
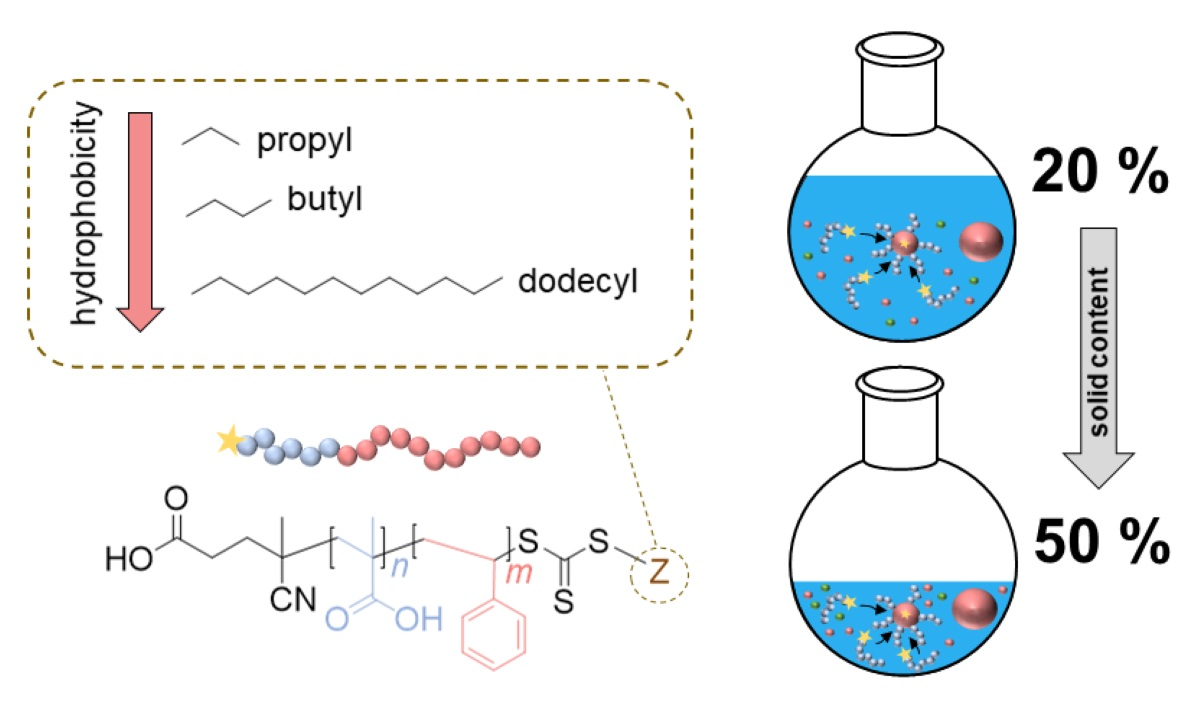
Synthesis of Poly(methacrylic acid)-block-Polystyrene Diblock Copolymers at High Solid Contents via RAFT Emulsion Polymerization



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. General Methods
2.3. Synthesis of 4-Cyano-4-Thioylthiopropylsulfanyl Pentanoic Acid (CPP)
2.4. Synthesis of 4-Cyano-4-Thioylthiobutylsulfanyl Pentanoic Acid (CBP)
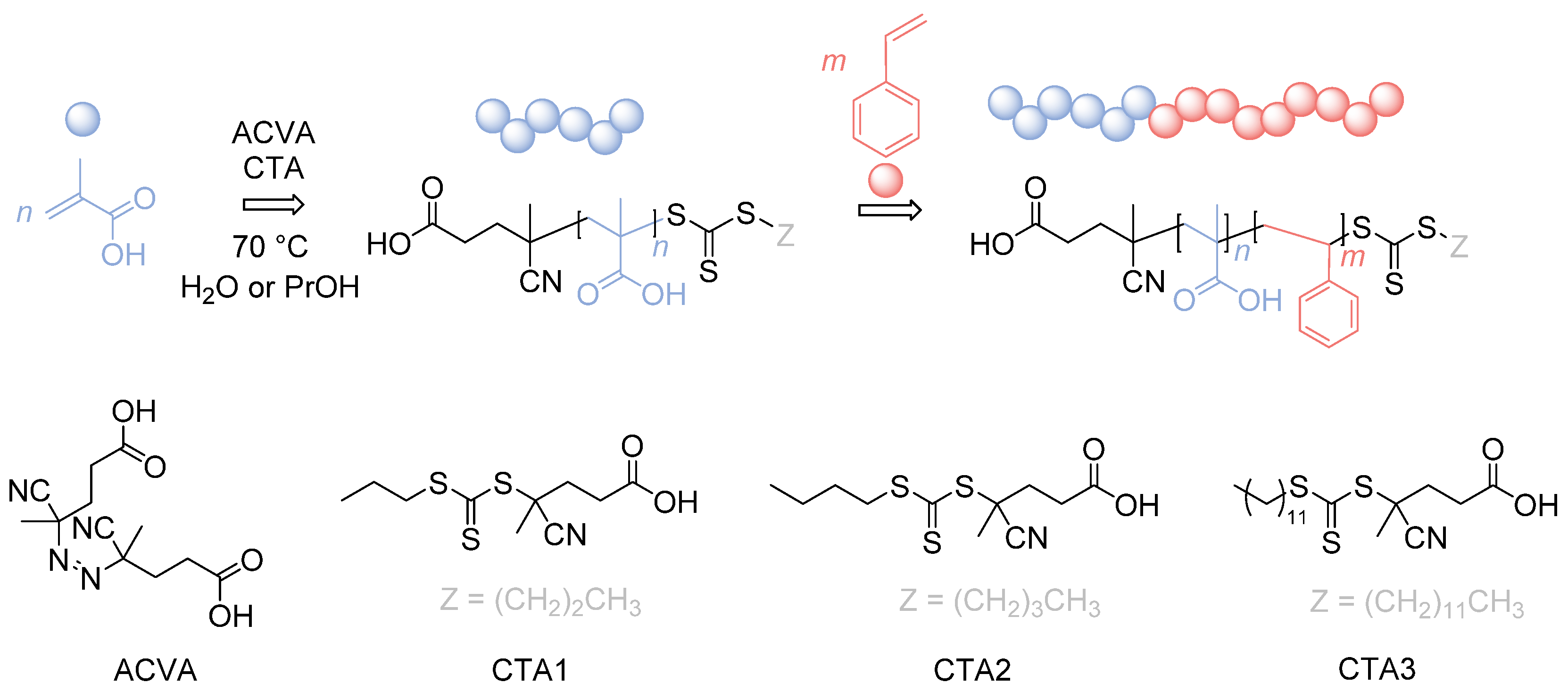
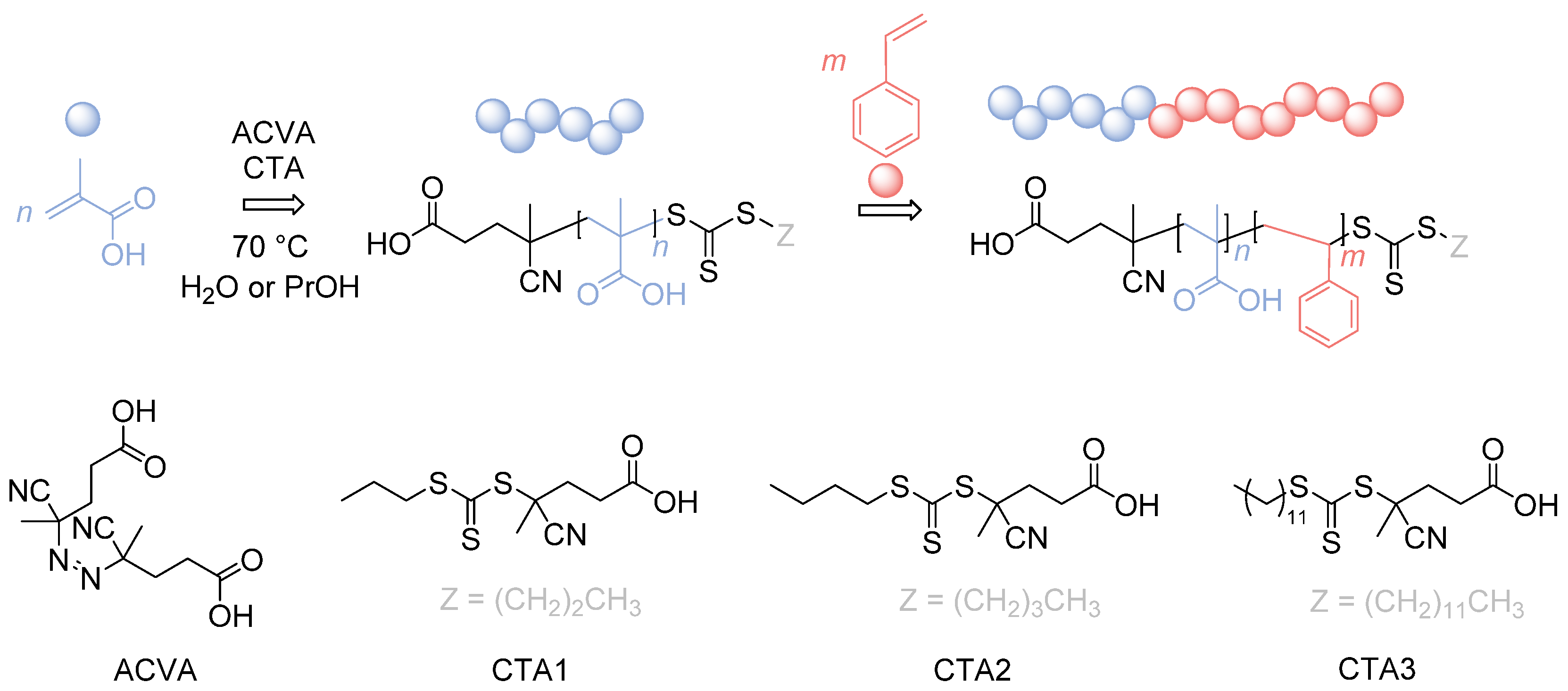
2.5. Synthesis of PMAA with CPP or CBP
2.6. Synthesis of PMAA with CDTPA
2.7. One-Pot Synthesis of PMAA-b-PS with CPP or CBP
2.8. Synthesis of PMAA-b-PS with CDTPA
2.9. Determination of the Molecular Weight
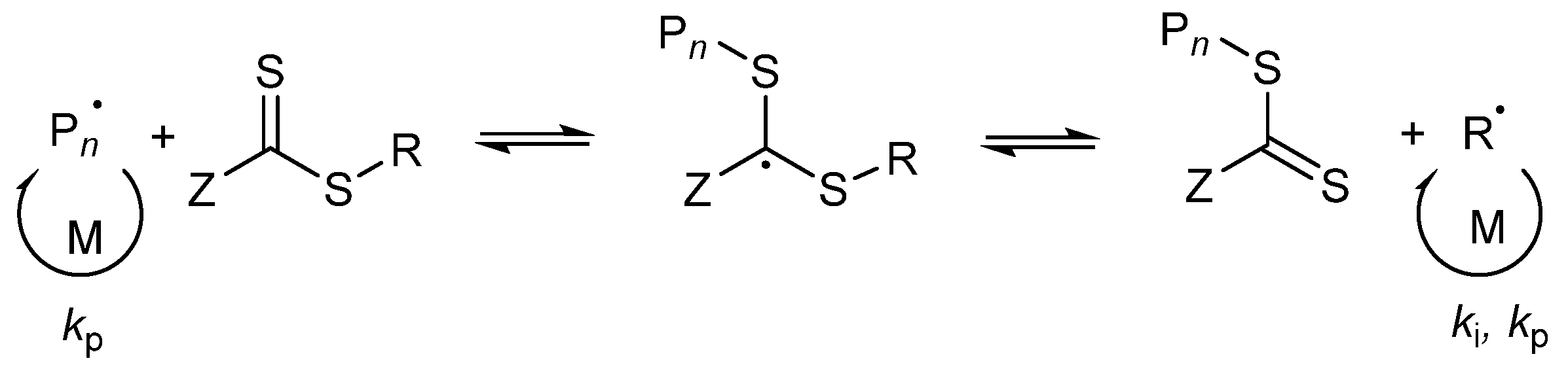
3. Results and Discussion
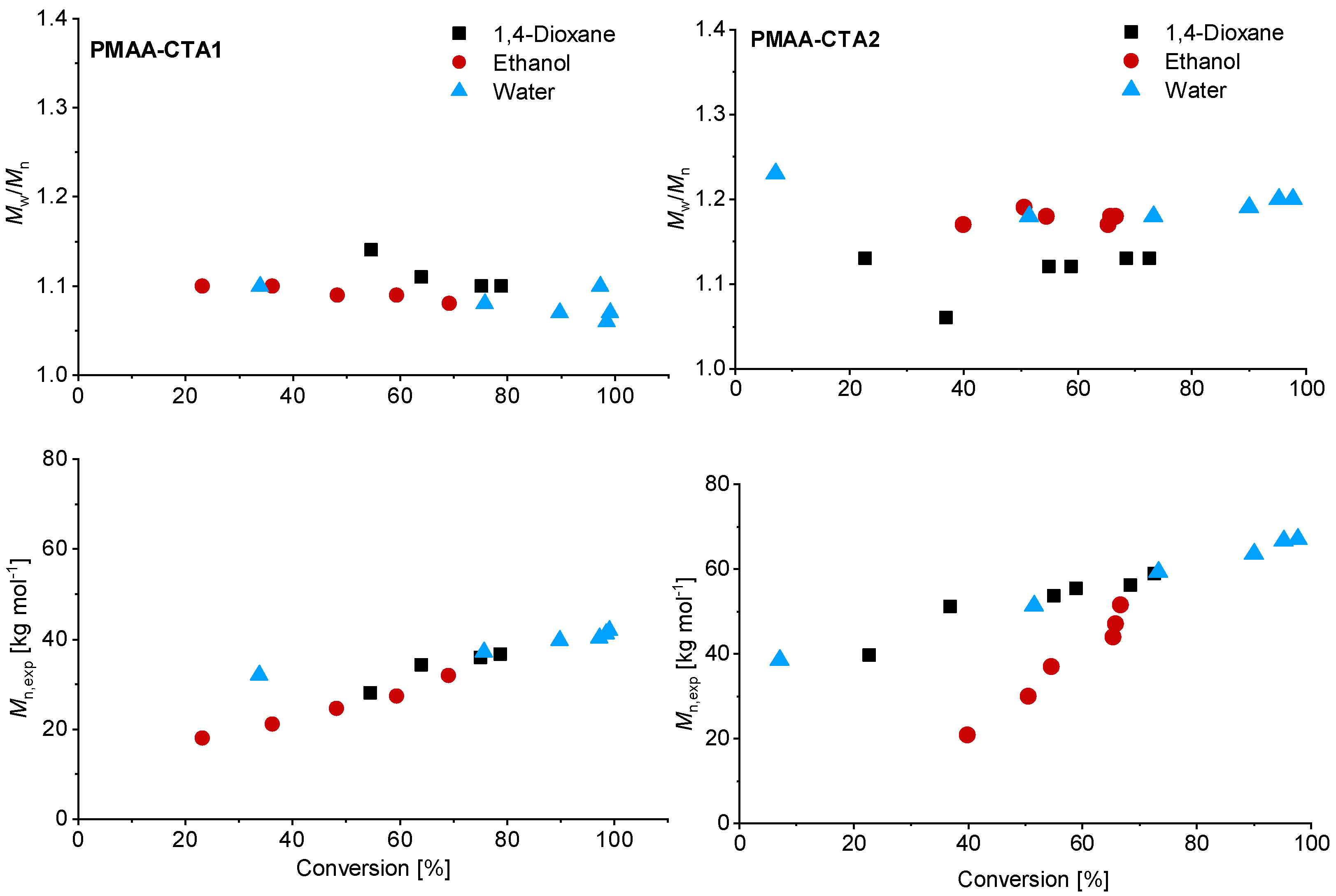
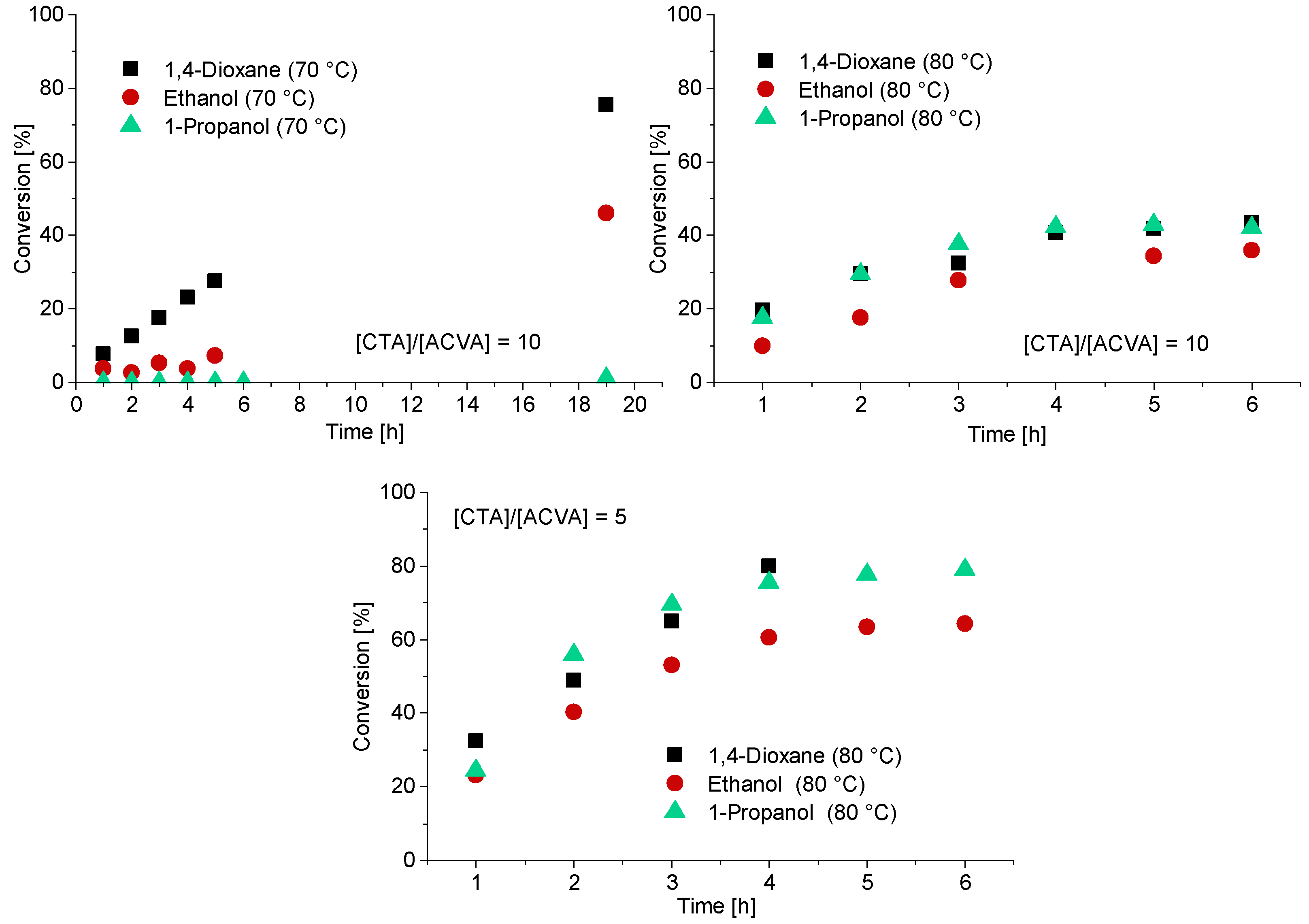
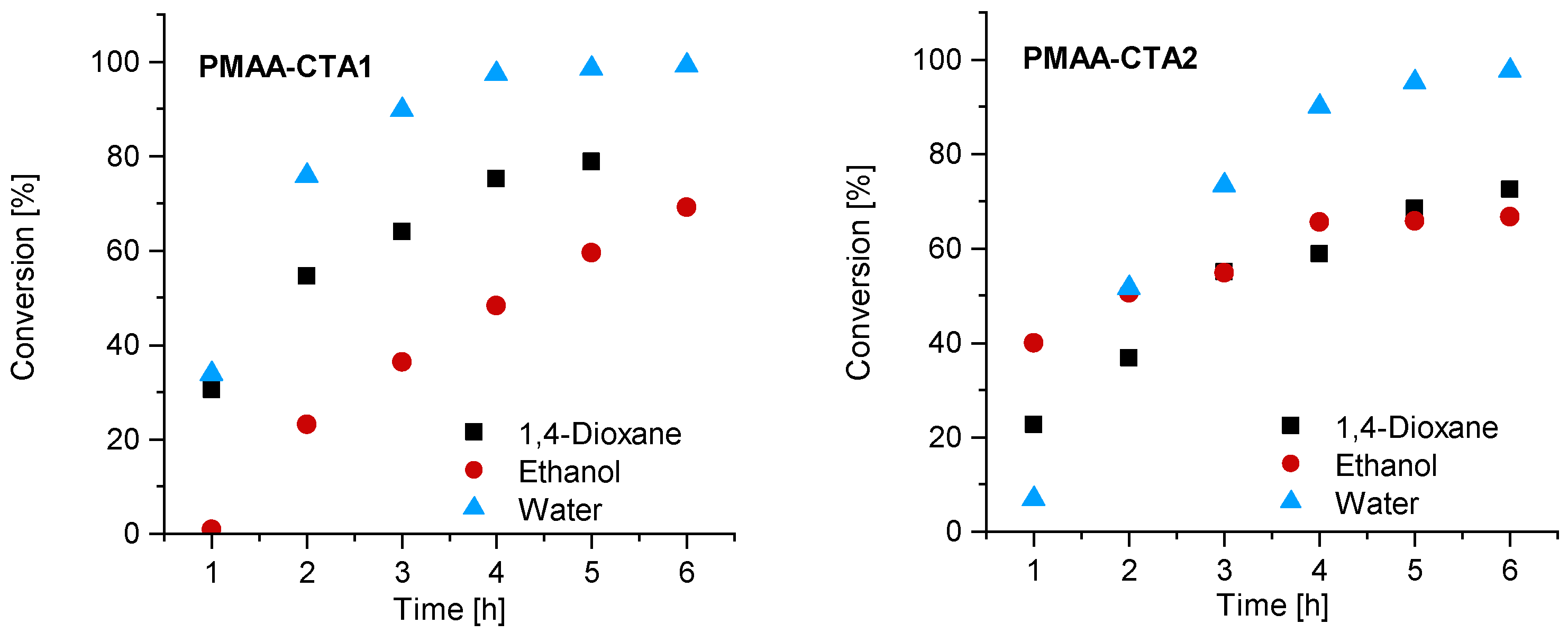
3.1. Kinetics of the Homopolymerization of PMAA
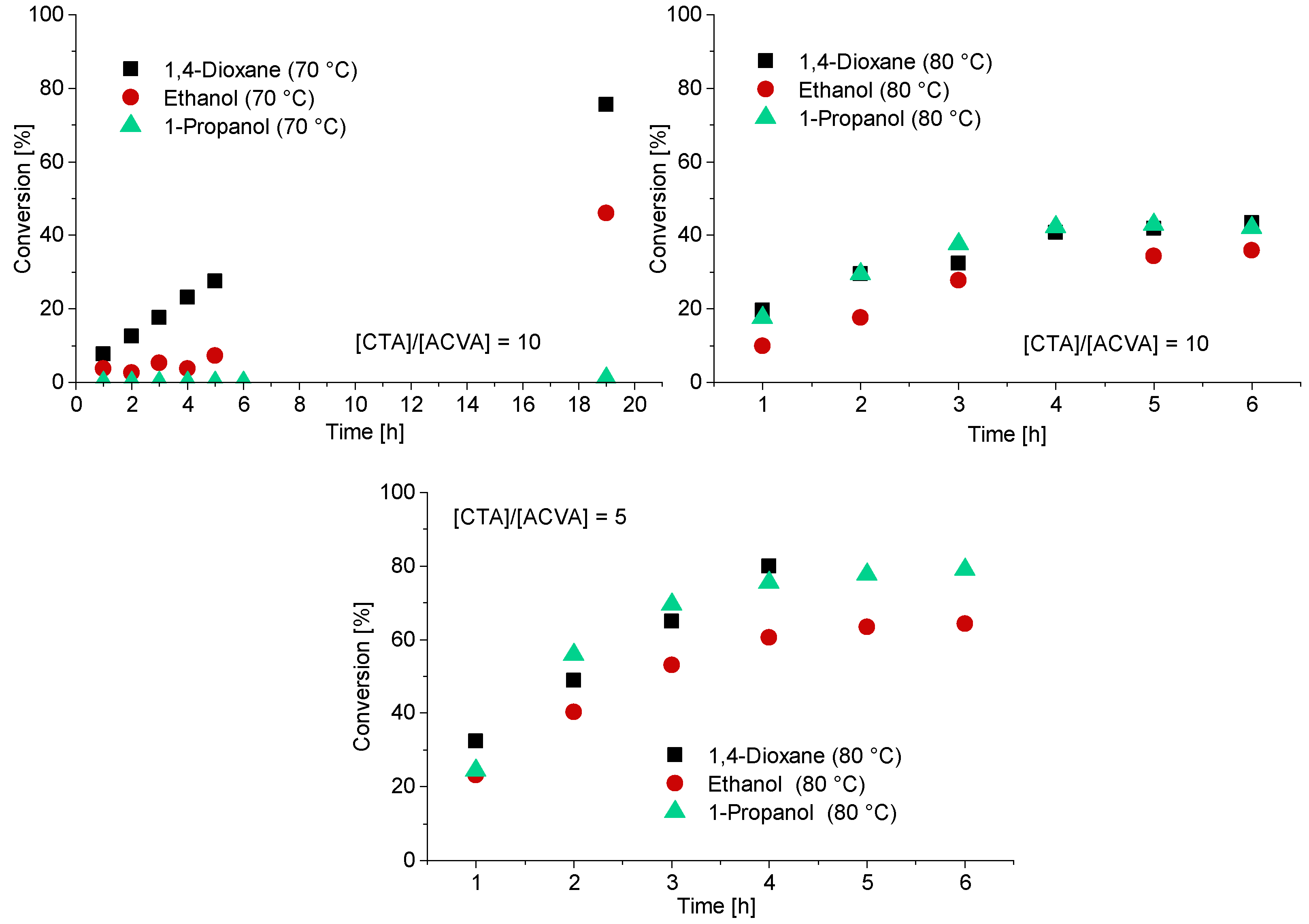
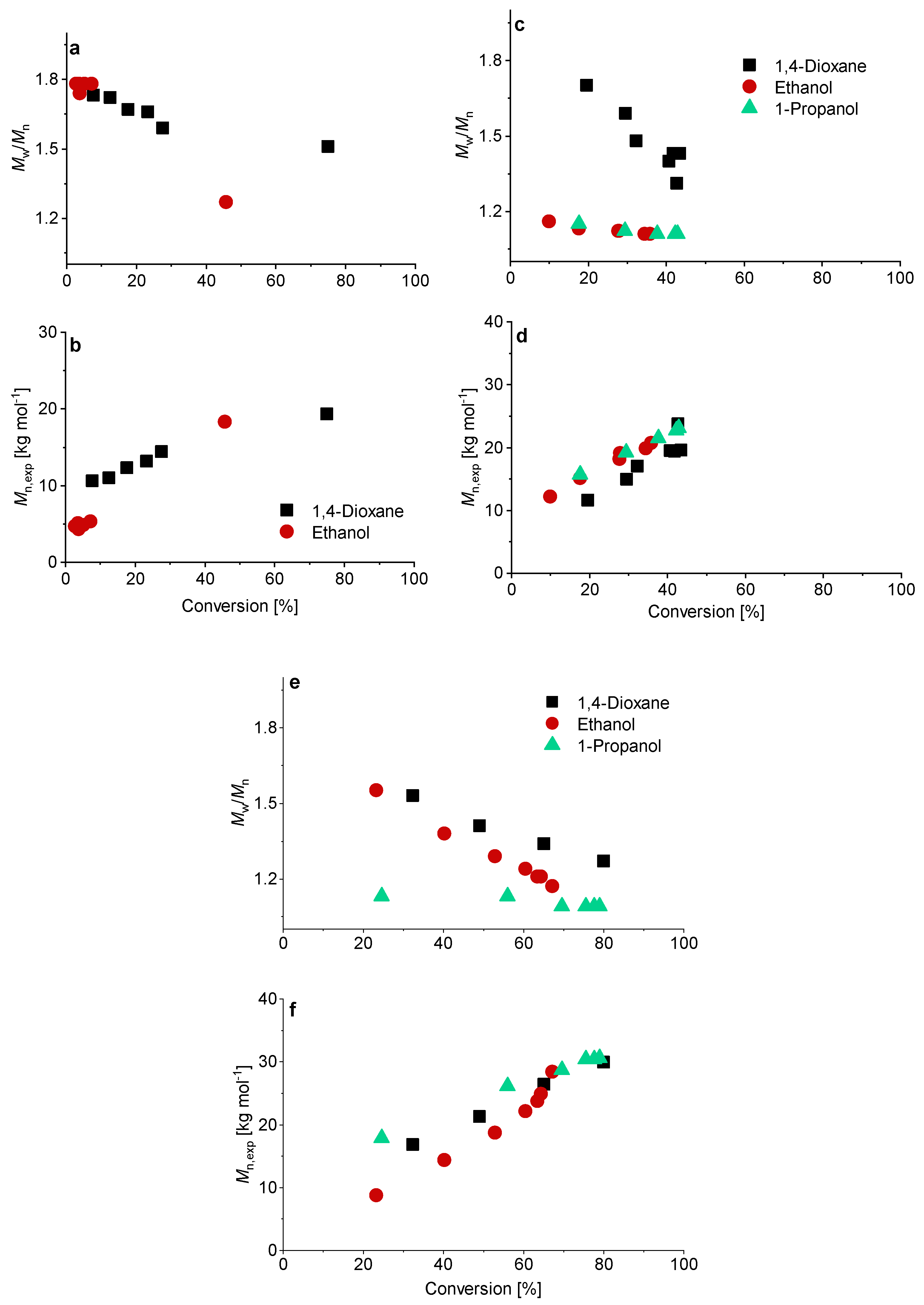
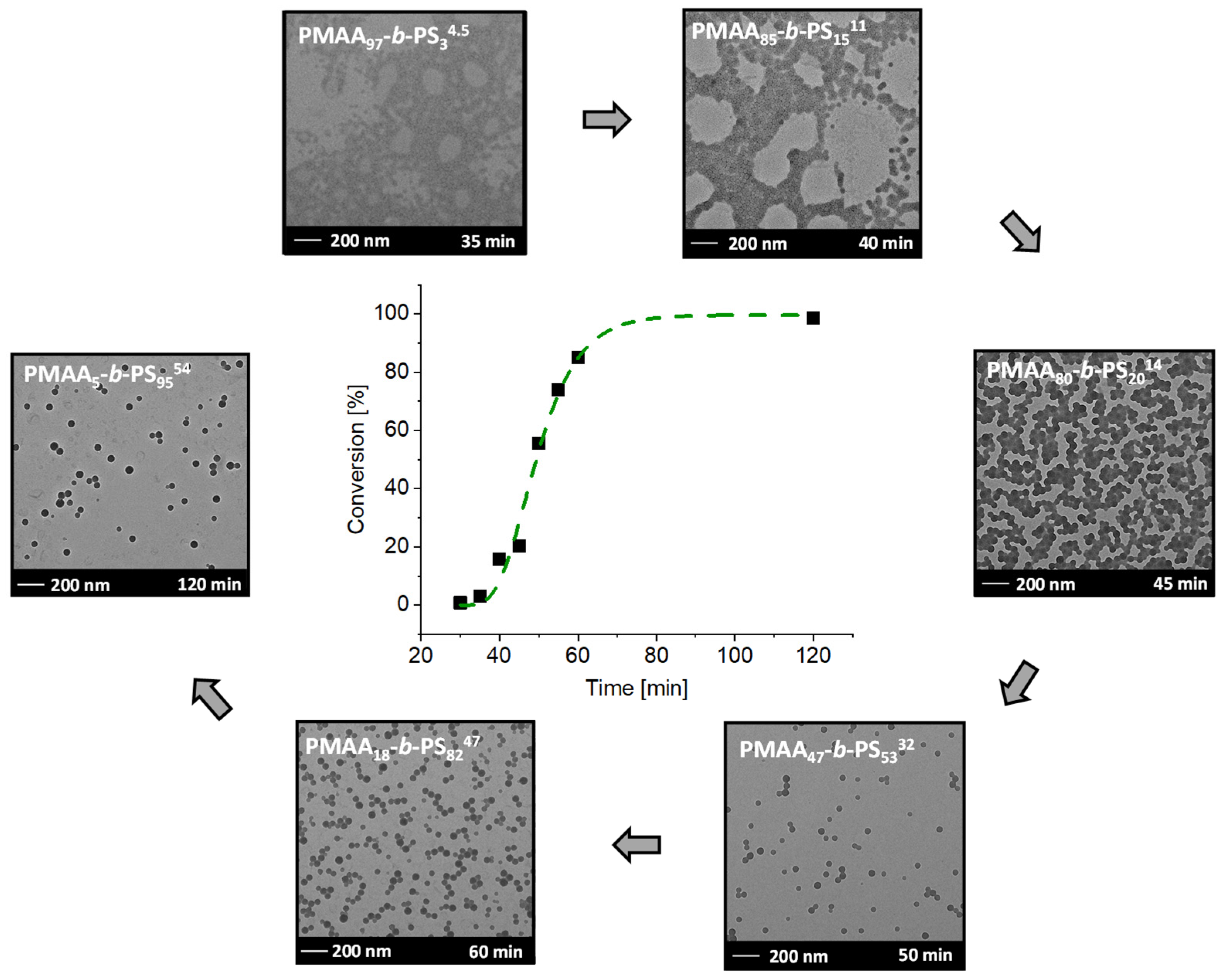
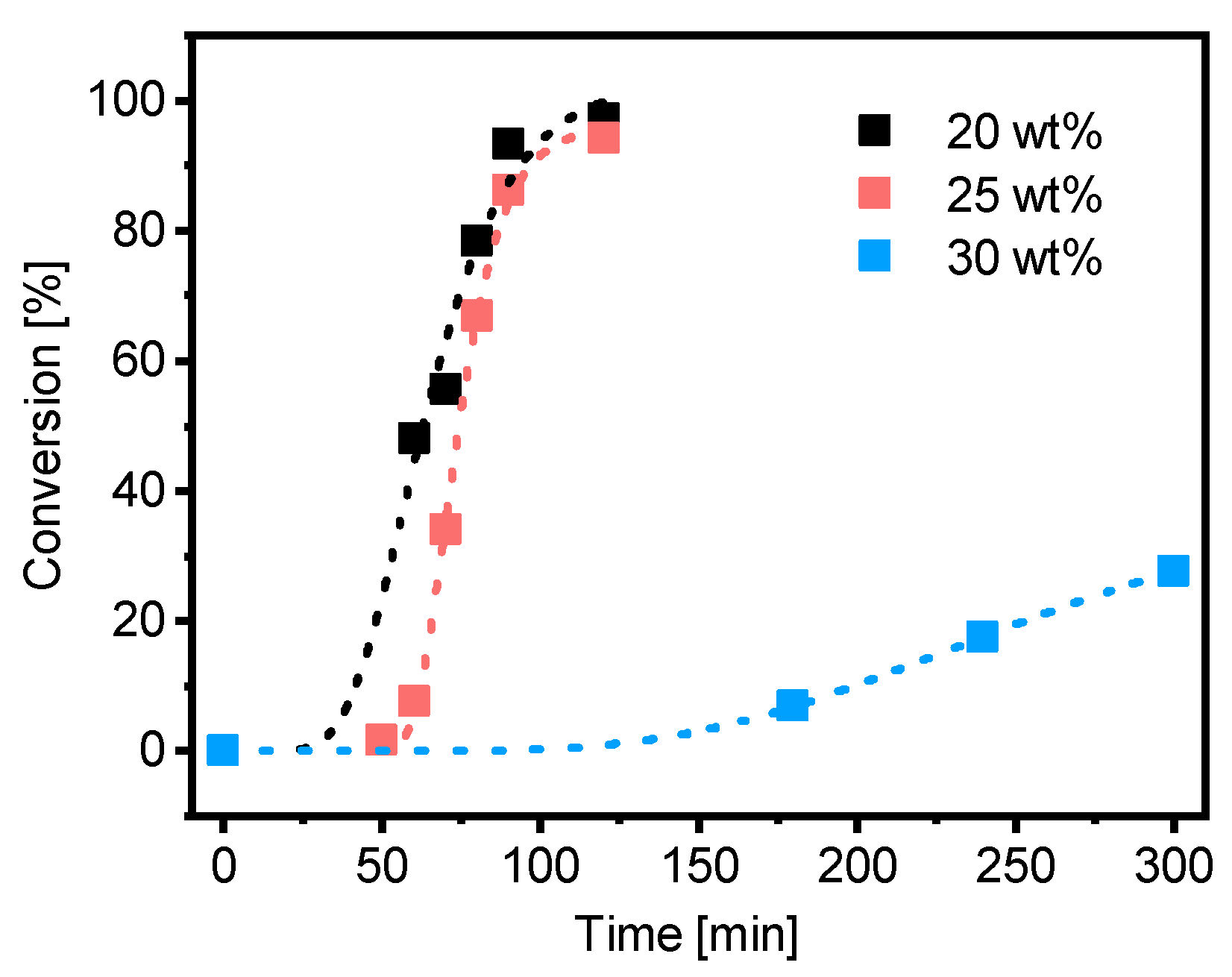
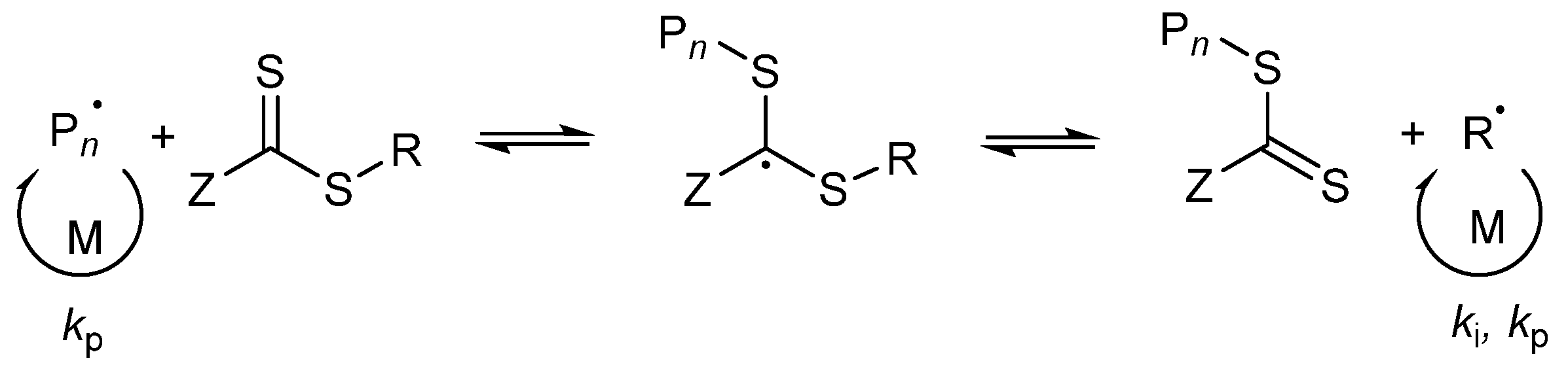
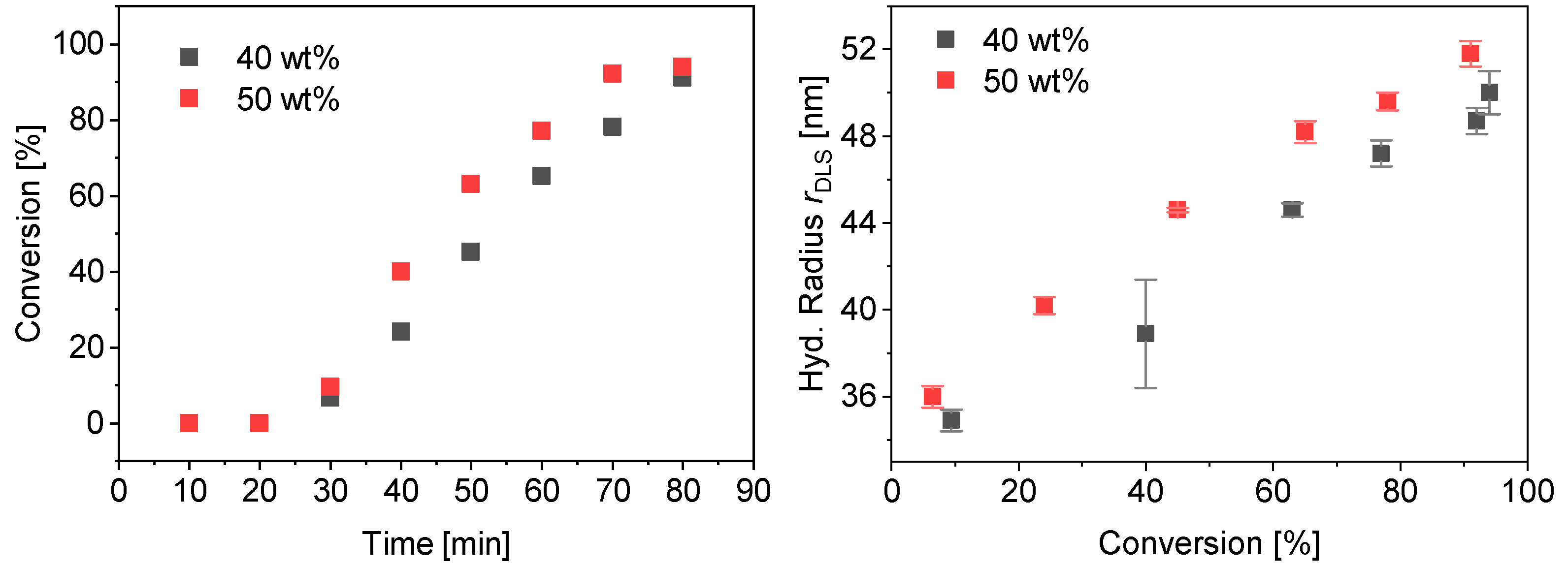
3.2. Kinetics of the Synthesis of PMAA-b-PS with CTA1 via RAFT Emulsion Polymerization
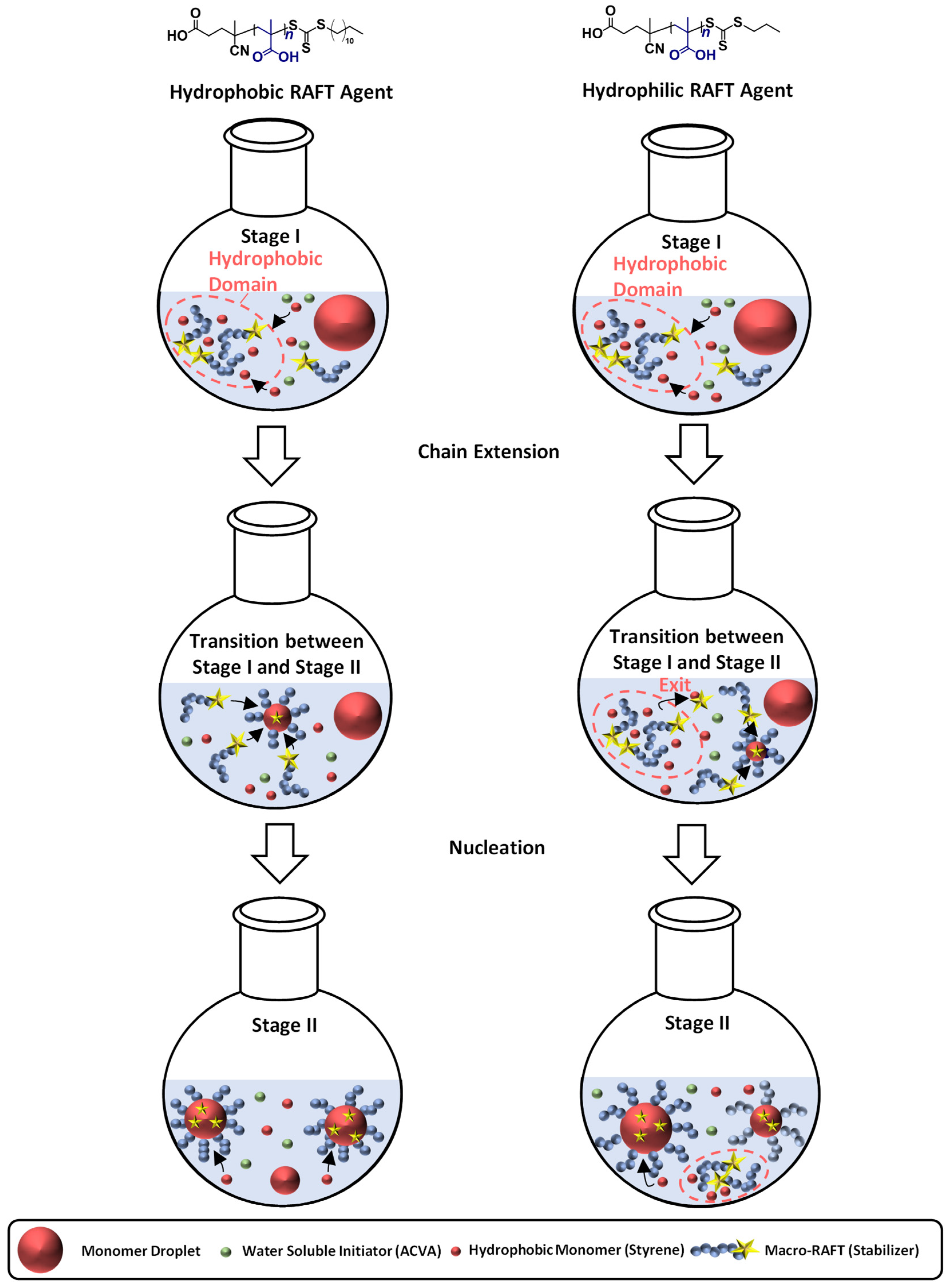
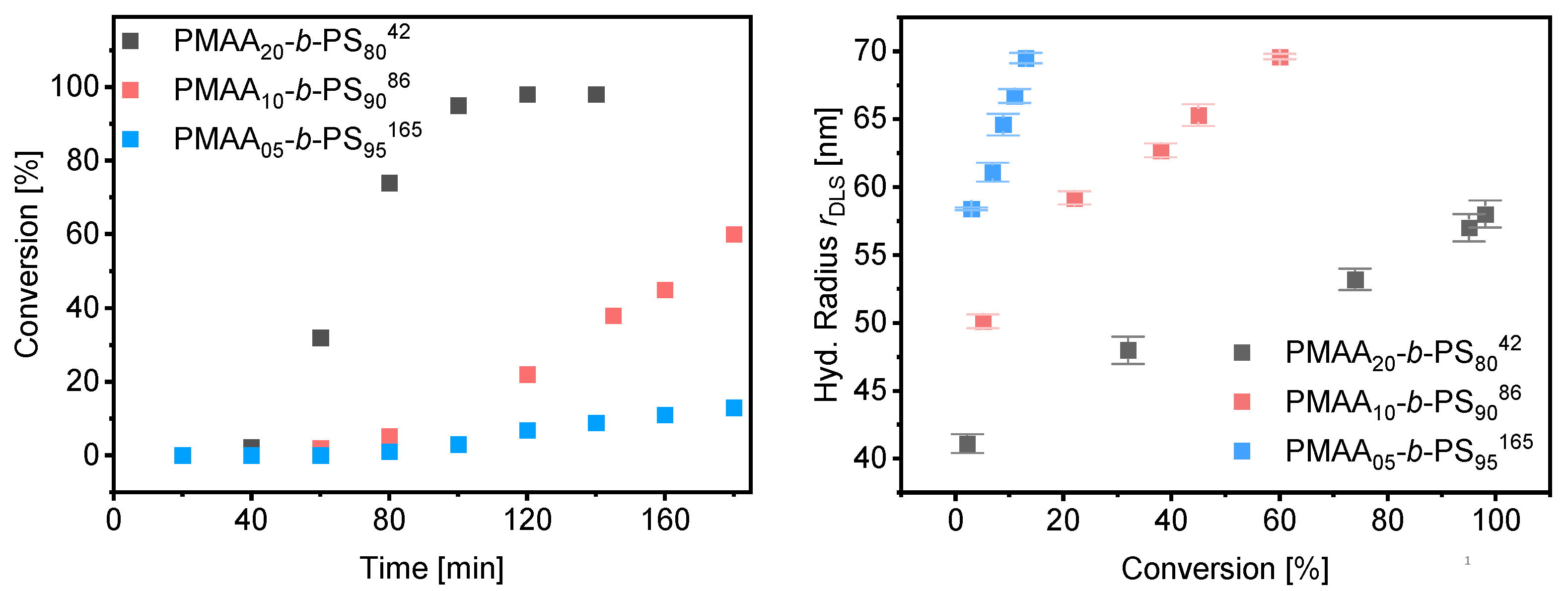
3.3. The Synthesis of PMAA-b-PS with CTA2 via RAFT Emulsion Polymerization
3.4. The Synthesis of PMAA-b-PS with CTA3 via RAFT Emulsion Polymerization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matyjaszewski, K.; Spanswick, J. Controlled/living radical polymerization. Mater. Today 2005, 8, 26–33. [Google Scholar] [CrossRef]
- Matyjaszewski, K. Atom Transfer Radical Polymerization (ATRP): Current status and future perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Acar, M.H.; Matyjaszewski, K. Block copolymers by transformation of living anionic polymerization into controlled/"living’ atom transfer radical polymerization. Macromol. Chem. Phys. 1999, 200, 1094–1100. [Google Scholar] [CrossRef]
