In Vitro Investigation of Thiol-Functionalized Cellulose Nanofibrils as a Chronic Wound Environment Modulator

 , ,
, ,  and
and
Abstract
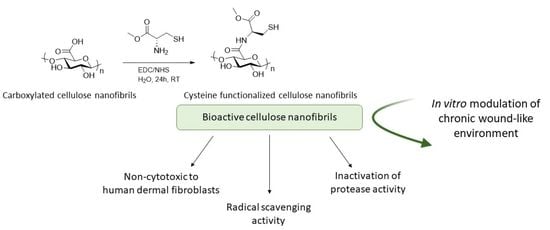
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
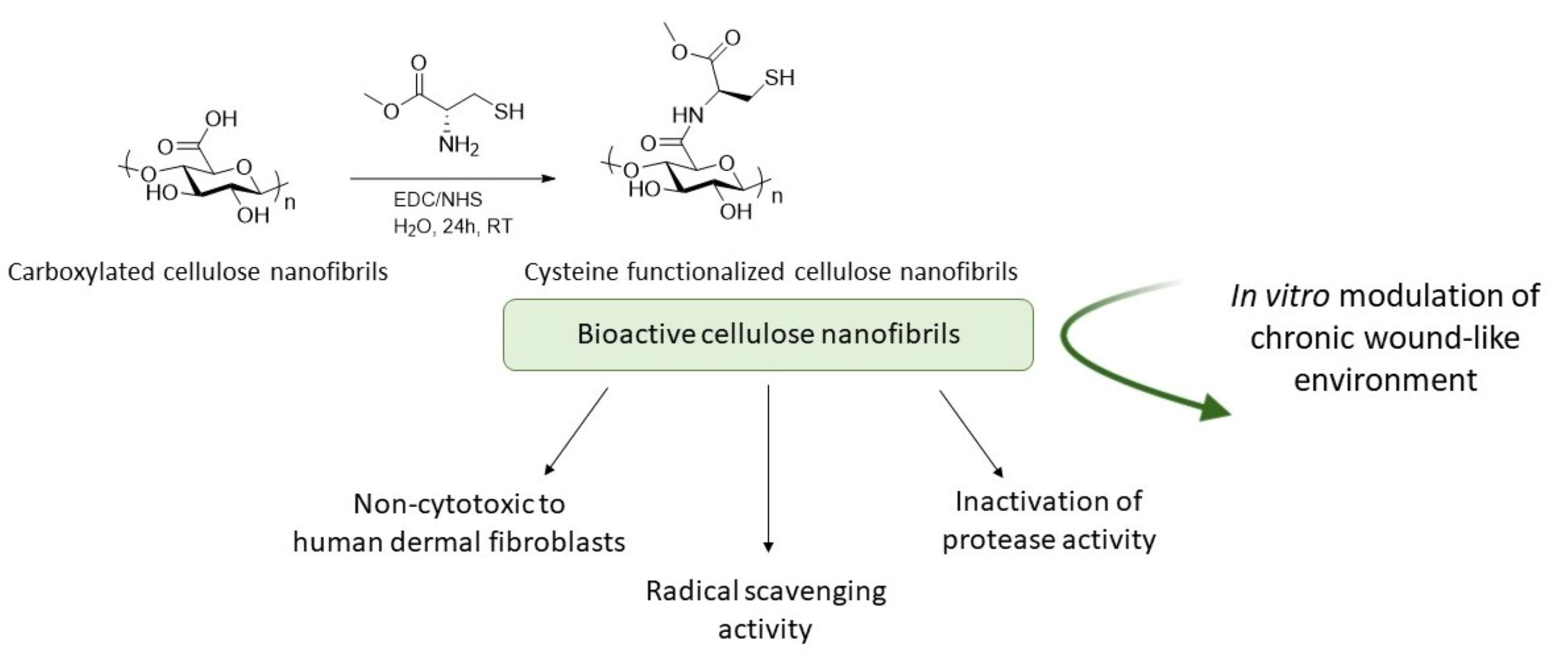
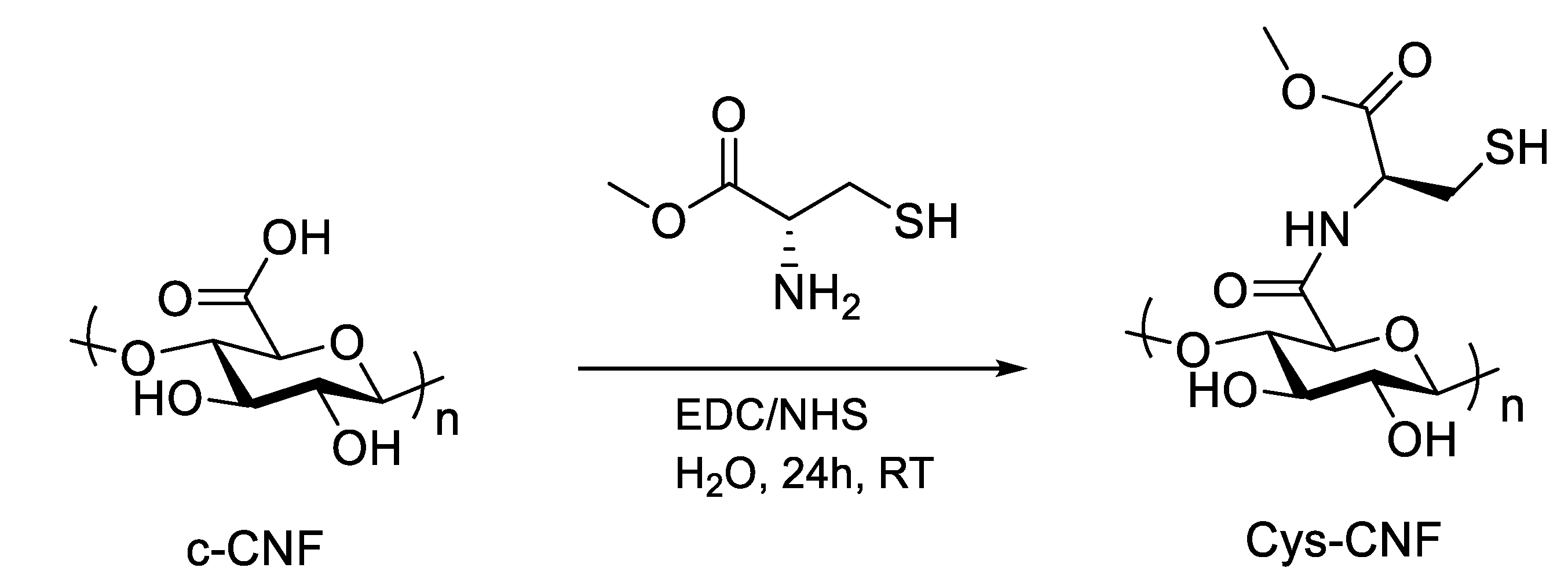
2.2. Covalent Incorporation of Methyl Cysteine to Cellulose Nanofibrils
2.3. Material Characterization
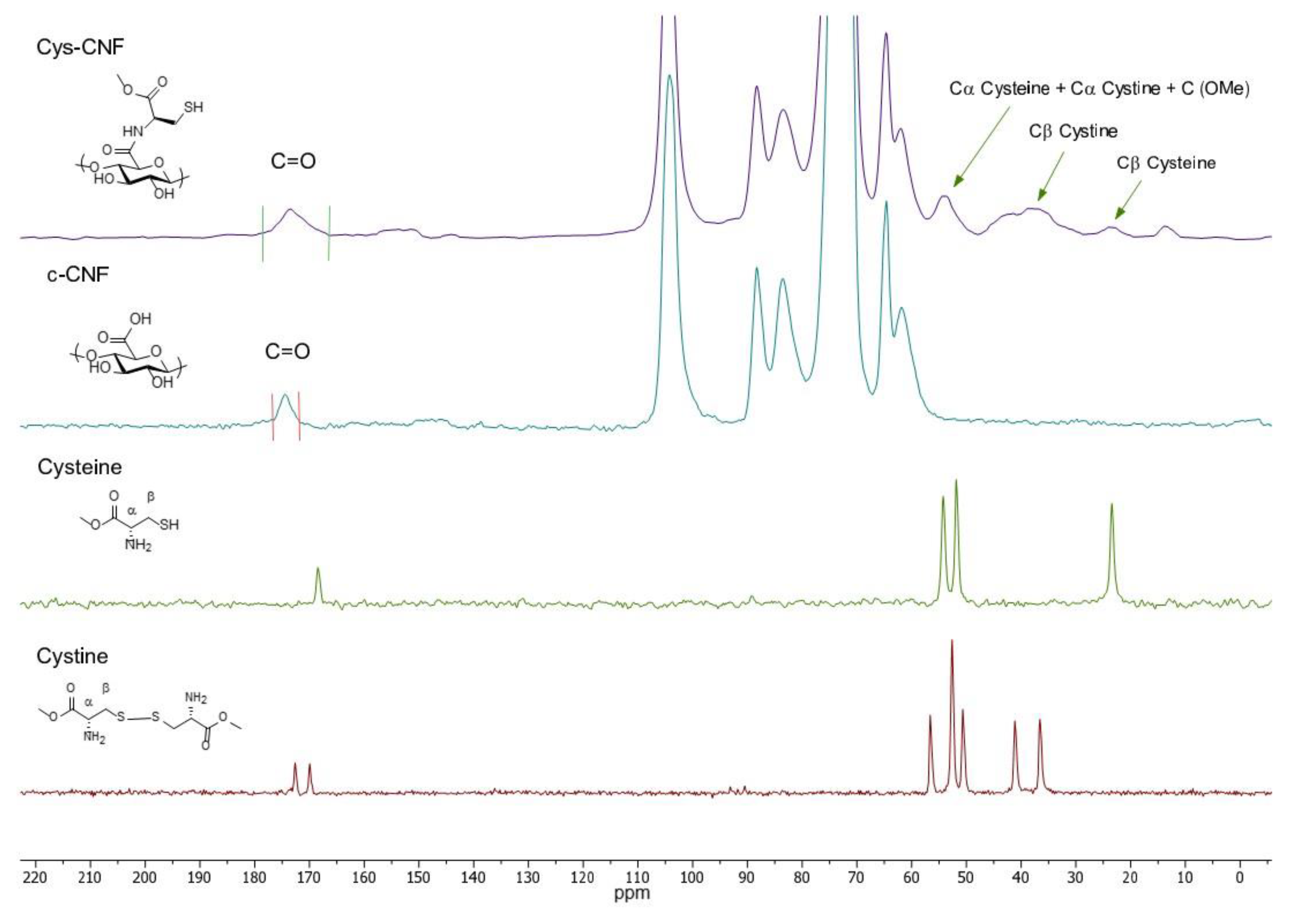
2.3.1. Characterization of the Molecular Structure
2.3.2. Degree of Substitution
2.3.3. Thiol Group Content
2.4. Radical Scavenging Capacity
2.5. Evaluation of the Material Interactions with the Metalloprotease Collagenase
2.5.1. Evaluation of Protease Inhibition
2.5.2. Evaluation of Protease Entrapment
2.5.3. Zn2+ Binding Assay
2.6. Material Stability over Time
2.7. Cell Studies
2.7.1. Cell Culture
2.7.2. Cytotoxicity Assessment
2.8. Statistical Analysis
3. Results and Discussion
3.1. Material Characterization
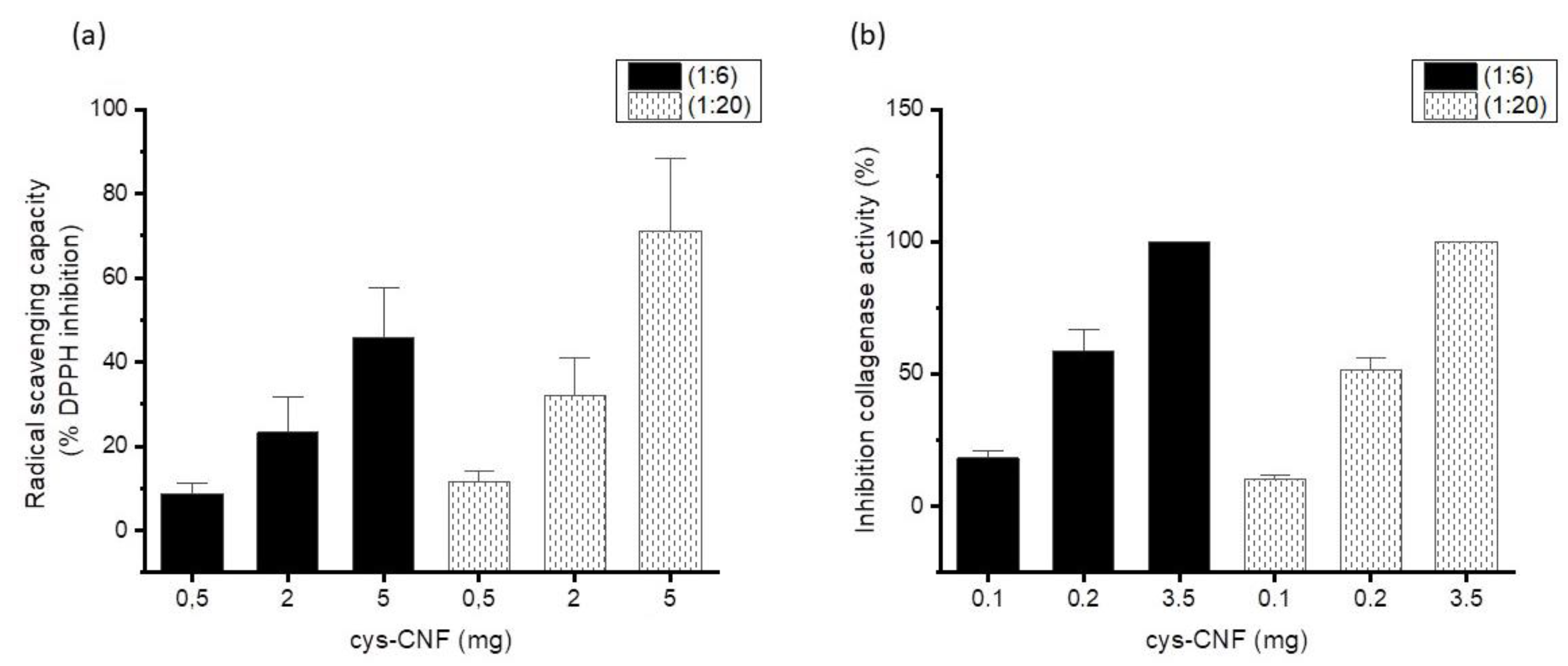
3.2. Radical Scavenging Activity
3.3. Interactions with the Metalloprotease Collagenase
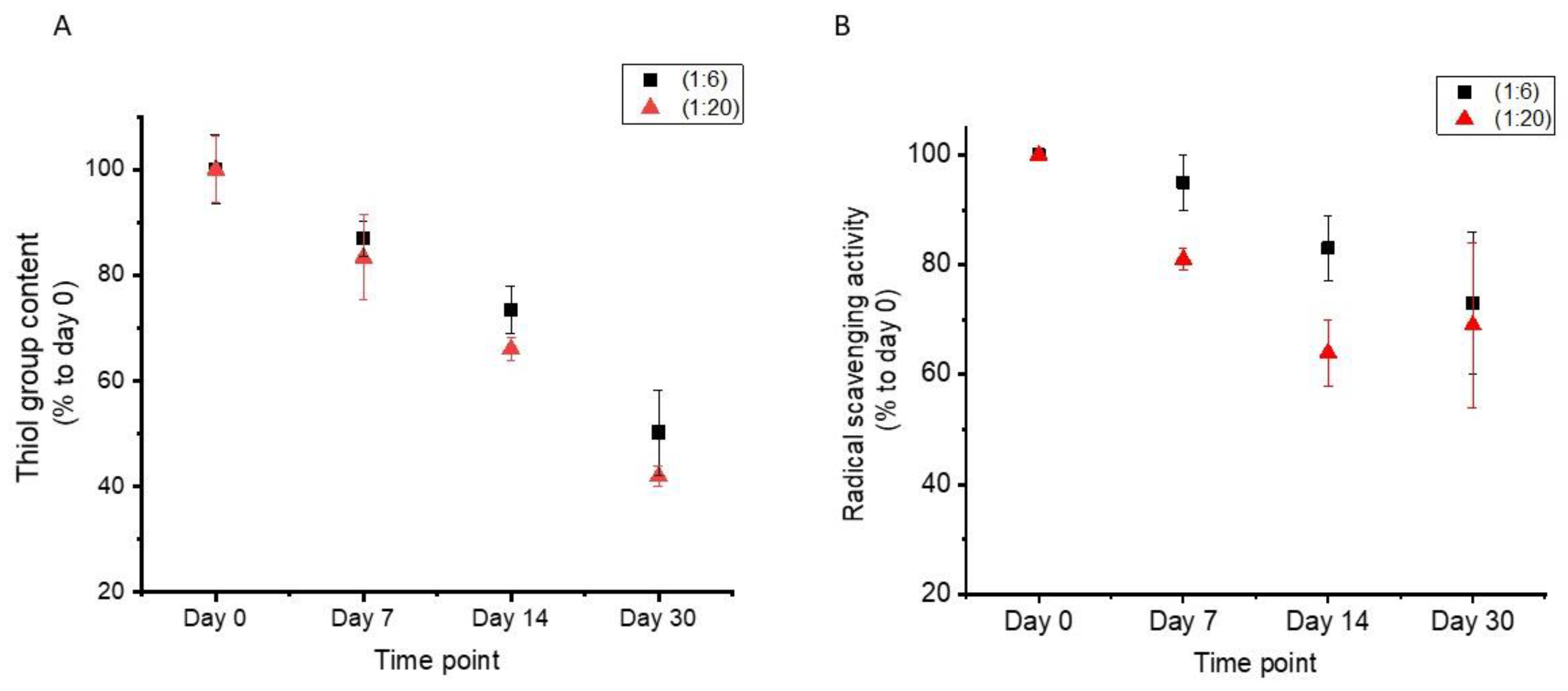
3.4. Material Stability over Time
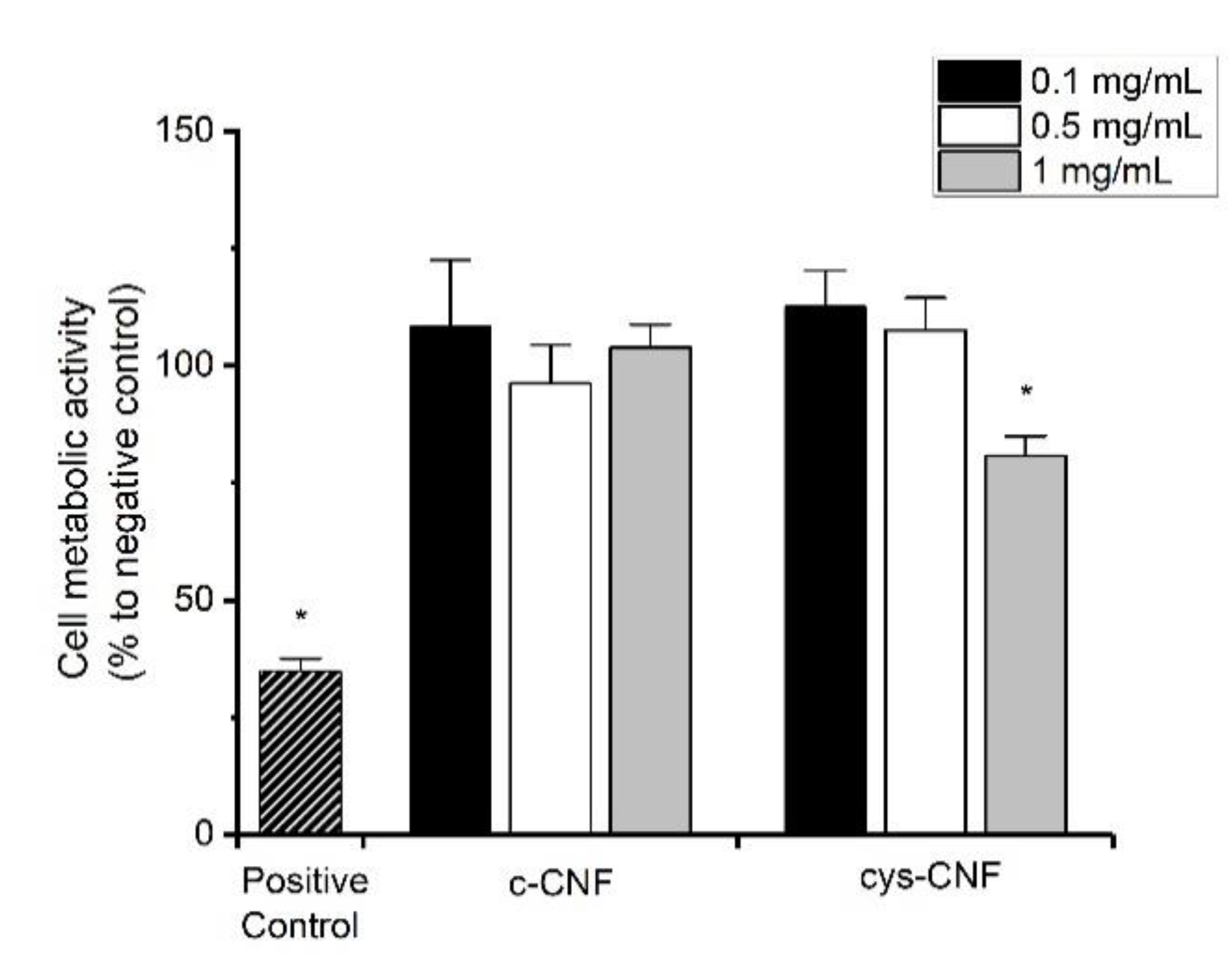
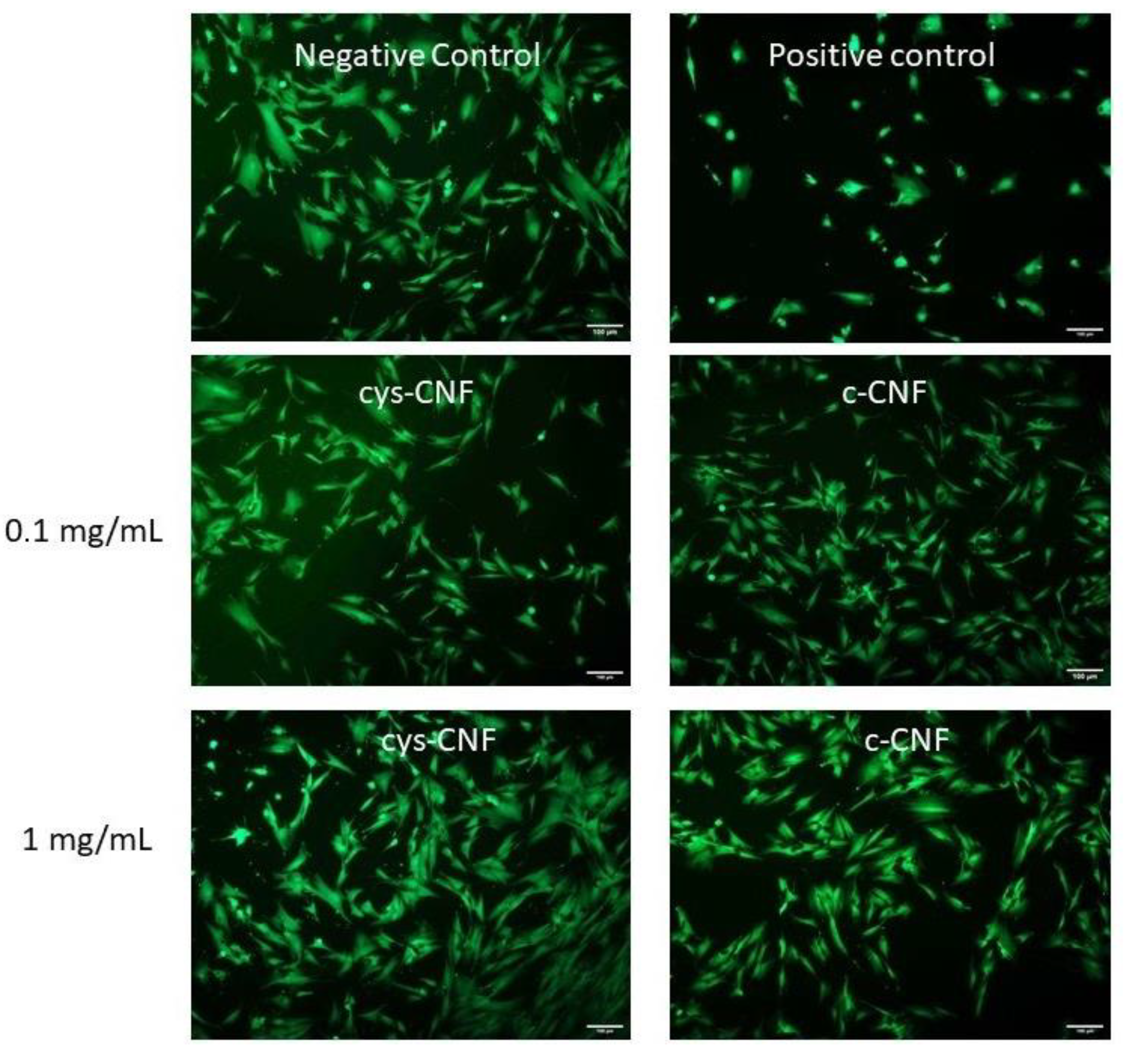
3.5. Cell Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stadelmann, W.K.; Digenis, A.G.; Tobin, G.R. Physiology and healing dynamics of chronic cutaneous wounds. Am. J. Surg. 1998, 176, 26S–38S. [Google Scholar] [CrossRef]
- Diegelmann, R.F.; Evans, M.C. Wound healing: An overview of acute, fibrotic and delayed healing. Front. Biosci. 2004, 9, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Hamdan, S.; Pastar, I.; Drakulich, S.; Dikici, E.; Tomic-Canic, M.; Deo, S.; Daunert, S. Nanotechnology-Driven Therapeutic Interventions in Wound Healing: Potential Uses and Applications. ACS Central Sci. 2017, 3, 163–175. [Google Scholar] [CrossRef]
