
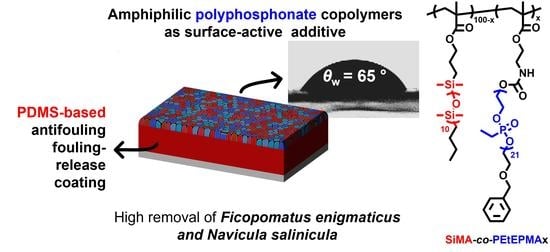
Amphiphilic Polyphosphonate Copolymers as New Additives for PDMS-Based Antifouling Coatings

 ,
,  , ,
, ,  , ,
, , 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis
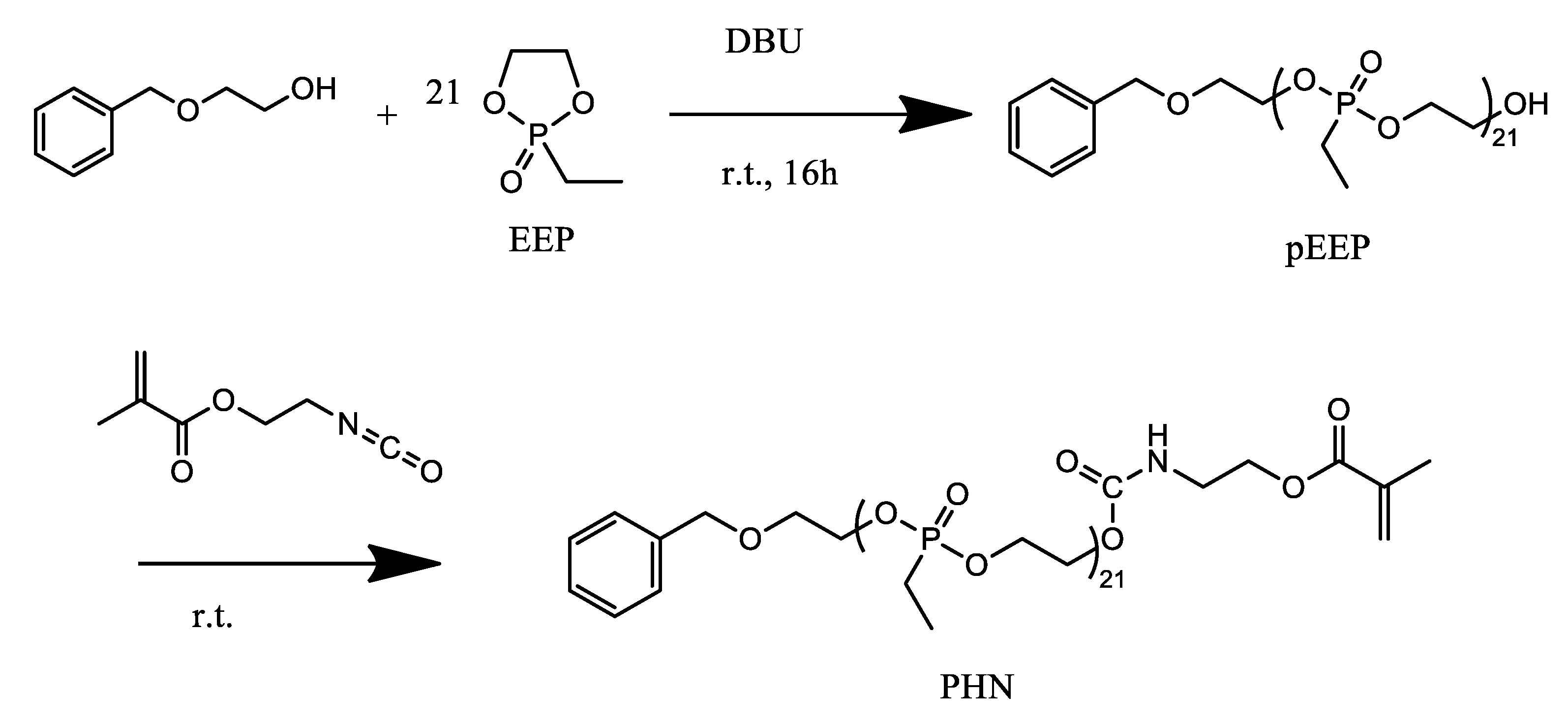
2.2.1. PEtEPMA Synthesis
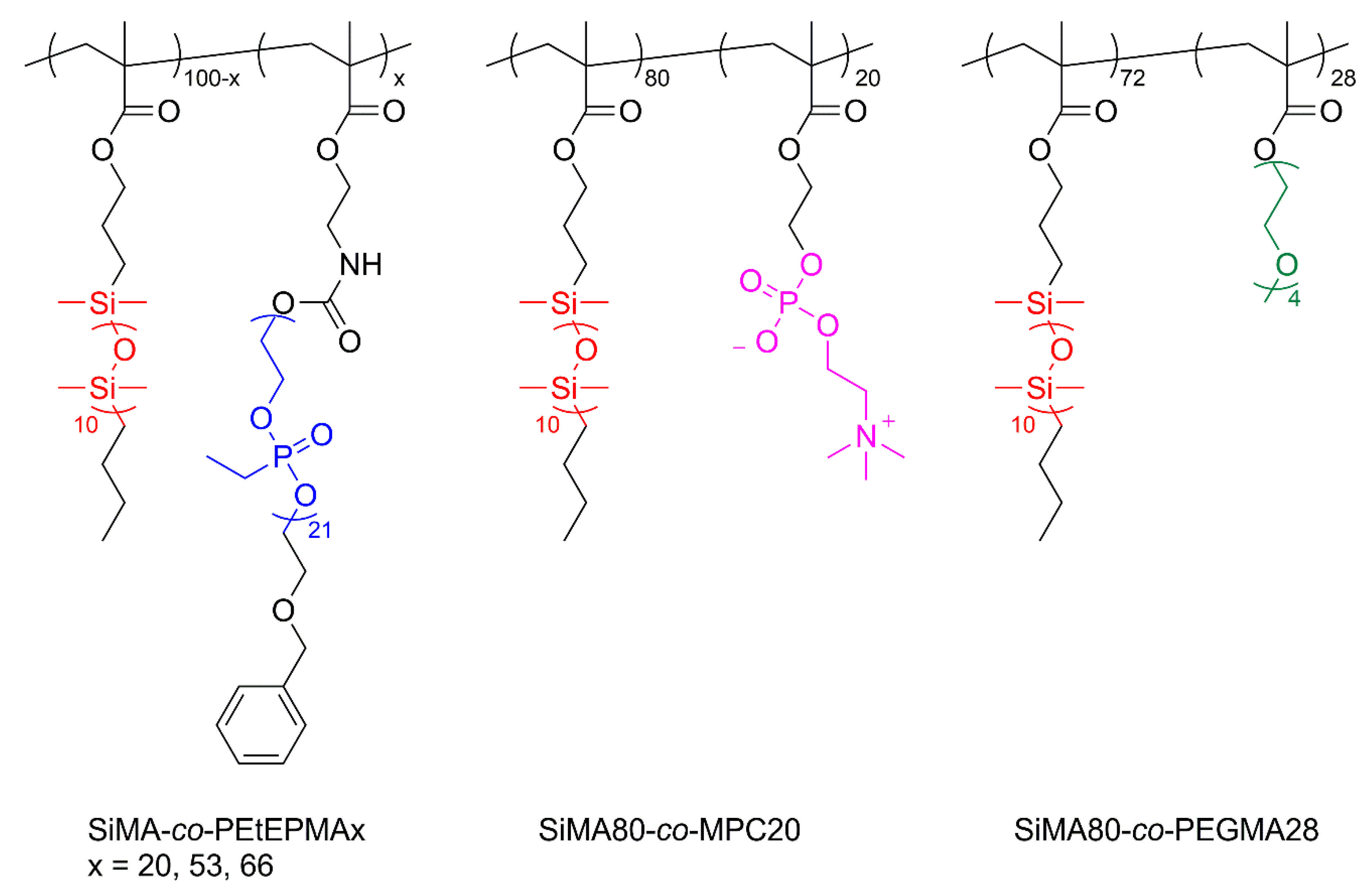
2.2.2. SiMA-co-PEtEPMAx Copolymer Synthesis
2.2.3. SiMA-co-MPC20 and SiMA-co-PEGMA28 Copolymer Synthesis
2.3. Film Preparation
2.3.1. Three-Layer PDMS-Based Polymer Films
2.3.2. Single Layer Polymer Films
2.4. Characterization
2.4.1. Biological Tests
Navicula salinicola Settlement and Detachment Assay
Settlement of Larvae and Adult Detachment of Ficopomatus enigmaticus
Statistical Analysis
3. Results and Discussion
3.1. PEtEPMA Macromonomer Synthesis
3.2. SiMA-co-PEtEPMAx Copolymerization and Chemical-Physical Characterization
3.3. SiMA-co-MPC20 and SiMA-co-PEGMA28 Copolymerizations
3.4. Thermal Analysis of the Copolymers
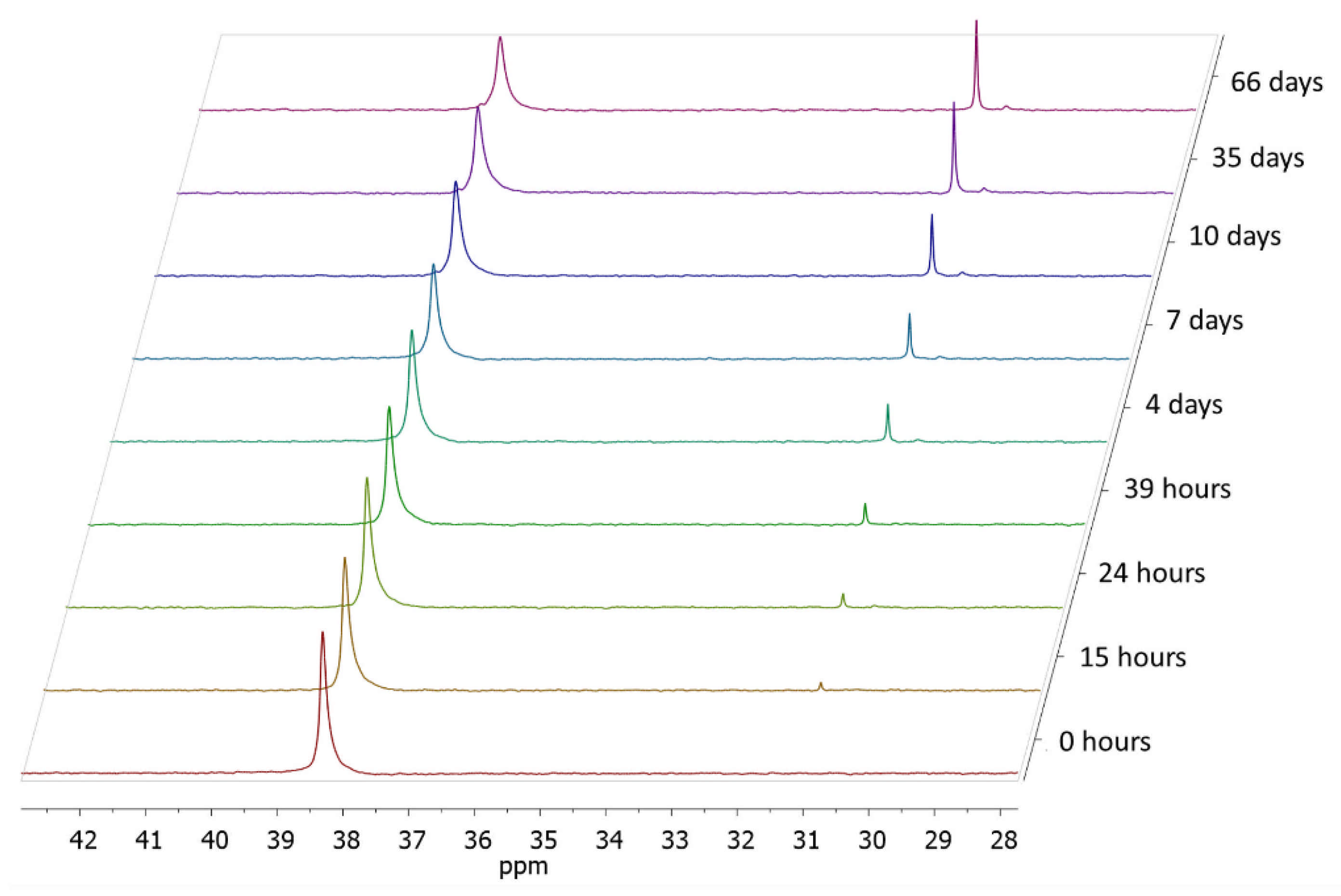
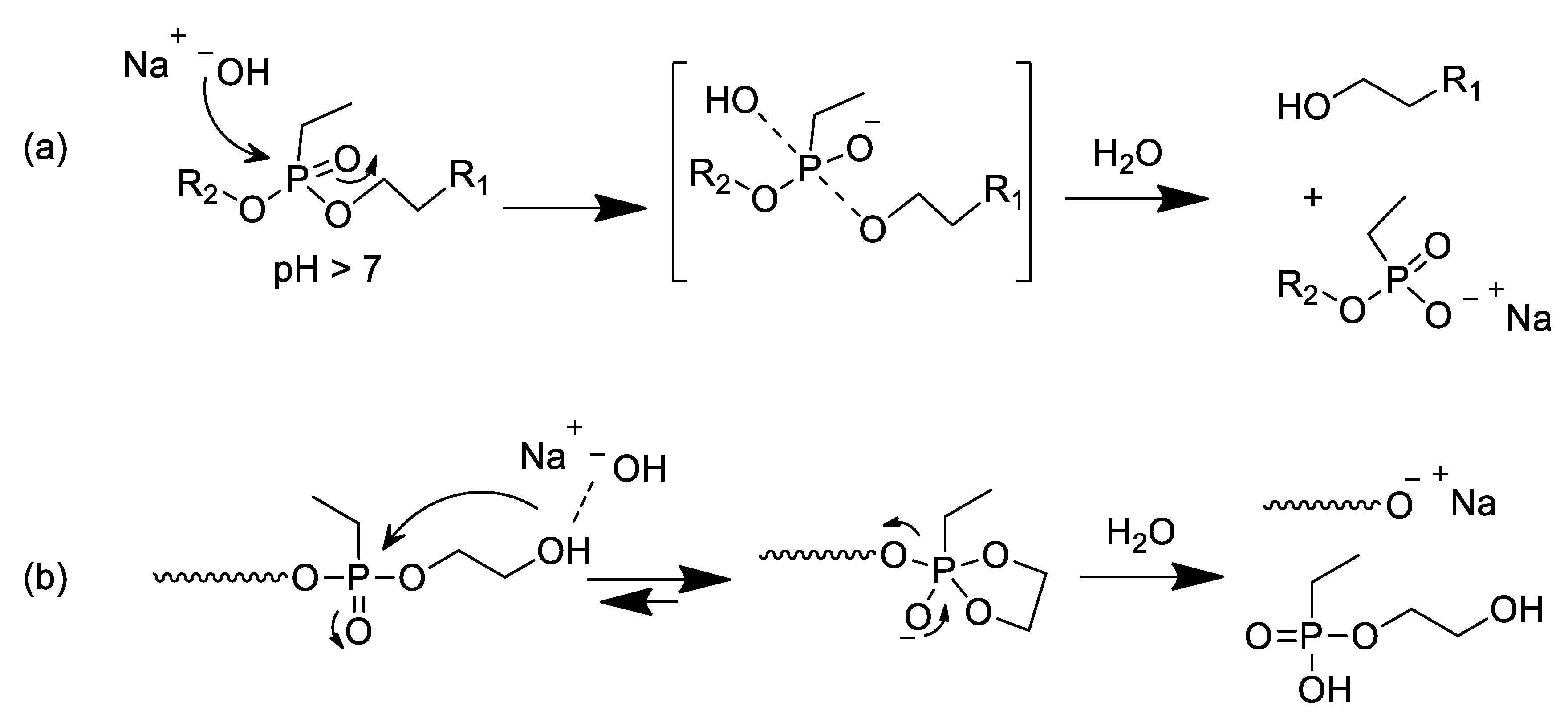
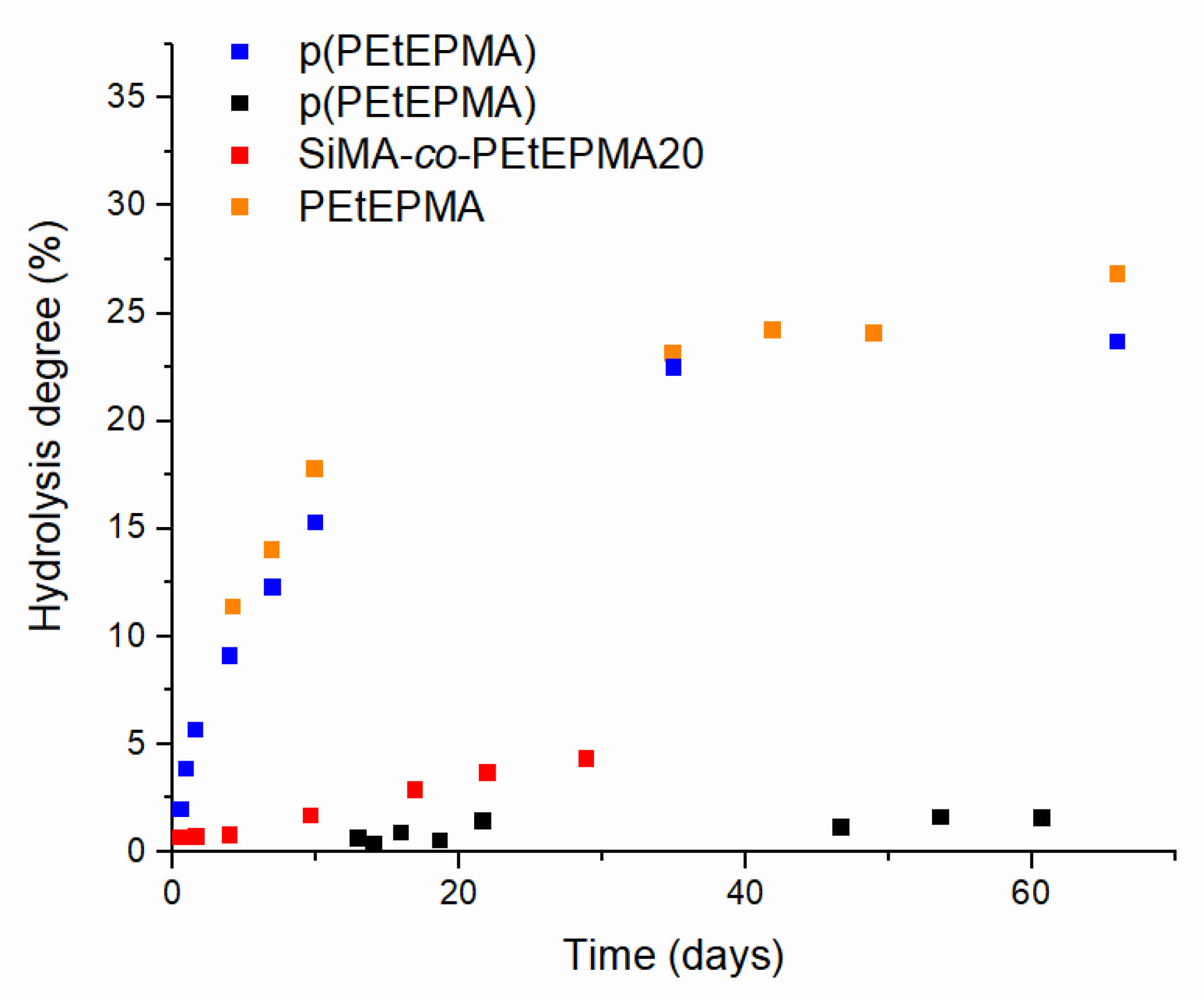
3.5. Hydrolisis of Poly(ethylphosphonate)-Based Polymers in Alkaline Conditions
3.6. Preparation of Polymer Films
3.7. Wettability and Surface Reconstruction
3.8. Biological Test with Marine Organisms
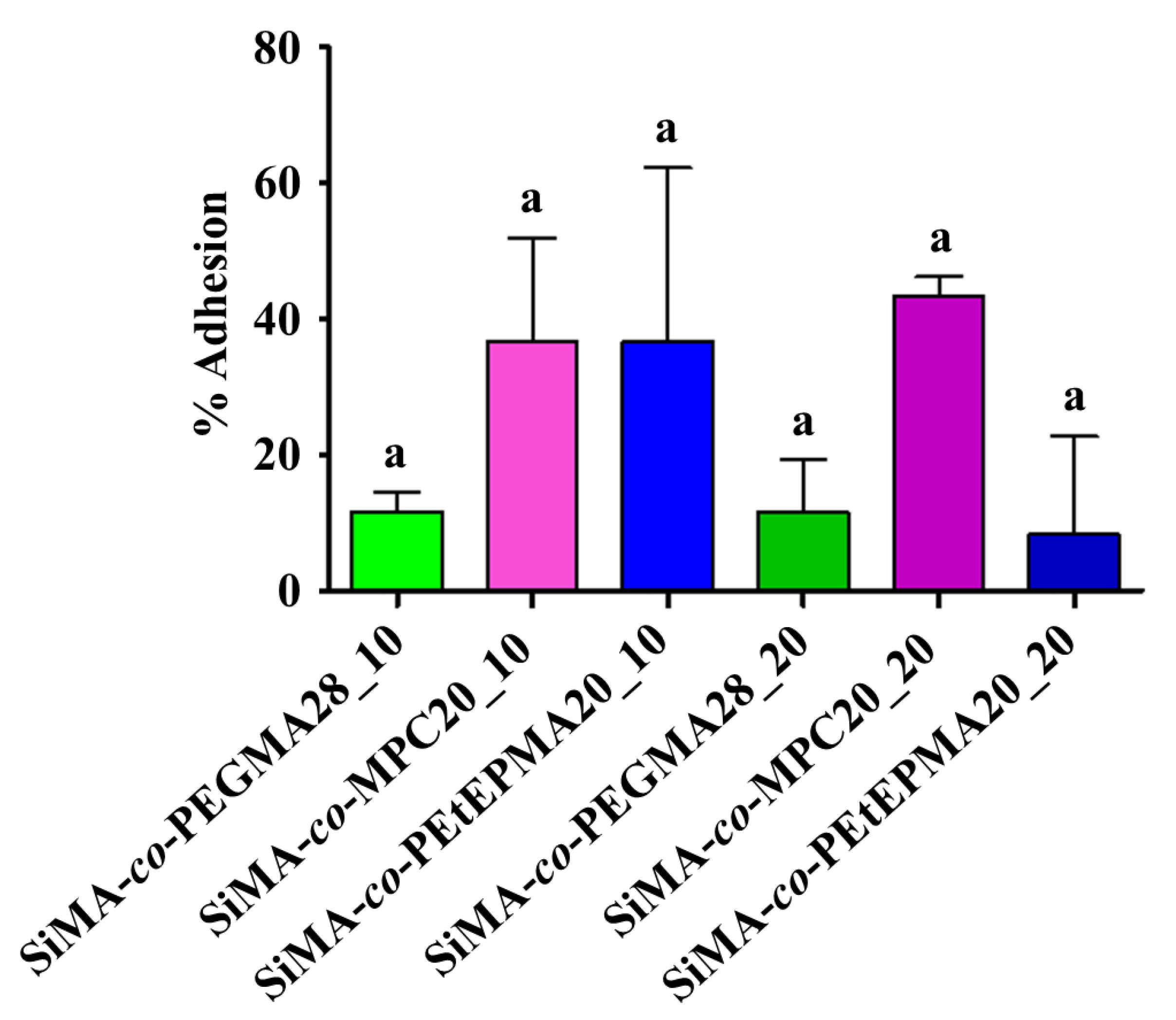
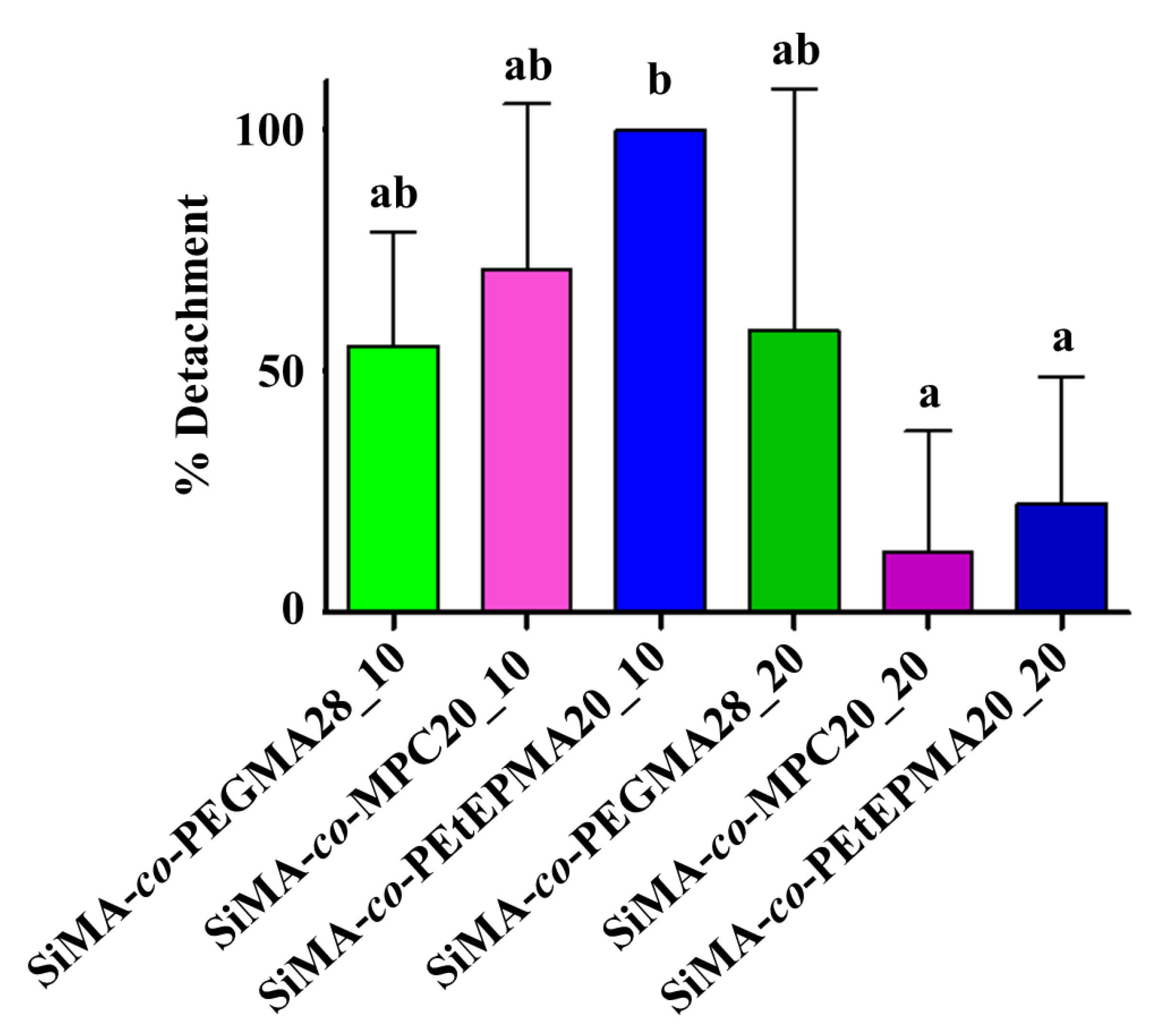
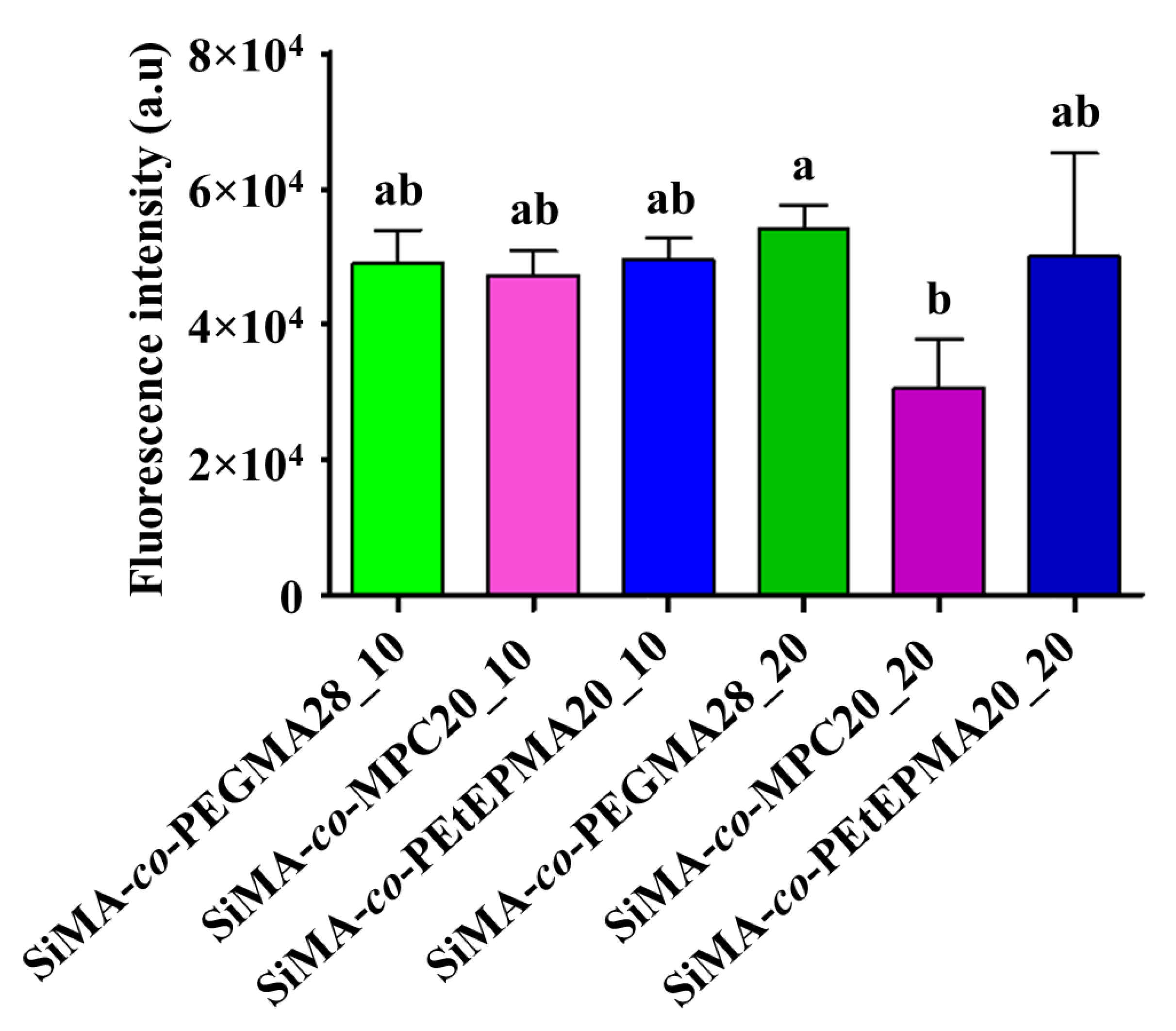
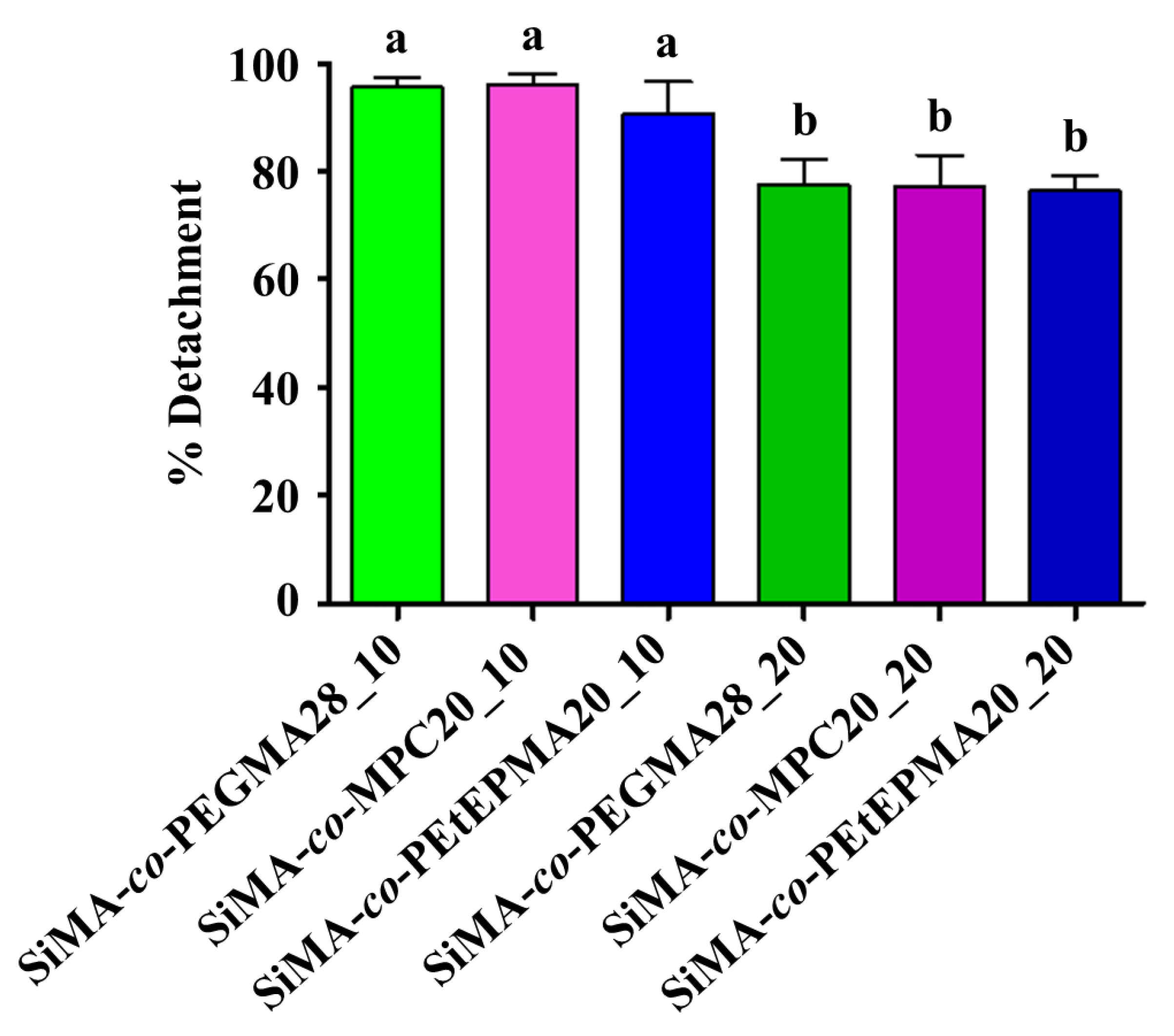
3.8.1. Ficopomatus Enigmaticus Settlement and Detachment Assay
3.8.2. Navicula Salinicola Settlement and Detachment Assay
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pelosi, C.; Tinè, M.R.; Wurm, F.R. Main-chain water-soluble polyphosphoesters: Multi-functional polymers as degradable PEG-alternatives for biomedical applications. Eur. Polym. J. 2020, 141, 110079. [Google Scholar] [CrossRef]
