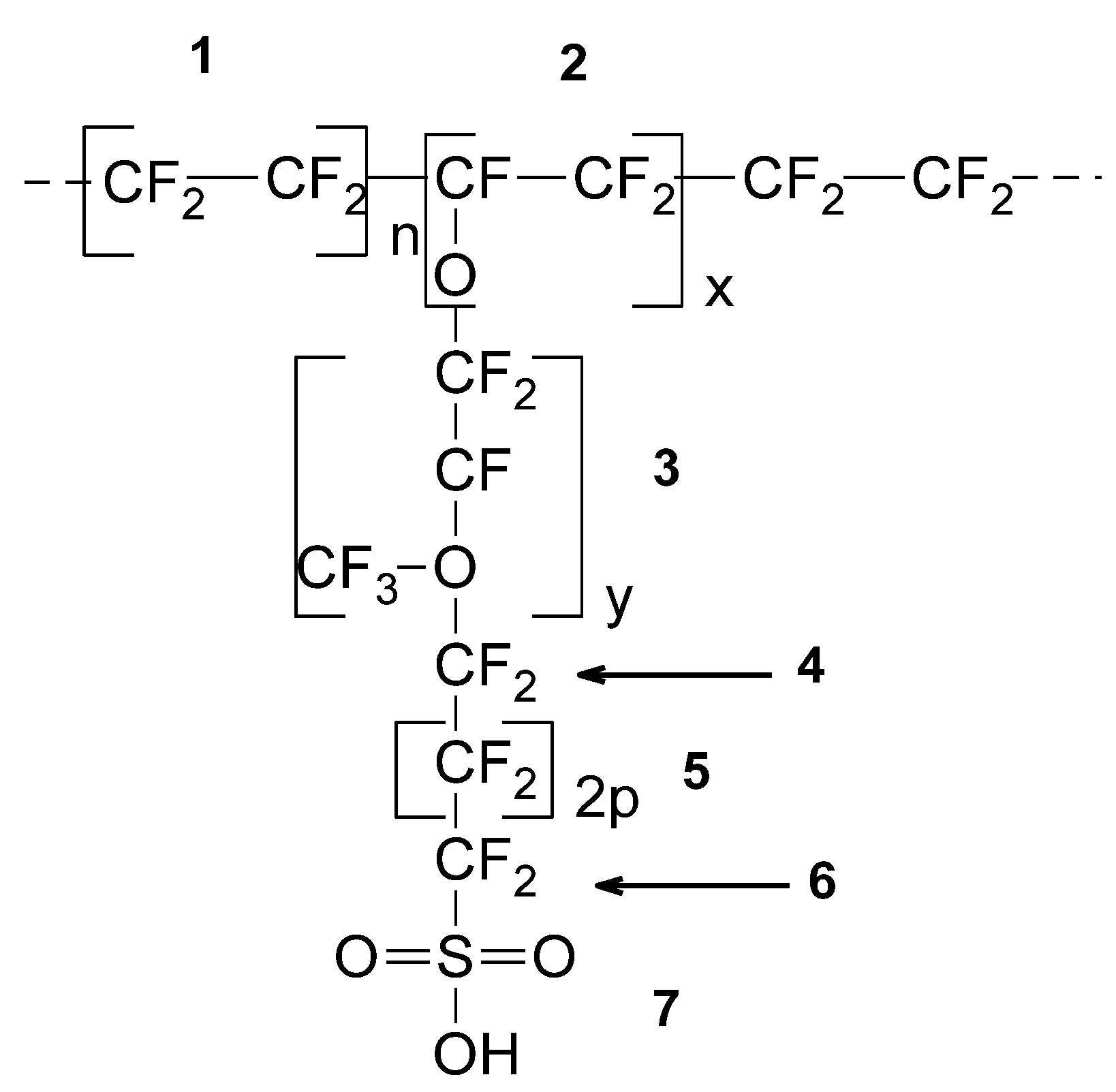
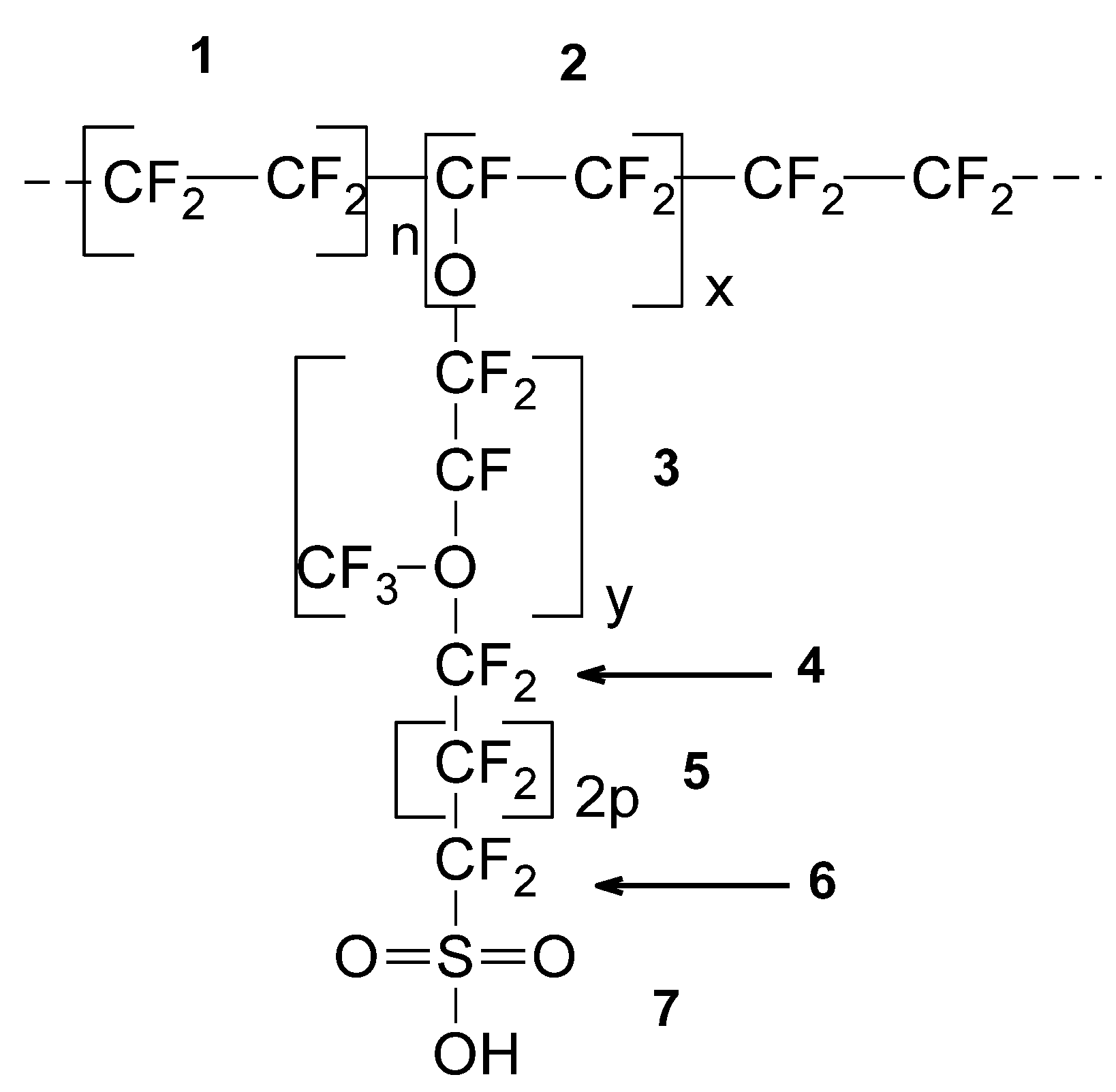
| SRU (See Figure 1) | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Mwu (g/mol) | 100 | 97 | 166 | 50 | 50 | 50 | 81 |
| VVWu (Å3) | 54.20 | 49.60 | 90.40 | 30.20 | 27.10 | 27.98 | 37.93 |
| Molecular weight copolymer: Mwc (x, y, p) = 97x + 166y + 100p + 181 Van der Waals volume: Vwc (x, y, p) = 49.6x + 90.4y + 54.2p + 96.11 Molecular weight SRU: Mw = 100n + Mwc (x, y, p) Van der Waals volume SRU: Vw = 54.2n + Vwc (x, y, p) | |||||||
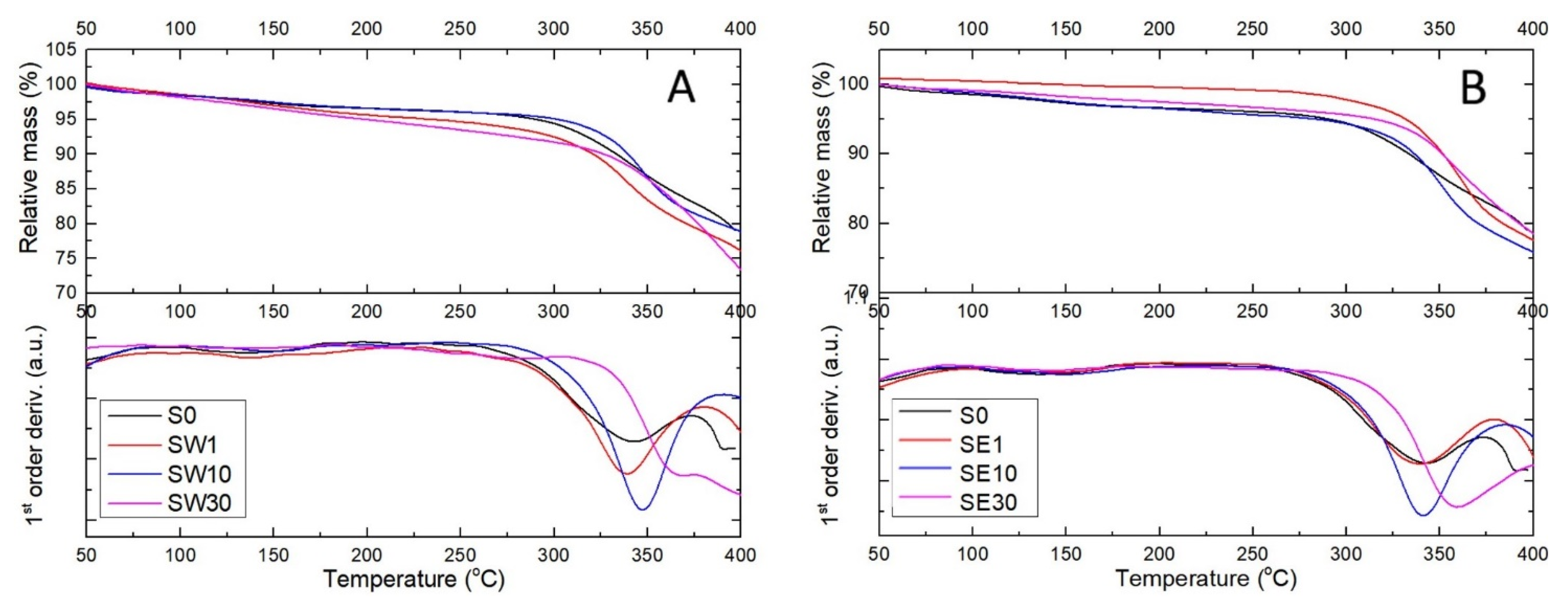