- Eming, S.A.; Martin, P.; Tomic-Canic, M. Wound repair and regeneration: Mechanisms, signaling, and translation. Sci. Transl. Med. 2014, 6, 265sr6. [Google Scholar] [CrossRef] [PubMed]
- Jorfi, M.; Foster, E.J. Recent advances in nanocellulose for biomedical applications. J. Appl. Polym. Sci. 2015, 132, 132. [Google Scholar] [CrossRef]
- Endes, C.; Camarero-Espinosa, S.; Mueller, S.; Foster, E.J.; Petri-Fink, A.; Rothen-Rutishauser, B.; Weder, C.; Clift, M.J.D. A critical review of the current knowledge regarding the biological impact of nanocellulose. J. Nanobiotechnol. 2016, 14, 1–14. [Google Scholar] [CrossRef]
- Klemm, D.O.; Kramer, F.; Moritz, S.; Lindström, T.; Ankerfors, M.; Gray, D.G.; Dorris, A. Nanocelluloses: A New Family of Nature-Based Materials. Angew. Chem. Int. Ed. 2011, 50, 5438–5466. [Google Scholar] [CrossRef]
- Lavoine, N.; Desloges, I.; Dufresne, A.; Bras, J. Microfibrillated cellulose—Its barrier properties and applications in cellulosic materials: A review. Carbohydr. Polym. 2012, 90, 735–764. [Google Scholar] [CrossRef]
- Lin, N.; Dufresne, A. Nanocellulose in biomedicine: Current status and future prospect. Eur. Polym. J. 2014, 59, 302–325. [Google Scholar] [CrossRef]
- Moohan, J.; Stewart, S.A.; Espinosa, E.; Rosal, A.; Rodríguez, A.; Larrañeta, E.; Donnelly, R.F.; Domínguez-Robles, J. Cellulose Nanofibers and Other Biopolymers for Biomedical Applications. A Review. Appl. Sci. 2020, 10, 65. [Google Scholar] [CrossRef]