- Beament, J.; Wolf, T.; Markwart, J.C.; Wurm, F.R.; Jones, M.D.; Buchard, A. Copolymerization of cyclic phosphonate and lactide: Synthetic strategies toward control of amphiphilic microstructure. Macromolecules 2019, 52, 1220–1226. [Google Scholar] [CrossRef]
- Lin, H.; Wolf, T.; Wurm, F.R.; Kelland, M.A. Poly(alkyl ethylene phosphonate)s: A new class of non-amide kinetic hydrate inhibitor polymers. Energy Fuels 2017, 31, 3843–3848. [Google Scholar] [CrossRef]
- Wolf, T.; Hunold, J.; Simon, J.; Rosenauer, C.; Hinderberger, D.; Wurm, F.R. Temperature responsive poly(phosphonate) copolymers: From single chains to macroscopic coacervates. Polym. Chem. 2018, 9, 490–498. [Google Scholar] [CrossRef] [Green Version]
- Wolf, T.; Rheinberger, T.; Wurm, F.R. Thermoresponsive coacervate formation of random poly(phosphonate) terpolymers. Eur. Polym. J. 2017, 95, 756–765. [Google Scholar] [CrossRef]
- Wolf, T.; Steinbach, T.; Wurm, F.R. A library of well-defined and water-soluble poly(alkyl phosphonate)s with adjustable hydrolysis. Macromolecules 2015, 48, 3853–3863. [Google Scholar] [CrossRef]
- Steinbach, T.; Alexandrino, E.M.; Wahlen, C.; Landfester, K.; Wurm, F.R. Poly(phosphonate)s via olefin metathesis: Adjusting hydrophobicity and morphology. Macromolecules 2014, 47, 4884–4893. [Google Scholar] [CrossRef]
- Simon, J.; Wolf, T.; Klein, K.; Landfester, K.; Wurm, F.R.; Mailänder, V. Hydrophilicity regulates the stealth properties of polyphosphoester-coated nanocarriers. Angew. Chem. Int. Ed. 2018, 57, 5548–5553. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, C.; Duce, C.; Wurm, F.R.; Tinè, M.R. Effect of polymer hydrophilicity and molar mass on the properties of the protein in protein–polymer conjugates: The case of PPEylated myoglobin. Biomacromolecules 2021, 22, 1932–1943. [Google Scholar] [CrossRef]