| Sample | tonset (°C) * | tmax (°C) ** | Ea (kJ/mol) |
|---|---|---|---|
| S0 | 294 | 338 | 15.78 |
| SW1 | 309 | 337 | 8.89 |
| SW10 | 323 | 347 | 7.06 |
| SW30 | 340 | 386 | 11.19 |
| SE1 | 305 | 336 | 4.46 |
| SE10 | 316 | 339 | 10.25 |
| SE30 | 331 | 355 | 11.47 |
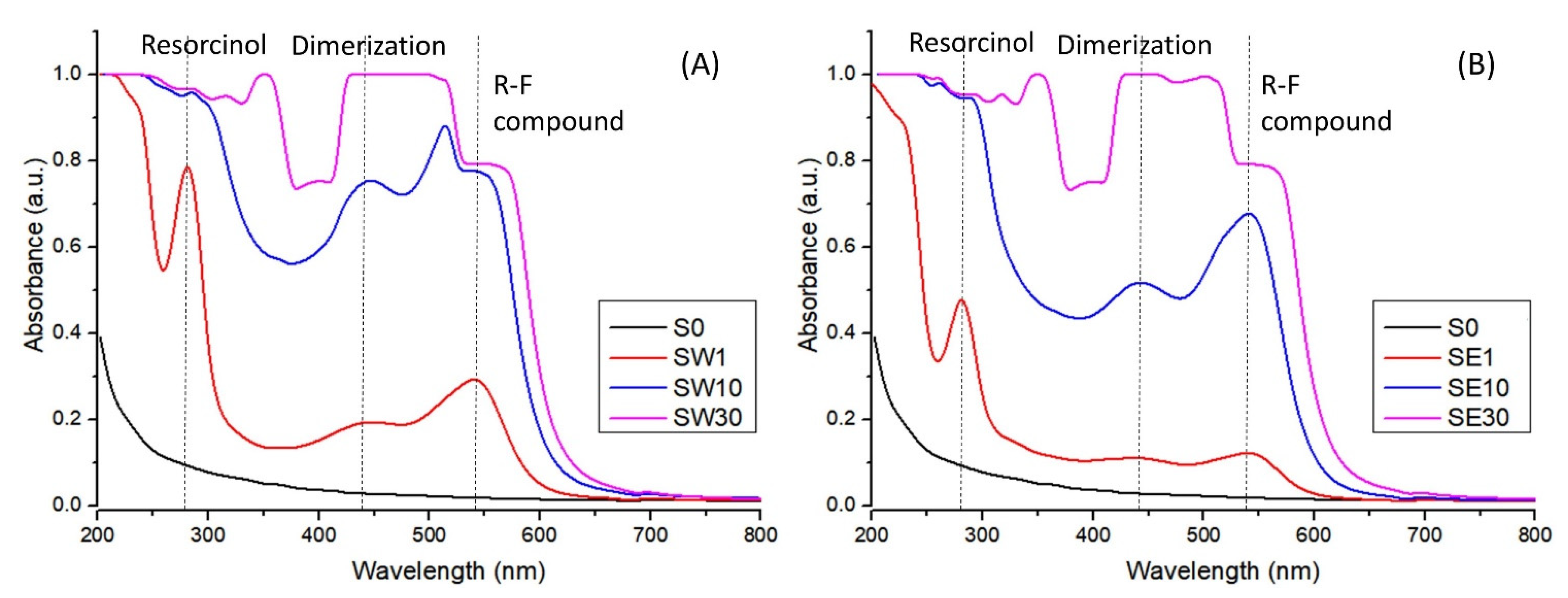
| Sample | Solvent | Immersion Time (min) | RF (%) | WU (%) | IEC (meq/g) | |
|---|---|---|---|---|---|---|
| S0 | - | 0 | 0.00 | 28.78 | 1.05 | 15 |
| SW1 | water | 1 | 0.575 | 28.42 | 1.32 | 12 |
| SW10 | water | 10 | 1.650 | 26.46 | 1.22 | 12 |