- Bacakova, L.; Pajorova, J.; Zikmundova, M.; Pajorova, J.; Kallio, P.; Kolarova, K.; Švorčík, V. Versatile Application of Nanocellulose: From Industry to Skin Tissue Engineering and Wound Healing. Nanomaterials 2019, 9, 164. [Google Scholar] [CrossRef] [PubMed]
- Tehrani, Z.; Nordli, H.R.; Pukstad, B.; Gethin, D.T.; Chinga-Carrasco, G. Translucent and ductile nanocellulose-PEG bionanocomposites—A novel substrate with potential to be functionalized by printing for wound dressing applications. Ind. Crop. Prod. 2016, 93, 193–202. [Google Scholar] [CrossRef]
- Liu, J.; Chinga-Carrasco, G.; Cheng, F.; Xu, W.; Willför, S.; Syverud, K.; Xu, C. Hemicellulose-reinforced nanocellulose hydrogels for wound healing application. Cellulose 2016, 23, 3129–3143. [Google Scholar] [CrossRef]
- Nordli, H.R.; Chinga-Carrasco, G.; Rokstad, A.M.; Pukstad, B. Producing ultrapure wood cellulose nanofibrils and evaluating the cytotoxicity using human skin cells. Carbohydr. Polym. 2016, 150, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Hakkarainen, T.; Koivuniemi, R.; Kosonen, M.; Escobedo-Lucea, C.; Sanz-Garcia, A.; Vuola, J.; Valtonen, J.; Tammela, P.; Mäkitie, A.; Luukko, K.; et al. Nanofibrillar cellulose wound dressing in skin graft donor site treatment. J. Control. Release 2016, 244, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Koivuniemi, R.; Hakkarainen, T.; Kiiskinen, J.; Kosonen, M.; Vuola, J.; Valtonen, J.; Luukko, K.; Kavola, H.; Yliperttula, M. Clinical Study of Nanofibrillar Cellulose Hydrogel Dressing for Skin Graft Donor Site Treatment. Adv. Wound Care 2020, 9, 199–210. [Google Scholar] [CrossRef]
- Basu, A.; Lindh, J.; Ålander, E.; Strømme, M.; Ferraz, N. On the use of ion-crosslinked nanocellulose hydrogels for wound healing solutions: Physicochemical properties and application-oriented biocompatibility studies. Carbohydr. Polym. 2017, 174, 299–308. [Google Scholar] [CrossRef]