- Iwasaki, Y.; Yamaguchi, E. Synthesis of well-defined thermoresponsive polyphosphoester macroinitiators using organocatalysts. Macromolecules 2010, 43, 2664–2666. [Google Scholar] [CrossRef]
- Steinbach, T.; Ritz, S.; Wurm, F.R. Water-soluble poly(phosphonate)s via living ring-opening polymerization. ACS Macro Lett. 2014, 3, 244–248. [Google Scholar] [CrossRef]
- Sauty, N.F.; da Silva, L.C.; Schulz, M.D.; Few, C.S.; Wagener, K.B. Acyclic diene metathesis polymerization and precision polymers. Appl. Petrochem. Res. 2014, 4, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, S.; Kumar, S.; Mohanty, S.; Nayak, S.K. Environmentally benign fouling-resistant marine coatings: A review. Polym.-Plast. Technol. Mater. 2019, 58, 498–518. [Google Scholar] [CrossRef]
- Lejars, M.; Margaillan, A.; Bressy, C. Fouling release coatings: A nontoxic alternative to biocidal antifouling coatings. Chem. Rev. 2012, 112, 4347–4390. [Google Scholar] [CrossRef] [PubMed]
- Galli, G.; Martinelli, E. Amphiphilic polymer platforms: Surface engineering of films for marine antibiofouling. Macromol. Rapid Commun. 2017, 38, 1600704. [Google Scholar] [CrossRef]
- Pocivavsek, L.; Pugar, J.; O’Dea, R.; Ye, S.H.; Wagner, W.; Tzeng, E.; Velankar, S.; Cerda, E. Topography-driven surface renewal. Nat. Phys. 2018, 14, 948–953. [Google Scholar] [CrossRef]
- Leonardi, A.K.; Ober, C.K. Polymer-based marine antifouling and fouling release surfaces: Strategies for synthesis and modification. Annu. Rev. Chem. Biomol. Eng. 2019, 10, 241–264. [Google Scholar] [CrossRef]
- Li, D.; Zheng, Q.; Wang, Y.; Chen, H. Combining surface topography with polymer chemistry: Exploring new interfacial biological phenomena. Polym. Chem. 2014, 5, 14–24. [Google Scholar] [CrossRef]
- Martinelli, E.; Guazzelli, E.; Glisenti, A.; Galli, G. Surface segregation of amphiphilic PDMS-based films containing terpolymers with siloxane, fluorinated and ethoxylated side chains. Coatings 2019, 9, 153. [Google Scholar] [CrossRef] [Green Version]