- Matyjaszewski, K.; Müller, A.H.E. 50 Years of Living Polymerization. Prog. Polym. Sci. 2006, 31, 1039–1040. [Google Scholar] [CrossRef]
- Perrier, S. 50th Anniversary Perspective: RAFT Polymerization—A User Guide. Macromolecules 2017, 50, 7433–7447. [Google Scholar] [CrossRef]
- Fielding, L.A.; Derry, M.J.; Ladmiral, V.; Rosselgong, J.; Rodrigues, A.M.; Ratcliffe, L.P.D.; Sugihara, S.; Armes, S.P. RAFT dispersion polymerization in non-polar solvents: Facile production of block copolymer spheres, worms and vesicles in n-alkanes. Chem. Sci. 2013, 4, 2081–2087. [Google Scholar] [CrossRef] [Green Version]
- Eggers, S.; Abetz, V. Surfactant-Free RAFT emulsion polymerization of styrene using thermoresponsive macroRAFT agents: Towards smartwell-defined block copolymers with high molecular weights. Polymers 2017, 9, 668. [Google Scholar] [CrossRef] [Green Version]
- Lauterbach, F.; Abetz, V. An eco-friendly pathway to thermosensitive micellar nanoobjects: Via photoRAFT PISA: The full guide to poly(N -acryloylpyrrolidin)- block -polystyrene diblock copolymers. Soft Matter 2020, 16, 2321–2331. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zheng, J.; Man, S.; Sun, X.; An, Z. Synthesis of poly(ionic liquid)-based nano-objects with morphological transitions: Via RAFT polymerization-induced self-assembly in ethanol. Polym. Chem. 2018, 9, 824–827. [Google Scholar] [CrossRef]
- Ni, H.; Liu, J.; Shi, K.; Wu, M.; Yang, Y.; Zhang, L. PMAA-based RAFT dispersion polymerization of MMA in ethanol: Conductivity, block length and self-assembly. RSC Adv. 2016, 6, 58218–58225. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhu, S. Ionic Liquids: Versatile Media for Preparation of Vesicles from Polymerization-Induced Self-Assembly. ACS Macro Lett. 2015, 4, 755–758. [Google Scholar] [CrossRef]
- Perrier, S.; Takolpuckdee, P. Macromolecular design via reversible addition-fragmentation chain transfer (RAFT)/xanthates (MADIX) polymerization. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 5347–5393. [Google Scholar] [CrossRef]