- Basu, A.; Hong, J.; Ferraz, N. Hemocompatibility of Ca2+ -Crosslinked Nanocellulose Hydrogels: Toward Efficient Management of Hemostasis. Macromol. Biosci. 2017, 17, 17. [Google Scholar] [CrossRef]
- Basu, A.; Celma, G.; Strømme, M.; Ferraz, N. In Vitro and in Vivo Evaluation of the Wound Healing Properties of Nanofibrillated Cellulose Hydrogels. ACS Appl. Bio Mater. 2018, 1, 1853–1863. [Google Scholar] [CrossRef]
- Basu, A.; Heitz, K.; Strømme, M.; Welch, K.; Ferraz, N. Ion-crosslinked wood-derived nanocellulose hydrogels with tunable antibacterial properties: Candidate materials for advanced wound care applications. Carbohydr. Polym. 2018, 181, 345–350. [Google Scholar] [CrossRef]
- Basu, A.; Strømme, M.; Ferraz, N. Towards Tunable Protein-Carrier Wound Dressings Based on Nanocellulose Hydrogels Crosslinked with Calcium Ions. Nanomaterials 2018, 8, 550. [Google Scholar] [CrossRef]
- Sabino, F.; auf dem Keller, U. Matrix metalloproteinases in impaired wound healing. Met. Med. 2015. [Google Scholar] [CrossRef]
- Dunnill, C.; Patton, T.; Brennan, J.; Barrett, J.; Dryden, M.; Cooke, J.; Leaper, D.; Georgopoulos, N.T. Reactive oxygen species (ROS) and wound healing: The functional role of ROS and emerging ROS-modulating technologies for augmentation of the healing process. Int. Wound J. 2017, 14, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Jang, H.-J.; Cho, W.-Y.; Yeon, S.-J.; Lee, C.-H. In vitro antioxidant actions of sulfur-containing amino acids. Arab. J. Chem. 2020, 13, 1678–1684. [Google Scholar] [CrossRef]