- Guazzelli, E.; Perondi, F.; Criscitiello, F.; Pretti, C.; Oliva, M.; Casu, V.; Maniero, F.; Gazzera, L.; Galli, G.; Martinelli, E. New amphiphilic copolymers for PDMS-based nanocomposite films with long-term marine antifouling performance. J. Mater. Chem. B 2020, 8, 9764–9776. [Google Scholar] [CrossRef]
- Mo, Y.; Xue, P.; Yang, Q.; Liu, H.; Zhao, X.; Wang, J.; Jin, M.; Qi, Y. Composite slow-release fouling release coating inspired by synergistic anti-fouling effect of scaly fish. Polymers 2021, 13, 2602. [Google Scholar] [CrossRef]
- Galli, G.; Martinelli, E.; Chiellini, E.; Ober, C.K.; Glisenti, A. Low surface energy characteristics of mesophase-forming ABC and ACB triblock copolymers with fluorinated B blocks. Mol. Cryst. Liq. Cryst. 2005, 441, 211–226. [Google Scholar] [CrossRef]
- Maan, A.M.C.; Hofman, A.H.; Vos, W.M.; Kamperman, M. Recent developments and practical feasibility of polymer-based antifouling coatings. Adv. Funct. Mater. 2020, 30, 2000936. [Google Scholar] [CrossRef]
- Martinelli, E.; Guazzelli, E.; Galli, G. Recent advances in designed non-toxic polysiloxane coatings to combat marine biofouling. In Marine Coatings and Membranes; Mittal, V., Ed.; Central West Publishing: Orange, Australia, 2019; pp. 137–180. ISBN 978-1-925823-47-9. [Google Scholar]
- Pretti, C.; Oliva, M.; Mennillo, E.; Barbaglia, M.; Funel, M.; Reddy Yasani, B.; Martinelli, E.; Galli, G. An ecotoxicological study on tin- and bismuth-catalysed PDMS based coatings containing a surface-active polymer. Ecotoxicol. Environ. Saf. 2013, 98, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Xie, Q.; Ma, C.; Zhang, G. Silicone-based fouling-release coatings for marine antifouling. Langmuir 2020, 36, 2170–2183. [Google Scholar] [CrossRef] [Green Version]