- Zamfir, M.; Patrickios, C.S.; Montagne, F.; Abetz, C.; Abetz, V.; Oss-Ronen, L.; Talmon, Y. Styrene-vinyl pyridine diblock copolymers: Synthesis by RAFT polymerization and self-assembly in solution and in the bulk. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 1636–1644. [Google Scholar] [CrossRef] [Green Version]
- Grazon, C.; Rieger, J.; Sanson, N.; Charleux, B. Study of Poly(N,N-diethylacrylamide) Nanogel Formation by Aqueous Dispersion Polymerization of N,N-Diethylacrylamide in the Presence of Poly(ethylene oxide)-b-Poly(N,N-dimethylacrylamide) Amphiphilic Macromolecular RAFT Agents. Soft Matter 2011, 7, 3482–3490. [Google Scholar] [CrossRef]
- Wu, J.; Tian, C.; Zhang, L.; Cheng, Z.; Zhu, X. Synthesis of soap-free emulsion with high solid content by differential dripping RAFT polymerization-induced self-assembly. RSC Adv. 2017, 7, 6559–6564. [Google Scholar] [CrossRef] [Green Version]
- Siirilä, J.; Häkkinen, S.; Tenhu, H. The emulsion polymerization induced self-assembly of a thermoresponsive polymer poly(: N -vinylcaprolactam). Polym. Chem. 2019, 10, 766–775. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, J.; Wang, C.Y.; Ren, Q. RAFT surfactant-free cationic emulsion polymerization of styrene: Effect of hydrophobicity of block macro-RAFT agent. J. Macromol. Sci. Part A Pure Appl. Chem. 2020, 58, 232–242. [Google Scholar] [CrossRef]
- Sprong, E.; Leswin, J.S.K.; Lamb, D.J.; Ferguson, C.J.; Hawkett, B.S.; Pham, B.T.T.; Nguyen, D.; Such, C.H.; Serelis, A.K.; Gilbert, R.G. Molecular watchmaking: Ab initio emulsion polymerization by RAFT-controlled self-assembly. Macromol. Symp. 2006, 231, 84–93. [Google Scholar] [CrossRef]
- Ferguson, C.J.; Hughes, R.J.; Pham, B.T.T.; Hawkett, B.S.; Gilbert, R.G.; Serelis, A.K.; Such, C.H. Effective ab initio emulsion polymerization under RAFT control. Macromolecules 2002, 35, 9243–9245. [Google Scholar] [CrossRef]
- Chaduc, I.; Crepet, A.; Boyron, O.; Charleux, B.; D’Agosto, F.; Lansalot, M. Effect of the ph on the raft polymerization of acrylic acid in water. Application to the synthesis of poly(acrylic acid)-stabilized polystyrene particles by RAFT emulsion polymerization. Macromolecules 2013, 46, 6013–6023. [Google Scholar] [CrossRef]