- Guidea, A.; Zăgrean-Tuza, C.; Mot, A.C.; Sârbu, C. Comprehensive evaluation of radical scavenging, reducing power and chelating capacity of free proteinogenic amino acids using spectroscopic assays and multivariate exploratory techniques. Spectrochim. Acta Part. A: Mol. Biomol. Spectrosc. 2020, 233, 118158. [Google Scholar] [CrossRef]
- Wang, H.; Liu, R.; Liu, Y.; Meng, Y.; Liu, Y.; Zhai, H.; Di, D. Investigation on Adsorption Mechanism of Peptides with Surface-Modified Super-Macroporous Resins. Langmuir 2019, 35, 4471–4480. [Google Scholar] [CrossRef]
- Rol, F.; Belgacem, M.N.; Gandini, A.; Bras, J. Recent advances in surface-modified cellulose nanofibrils. Prog. Polym. Sci. 2019, 88, 241–264. [Google Scholar] [CrossRef]
- Chen, H.; Sharma, S.K.; Sharma, P.R.; Yeh, H.; Johnson, K.; Hsiao, B.S. Arsenic(III) Removal by Nanostructured Dialdehyde Cellulose–Cysteine Microscale and Nanoscale Fibers. ACS Omega 2019, 4, 22008–22020. [Google Scholar] [CrossRef]
- Hernández, M.; Leyva, G.; Magaña, J.J.; Guzmán-Vargas, A.; Felipe-Mendoza, C.; Lara, V.; Lima, E. New copolymers as hosts of ribosomal RNA. BMC Chem. 2019, 13, 33. [Google Scholar] [CrossRef]
- Nouvong, A.; Ambrus, A.M.; Zhang, E.R.; Hultman, L.; Coller, H.A. Reactive oxygen species and bacterial biofilms in diabetic wound healing. Physiol. Genom. 2016, 48, 889–896. [Google Scholar] [CrossRef]
- Comino-Sanz, I.M.; López-Franco, M.D.; Castro, B.; Pancorbo-Hidalgo, P.L. Antioxidant dressing therapy versus standard wound care in chronic wounds (the REOX study): Study protocol for a randomized controlled trial. Trials 2020, 21, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Han, S.; Gu, Z.; Wu, J. Advances and Impact of Antioxidant Hydrogel in Chronic Wound Healing. Adv. Health Mater. 2020, 9, 1901502. [Google Scholar] [CrossRef] [PubMed]