- Holland, R.; Dugdale, T.M.; Wetherbee, R.; Brennan, A.B.; Finlay, J.A.; Callow, J.A.; Callow, M.E. Adhesion and motility of fouling diatoms on a silicone elastomer. Biofouling 2004, 20, 323–329. [Google Scholar] [CrossRef]
- Stafslien, S.J.; Christianson, D.; Daniels, J.; VanderWal, L.; Chernykh, A.; Chisholm, B.J. Combinatorial materials research applied to the development of new surface coatings XVI: Fouling-release properties of amphiphilic polysiloxane coatings. Biofouling 2015, 31, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, E.; Sarvothaman, M.K.; Galli, G.; Pettitt, M.E.; Callow, M.E.; Callow, J.A.; Conlan, S.L.; Clare, A.S.; Sugiharto, A.B.; Davies, C.; et al. Poly(dimethyl siloxane) (PDMS) network blends of amphiphilic acrylic copolymers with Poly(ethylene glycol)-fluoroalkyl side chains for fouling-release coatings. II. Laboratory assays and field immersion trials. Biofouling 2012, 28, 571–582. [Google Scholar] [CrossRef]
- Galli, G.; Barsi, D.; Martinelli, E.; Glisenti, A.; Finlay, J.A.; Callow, M.E.; Callow, J.A. Copolymer films containing amphiphilic side chains of well-defined fluoroalkyl-segment length with biofouling-release potential. RSC Adv. 2016, 6, 67127–67135. [Google Scholar] [CrossRef]
- Wenning, B.M.; Martinelli, E.; Mieszkin, S.; Finlay, J.A.; Fischer, D.; Callow, J.A.; Callow, M.E.; Leonardi, A.K.; Ober, C.K.; Galli, G. Model amphiphilic block copolymers with tailored molecular weight and composition in PDMS-based films to limit soft biofouling. ACS Appl. Mater. Interfaces 2017, 9, 16505–16516. [Google Scholar] [CrossRef]
- Goda, T.; Matsuno, R.; Konno, T.; Takai, M.; Ishihara, K. Photografting of 2-methacryloyloxyethyl phosphorylcholine from polydimethylsiloxane: Tunable protein repellency and lubrication property. Colloids Surf. B Biointerfaces 2008, 63, 64–72. [Google Scholar] [CrossRef]