- Chaduc, I.; Girod, M.; Antoine, R.; Charleux, B.; D’Agosto, F.; Lansalot, M. Batch emulsion polymerization mediated by poly(methacrylic acid) macroRAFT agents: One-pot synthesis of self-stabilized particles. Macromolecules 2012, 45, 5881–5893. [Google Scholar] [CrossRef]
- Zhang, W.; D’Agosto, F.; Boyron, O.; Rieger, J.; Charleux, B. One-pot synthesis of poly(methacrylic acid-co-poly(ethylene oxide) methyl ether methacrylate)-b-polystyrene amphiphilic block copolymers and their self-assemblies in water via RAFT-mediated radical emulsion polymerization. A kinetic study. Macromolecules 2011, 44, 7584–7593. [Google Scholar] [CrossRef]
- Chaduc, I.; Zhang, W.; Rieger, J.; Lansalot, M.; D’Agosto, F.; Charleux, B. Amphiphilic block copolymers from a direct and one-pot RAFT synthesis in water. Macromol. Rapid Commun. 2011, 32, 1270–1276. [Google Scholar] [CrossRef] [PubMed]
- Rieger, J.; Zhang, W.; Stoffelbach, F.; Charleux, B. Surfactant-free RAFT emulsion polymerization using poly(N,N-dimethylacrylamide) trithiocarbonate macromolecular chain transfer agents. Macromolecules 2010, 43, 6302–6310. [Google Scholar] [CrossRef]
- Zhang, X.; Rieger, J.; Charleux, B. Effect of the solvent composition on the morphology of nano-objects synthesized via RAFT polymerization of benzyl methacrylate in dispersed systems. Polym. Chem. 2012, 3, 1502–1509. [Google Scholar] [CrossRef]
- Manguian, M.; Save, M.; Charleux, B. Batch emulsion polymerization of styrene stabilized by a hydrophilic macro-RAFT agenta. Macromol. Rapid Commun. 2006, 27, 399–404. [Google Scholar] [CrossRef]
- Charleux, B.; Delaittre, G.; Rieger, J.; D’Agosto, F. Polymerization-induced self-assembly: From soluble macromolecules to block copolymer nano-objects in one step. Macromolecules 2012, 45, 6753–6765. [Google Scholar] [CrossRef]
- Wi, Y.; Lee, K.; Lee, B.H.; Choe, S. Soap-free emulsion polymerization of styrene using poly(methacrylic acid) macro-RAFT agent. Polymer 2008, 49, 5626–5635. [Google Scholar] [CrossRef]