- Moseley, R.; Leaver, M.; Walker, M.; Waddington, R.; Parsons, D.; Chen, W.; Embery, G. Comparison of the antioxidant properties of HYAFF®-11p75, AQUACEL® and hyaluronan towards reactive oxygen species in vitro. Biomaterials 2002, 23, 2255–2264. [Google Scholar] [CrossRef]
- Moseley, R.; Walker, M.; Waddington, R.J.; Chen, W. Comparison of the antioxidant properties of wound dressing materials–carboxymethylcellulose, hyaluronan benzyl ester and hyaluronan, towards polymorphonuclear leukocyte-derived reactive oxygen species. Biomaterials 2003, 24, 1549–1557. [Google Scholar] [CrossRef]
- Ataide, J.A.; De Carvalho, N.M.; Rebelo, M.D.A.; Chaud, M.V.; Grotto, D.; Gerenutti, M.; Rai, M.; Mazzola, P.G.; Jozala, A.F. Bacterial Nanocellulose Loaded with Bromelain: Assessment of Antimicrobial, Antioxidant and Physical-Chemical Properties. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Morais, E.S.; Silva, N.H.; Sintra, T.E.; Santos, S.A.; Neves, B.M.; Almeida, I.F.; Costa, P.; Correia-Sá, I.; Ventura, S.P.M.; Silvestre, A.J.D.; et al. Anti-inflammatory and antioxidant nanostructured cellulose membranes loaded with phenolic-based ionic liquids for cutaneous application. Carbohydr. Polym. 2019, 206, 187–197. [Google Scholar] [CrossRef]
- Cullen, B.; Watt, P.W.; Lundqvist, C.; Silcock, D.; Schmidt, R.J.; Bogan, D.; Light, N.D. The role of oxidised regenerated cellulose/collagen in chronic wound repair and its potential mechanism of action. Int. J. Biochem. Cell Biol. 2002, 34, 1544–1556. [Google Scholar] [CrossRef]
- Mccarty, S.M.; Percival, S.L. Proteases and Delayed Wound Healing. Adv. Wound Care 2013, 2, 438–447. [Google Scholar] [CrossRef]