- Keefe, A.J.; Brault, N.D.; Jiang, S. Suppressing surface reconstruction of superhydrophobic PDMS using a superhydrophilic zwitterionic polymer. Biomacromolecules 2012, 13, 1683–1687. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Zhang, J.; Wang, Z.; Chen, S. Development of robust biocompatible silicone with high resistance to protein adsorption and bacterial adhesion. Acta Biomater. 2011, 7, 2053–2059. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Chen, P.; Tian, S.; Ma, Y.; Li, Q.; Wen, C.; Yang, J.; Zhang, L. Amphiphilic marine antifouling coatings based on a hydrophilic polyvinylpyrrolidone and hydrophobic fluorine–silicon-containing block copolymer. Langmuir 2020, 36, 14573–14581. [Google Scholar] [CrossRef]
- Knop, K.; Hoogenboom, R.; Fischer, D.; Schubert, U.S. poly(ethylene glycol) in drug delivery: Pros and cons as well as potential alternatives. Angew. Chem. Int. Ed. 2010, 49, 6288–6308. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Vancso, G.J.; de Beer, S. Substantially enhanced stability against degrafting of zwitterionic PMPC brushes by utilizing PGMA-linked initiators. Eur. Polym. J. 2017, 89, 221–229. [Google Scholar] [CrossRef]
- Laschewsky, A. Structures and synthesis of zwitterionic polymers. Polymers 2014, 6, 1544–1601. [Google Scholar] [CrossRef]
- Bernhard, C.; Bauer, K.N.; Bonn, M.; Wurm, F.R.; Gonella, G. Interfacial conformation of hydrophilic polyphosphoesters affects blood protein adsorption. ACS Appl. Mater. Interfaces 2019, 11, 1624–1629. [Google Scholar] [CrossRef] [PubMed]
- Guazzelli, E.; Galli, G.; Martinelli, E.; Margaillan, A.; Bressy, C. Amphiphilic hydrolyzable polydimethylsiloxane-b-poly(ethyleneglycol methacrylate-co-trialkylsilyl methacrylate) block copolymers for marine coatings. I. Synthesis, hydrolysis and surface wettability. Polymer 2020, 186, 121954. [Google Scholar] [CrossRef]