- Oral, I.; Abetz, V. A Highly Selective Polymer Material using Benzo-9-Crown-3 for the Extraction of Lithium in Presence of Other Interfering Alkali Metal Ions. Macromol. Rapid Commun. 2021, 42, 2000746. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, F.; Muhamad, I.I. Physical and Chemical Characterisation of Acrylamide-Based Hydrogels, Aam, Aam/NaCMC and Aam/NaCMC/MgO. J. Inorg. Organomet. Polym. Mater. 2017, 27, 1439–1449. [Google Scholar] [CrossRef]
- Lacík, I.; Stach, M.; Kasák, P.; Semak, V.; Uhelská, L.; Chovancová, A.; Reinhold, G.; Kilz, P.; Delaittre, G.; Charleux, B.; et al. SEC Analysis of Poly (Acrylic Acid) and Poly (Methacrylic Acid). Macromol. Chem. Phys. 2015, 216, 23–37. [Google Scholar] [CrossRef]
- Xu, E.; Smith, A.E.; Kirkland, S.E.; McCormick, C.L. Aqueous RAFT synthesis of pH-responsive triblock copolymer mPEO-PAPMA-PDPAEMA and formation of shell cross-linked micelles. Macromolecules 2008, 41, 8429–8435. [Google Scholar] [CrossRef]
- Lowe, A.B.; McCormick, C.L. Reversible addition-fragmentation chain transfer (RAFT) radical polymerization and the synthesis of water-soluble (co)polymers under homogeneous conditions in organic and aqueous media. Prog. Polym. Sci. 2007, 32, 283–351. [Google Scholar] [CrossRef]
- Chaduc, I.; Lansalot, M.; D’Agosto, F.; Charleux, B. RAFT polymerization of methacrylic acid in water. Macromolecules 2012, 45, 1241–1247. [Google Scholar] [CrossRef]
- Fuchs, A.V.; Thurecht, K.J. Stability of trithiocarbonate RAFT agents containing both a cyano and a carboxylic acid functional group. ACS Macro Lett. 2017, 6, 287–291. [Google Scholar] [CrossRef]
- Vshivkov, S.A.; Soliman, T.S.; Kluzhin, E.S.; Kapitanov, A.A. Structure of poly(acrylic acid), poly(methacrylic acid) and gelatin solutions. J. Mol. Liq. 2019, 294, 111551. [Google Scholar] [CrossRef]
- Kuchta, F.D.; Van Herk, A.M.; German, A.L. Propagation kinetics of acrylic and methacrylic acid in water and organic solvents studied by pulsed-laser polymerization. Macromolecules 2000, 33, 3641–3649. [Google Scholar] [CrossRef]