| SW30 | water | 30 | 4.910 | 15.28 | 1.14 | 7 |
| SE1 | ethanol | 1 | 0.695 | 30.22 | 1.25 | 13 |
| SE10 | ethanol | 10 | 2.033 | 25.93 | 1.16 | 12 |
| SE30 | ethanol | 30 | 6.849 | 20.54 | 1.11 | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trefilov, A.M.I.; Balan, A.; Stamatin, I. Hybrid Proton-Exchange Membrane Based on Perfluorosulfonated Polymers and Resorcinol–Formaldehyde Hydrogel. Polymers 2021, 13, 4123. https://doi.org/10.3390/polym13234123
Trefilov AMI, Balan A, Stamatin I. Hybrid Proton-Exchange Membrane Based on Perfluorosulfonated Polymers and Resorcinol–Formaldehyde Hydrogel. Polymers. 2021; 13(23):4123. https://doi.org/10.3390/polym13234123
Chicago/Turabian StyleTrefilov, Alexandra Maria Isabel, Adriana Balan, and Ioan Stamatin. 2021. "Hybrid Proton-Exchange Membrane Based on Perfluorosulfonated Polymers and Resorcinol–Formaldehyde Hydrogel" Polymers 13, no. 23: 4123. https://doi.org/10.3390/polym13234123
APA StyleTrefilov, A. M. I., Balan, A., & Stamatin, I. (2021). Hybrid Proton-Exchange Membrane Based on Perfluorosulfonated Polymers and Resorcinol–Formaldehyde Hydrogel. Polymers, 13(23), 4123. https://doi.org/10.3390/polym13234123