- Guazzelli, E.; Martinelli, E.; Pelloquet, L.; Briand, J.F.; Margaillan, A.; Bunet, R.; Galli, G.; Bressy, C. Amphiphilic hydrolyzable polydimethylsiloxane-b-poly(ethyleneglycol methacrylate-co-trialkylsilyl methacrylate) block copolymers for marine coatings. II. Antifouling laboratory tests and field trials. Biofouling 2020, 36, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Ma, C.; Zhang, G.; Bressy, C. Poly(ester)–poly(silyl methacrylate) copolymers: Synthesis and hydrolytic degradation kinetics. Polym. Chem. 2018, 9, 1448–1454. [Google Scholar] [CrossRef]
- Gevaux, L.; Lejars, M.; Margaillan, A.; Briand, J.-F.; Bunet, R.; Bressy, C. Hydrolyzable additive-based silicone elastomers: A new approach for antifouling coatings. Polymers 2019, 11, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gevaux, L.; Lejars, M.; Margaillan, A.; Bressy, C. Water erodible coatings based on a hydrolyzable PDMS/polyester network. Mater. Today Commun. 2018, 17, 517–526. [Google Scholar] [CrossRef]
- Xie, Q.; Pan, J.; Ma, C.; Zhang, G. Dynamic surface antifouling: Mechanism and systems. Soft Matter 2019, 15, 1087–1107. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, T.; Becker, G.; Spiegel, A.; Figueiredo, T.; Russo, D.; Wurm, F.R. Reversible bioconjugation: Biodegradable poly(phosphate)-protein conjugates. Macromol. Biosci. 2017, 17, 1600377. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, C.; Duce, C.; Russo, D.; Tiné, M.R.; Wurm, F.R. PPEylation of proteins: Synthesis, activity, and stability of myoglobin-polyphosphoester conjugates. Eur. Polym. J. 2018, 108, 357–363. [Google Scholar] [CrossRef]
- Wolf, T.; Naß, J.; Wurm, F.R. Cyclohexyl-substituted poly(phosphonate)-copolymers with adjustable glass transition temperatures. Polym. Chem. 2016, 7, 2934–2937. [Google Scholar] [CrossRef] [Green Version]