- Wan, W.-M.; Pan, C.-Y. One-pot synthesis of polymeric nanomaterials via RAFT dispersion polymerization induced self-assembly and re-organization. Polym. Chem. 2010, 1, 1475. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, S.; Gao, X.; Luo, Y.; Li, B. RAFT Ab Initio Emulsion Polymerization of Styrene Using Poly(acrylic acid)-b-polystyrene Trithiocarbonate of Various Structures as Mediator and Surfactant. Macromol. React. Eng. 2014, 8, 696–705. [Google Scholar] [CrossRef]
- Thickett, S.C.; Gilbert, R.G. Emulsion polymerization: State of the art in kinetics and mechanisms. Polymer 2007, 48, 6965–6991. [Google Scholar] [CrossRef] [Green Version]
- Canning, S.L.; Smith, G.N.; Armes, S.P. A Critical Appraisal of RAFT-Mediated Polymerization-Induced Self-Assembly. Macromolecules 2016, 49, 1985–2001. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, V.J.; Alswieleh, A.M.; Thompson, K.L.; Williams, M.; Leggett, G.J.; Armes, S.P.; Musa, O.M. Poly(glycerol monomethacrylate)-poly(benzyl methacrylate) diblock copolymer nanoparticles via RAFT emulsion polymerization: Synthesis, characterization, and interfacial activity. Macromolecules 2014, 47, 5613–5623. [Google Scholar] [CrossRef]
- Truong, N.P.; Dussert, M.V.; Whittaker, M.R.; Quinn, J.F.; Davis, T.P. Rapid synthesis of ultrahigh molecular weight and low polydispersity polystyrene diblock copolymers by RAFT-mediated emulsion polymerization. Polym. Chem. 2015, 6, 3865–3874. [Google Scholar] [CrossRef]
- Zhang, W.; D’Agosto, F.; Boyron, O.; Rieger, J.; Charleux, B. Toward a better understanding of the parameters that lead to the formation of nonspherical polystyrene particles via RAFT-mediated one-pot aqueous emulsion polymerization. Macromolecules 2012, 45, 4075–4084. [Google Scholar] [CrossRef]
- Zhang, X.; Boissé, S.; Zhang, W.; Beaunier, P.; D’Agosto, F.; Rieger, J.; Charleux, B. Well-defined amphiphilic block copolymers and nano-objects formed in situ via RAFT-mediated aqueous emulsion polymerization. Macromolecules 2011, 44, 4149–4158. [Google Scholar] [CrossRef]