- Jacobsen, J.A.; Jourden, J.L.M.; Miller, M.T.; Cohen, S.M. To bind zinc or not to bind zinc: An examination of innovative approaches to improved metalloproteinase inhibition. Biochim. Et Biophys. Acta (BBA)–Bioenerg. 2010, 1803, 72–94. [Google Scholar] [CrossRef]
- Pace, N.J.; Weerapana, E. Zinc-Binding Cysteines: Diverse Functions and Structural Motifs. Biomolecules 2014, 4, 419–434. [Google Scholar] [CrossRef]
- Benesch, R.E.; Benesch, R. The Acid Strength of the -SH Group in Cysteine and Related Compounds. J. Am. Chem. Soc. 1955, 77, 5877–5881. [Google Scholar] [CrossRef]
- Bagiyan, G.A.; Koroleva, I.K.; Soroka, N.V.; Ufimtsev, A.V. Oxidation of thiol compounds by molecular oxygen in aqueous solutions. Russ. Chem. Bull. 2003, 52, 1135–1141. [Google Scholar] [CrossRef]
- Part 5: Test for in vitro cytotoxicityIso. In Iso 10993-5: Biological Evaluation of Medical devices; ISO: Geneva, Switzerland.
- Lopes, V.R.; Sanchez-Martinez, C.; Strømme, M.; Ferraz, N. In vitro biological responses to nanofibrillated cellulose by human dermal, lung and immune cells: Surface chemistry aspect. Part. Fibre Toxicol. 2017, 14, 1–13. [Google Scholar] [CrossRef]
- Lopes, V.R.; Strømme, M.; Ferraz, N. In Vitro Biological Impact of Nanocellulose Fibers on Human Gut Bacteria and Gastrointestinal Cells. Nanomaterials 2020, 10, 1159. [Google Scholar] [CrossRef] [PubMed]
- Čolić, M.; Mihajlović, D.; Mathew, A.P.; Naseri, N.; Kokol, V. Cytocompatibility and immunomodulatory properties of wood based nanofibrillated cellulose. Cellulose 2015, 22, 763–778. [Google Scholar] [CrossRef]