- Blanazs, A.; Armes, S.P.; Ryan, A.J. Self-assembled block copolymer aggregates: From micelles to vesicles and their biological applications. Macromol. Rapid Commun. 2009, 30, 267–277. [Google Scholar] [CrossRef]
- Thompson, S.W.; Guimarães, T.R.; Zetterlund, P.B. RAFT Emulsion Polymerization: MacroRAFT Agent Self-Assembly Investigated Using a Solvachromatic Dye. Biomacromolecules 2020, 21, 4577–4590. [Google Scholar] [CrossRef]
- Asua, J.M. Emulsion Polymerization: From Fundamental Mechanisms to Process Developments. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 1025–1041. [Google Scholar] [CrossRef]
- Gilbert, R.G.; Prescott, S.W.; Smulders, W.; Monteiro, M.J.; Ballard, M.J.; Rizzardo, E. RAFT in emulsion polymerization: What makes it different. Am. Chem. Soc. Polym. Prepr. Div. Polym. Chem. 2002, 43, 130–131. [Google Scholar] [CrossRef]
- Semsarilar, M.; Abetz, V. Polymerizations by RAFT: Developments of the Technique and Its Application in the Synthesis of Tailored (Co)polymers. Macromol. Chem. Phys. 2020, 222, 2000311. [Google Scholar] [CrossRef]
- Butté, A.; Storti, G.; Morbidelli, M. Miniemulsion living free radical polymerization by RAFT. Macromolecules 2001, 34, 5885–5896. [Google Scholar] [CrossRef]
- Sanders, C.A.; George, S.R.; Deeter, G.A.; Campbell, J.D.; Reck, B.; Cunningham, M.F. Amphiphilic Block-Random Copolymers: Self-Folding Behavior and Stabilizers in Emulsion Polymerization. Macromolecules 2019, 52, 4510–4519. [Google Scholar] [CrossRef]
- Guimarães, T.R.; Bong, Y.L.; Thompson, S.W.; Moad, G.; Perrier, S.; Zetterlund, P.B. Polymerization-induced self-assembly via RAFT in emulsion: Effect of Z-group on the nucleation step. Polym. Chem. 2020, 12, 122–133. [Google Scholar] [CrossRef]
- Luo, Y.; Wang, X.; Li, B.G.; Zhu, S. Toward well-controlled ab initio RAFT emulsion polymerization of styrene mediated by 2-(((Dodecylsulfanyl)carbonothioyl)sulfanyl)propanoic acid. Macromolecules 2011, 44, 221–229. [Google Scholar] [CrossRef]