- Alexandrescu, L.; Syverud, K.; Gatti, A.; Chinga-Carrasco, G. Cytotoxicity tests of cellulose nanofibril-based structures. Cellulose 2013, 20, 1765–1775. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molar Reaction Ratio 1 | Cysteine Content (mmol/g CNF) 2 | Degree of Substitution 3 | Thiol Content (mmol/g CNF) 4 | Thiol/Sulfur (%) |
|---|---|---|---|---|
| 1:6 | 0.71 ± 0.07 | 0.54 ± 0.05 | 0.14 ± 0.02 | 20 |
| 1:20 | 0.78 ± 0.04 | 0.60 ± 0.05 | 0.20 ± 0.03 | 26 |
| Cys-CNF Sample | Collagenase Entrapment (% of Control) |
|---|---|
| (1:6) | 20 ± 5 |
| (1:20) | 30 ± 5 |
| Cys-CNF Sample | Suspension (Air) | Aerogel | Inert Atmosphere (N2) |
|---|---|---|---|
| (1:6) | 49 ± 13 | 67 ± 19 | 105 ± 4 |
| (1:20) | 43 ± 8 | 89 ± 11 | 76 ± 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blasi-Romero, A.; Palo-Nieto, C.; Sandström, C.; Lindh, J.; Strømme, M.; Ferraz, N. In Vitro Investigation of Thiol-Functionalized Cellulose Nanofibrils as a Chronic Wound Environment Modulator. Polymers 2021, 13, 249. https://doi.org/10.3390/polym13020249
Blasi-Romero A, Palo-Nieto C, Sandström C, Lindh J, Strømme M, Ferraz N. In Vitro Investigation of Thiol-Functionalized Cellulose Nanofibrils as a Chronic Wound Environment Modulator. Polymers. 2021; 13(2):249. https://doi.org/10.3390/polym13020249
Chicago/Turabian StyleBlasi-Romero, Anna, Carlos Palo-Nieto, Corine Sandström, Jonas Lindh, Maria Strømme, and Natalia Ferraz. 2021. "In Vitro Investigation of Thiol-Functionalized Cellulose Nanofibrils as a Chronic Wound Environment Modulator" Polymers 13, no. 2: 249. https://doi.org/10.3390/polym13020249
APA StyleBlasi-Romero, A., Palo-Nieto, C., Sandström, C., Lindh, J., Strømme, M., & Ferraz, N. (2021). In Vitro Investigation of Thiol-Functionalized Cellulose Nanofibrils as a Chronic Wound Environment Modulator. Polymers, 13(2), 249. https://doi.org/10.3390/polym13020249