- Monteiro, M.J.; Hodgson, M.; De Brouwer, H. Influence of RAFT on the rates and molecular weight distributions of styrene in seeded emulsion polymerizations. J. Polym. Sci. Part A Polym. Chem. 2000, 38, 3864–3874. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, S.; Gao, X.; Luo, Y.; Li, B. Ab initio RAFT emulsion copolymerization of styrene and acrylonitrile. Ind. Eng. Chem. Res. 2014, 53, 7688–7695. [Google Scholar] [CrossRef]
- Lansalot, M.; Davis, T.P.; Heuts, J.P.A. RAFT miniemulsion polymerization: Influence of the structure of the RAFT agent. Macromolecules 2002, 35, 7582–7591. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (w/w) Solids [%] | Conversion [%] | rTEM [nm] | rDLS [nm] | PDI | Mn,theo [kDa] | Mn,exp [kDa] | Mw/Mn |
|---|---|---|---|---|---|---|---|
| 35 | 99 | 61 ± 4 | 85.3 ± 0.5 | 0.09 ± 0.03 | 46 | 166 | 1.57 |
| 40 | 99 | 70 ± 5 | 96 ± 2 | 0.3 ± 0.2 | 46 | 158 | 1.61 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oral, I.; Grossmann, L.; Fedorenko, E.; Struck, J.; Abetz, V. Synthesis of Poly(methacrylic acid)-block-Polystyrene Diblock Copolymers at High Solid Contents via RAFT Emulsion Polymerization. Polymers 2021, 13, 3675. https://doi.org/10.3390/polym13213675
Oral I, Grossmann L, Fedorenko E, Struck J, Abetz V. Synthesis of Poly(methacrylic acid)-block-Polystyrene Diblock Copolymers at High Solid Contents via RAFT Emulsion Polymerization. Polymers. 2021; 13(21):3675. https://doi.org/10.3390/polym13213675
Chicago/Turabian StyleOral, Iklima, Larissa Grossmann, Elena Fedorenko, Jana Struck, and Volker Abetz. 2021. "Synthesis of Poly(methacrylic acid)-block-Polystyrene Diblock Copolymers at High Solid Contents via RAFT Emulsion Polymerization" Polymers 13, no. 21: 3675. https://doi.org/10.3390/polym13213675
APA StyleOral, I., Grossmann, L., Fedorenko, E., Struck, J., & Abetz, V. (2021). Synthesis of Poly(methacrylic acid)-block-Polystyrene Diblock Copolymers at High Solid Contents via RAFT Emulsion Polymerization. Polymers, 13(21), 3675. https://doi.org/10.3390/polym13213675