- Martini, F.; Guazzelli, E.; Martinelli, E.; Borsacchi, S.; Geppi, M.; Galli, G. Molecular dynamics of amphiphilic random copolymers in the bulk: A 1 H and 19 F NMR relaxometry study. Macromol. Chem. Phys. 2019, 220, 1900177. [Google Scholar] [CrossRef]
- Hatakeyama, T.; Tanaka, M.; Hatakeyama, H. Studies on bound water restrained by poly(2-methacryloyloxyethyl phosphorylcholine): Comparison with polysaccharide–water systems. Acta Biomater. 2010, 6, 2077–2082. [Google Scholar] [CrossRef]
- Inoue, Y.; Watanabe, J.; Takai, M.; Yusa, S.; Ishihara, K. Synthesis of sequence-controlled copolymers from extremely polar and apolar monomers by living radical polymerization and their phase-separated structures. J. Polym. Sci. Part. A Polym. Chem. 2005, 43, 6073–6083. [Google Scholar] [CrossRef]
- Boutevin, B.; Hervaud, Y.; Jeanmaire, T.; Boulahna, A.; Elasri, M. Monodealkylation des esters phosphoniques synthese de monosels et de monoacides phosphoniques. Phosphorus Sulfur Silicon Relat. Elem. 2001, 174, 1–14. [Google Scholar] [CrossRef]
- Olagnon-Bourgeot, S.; Chastrette, F.; Wilhelm, D. 31P NMR—Structure correlations for phosphonocarboxylic acids and esters. Magn. Reson. Chem. 1995, 33, 971–976. [Google Scholar] [CrossRef]
- Mabey, W.; Mill, T. Critical review of hydrolysis of organic compounds in water under environmental conditions. J. Phys. Chem. Ref. Data 1978, 7, 383–415. [Google Scholar] [CrossRef] [Green Version]
- Bauer, K.N.; Liu, L.; Wagner, M.; Andrienko, D.; Wurm, F.R. Mechanistic study on the hydrolytic degradation of polyphosphates. Eur. Polym. J. 2018, 108, 286–294. [Google Scholar] [CrossRef]
- Li, Y.; Liu, C.-M.; Yang, J.-Y.; Gao, Y.-H.; Li, X.-S.; Que, G.-H.; Lu, J.R. Anti-biofouling properties of amphiphilic phosphorylcholine polymer films. Colloids Surf. B Biointerfaces 2011, 85, 125–130. [Google Scholar] [CrossRef]
- Uilk, J.M.; Mera, A.E.; Fox, R.B.; Wynne, K.J. Hydrosilation-cured poly(dimethylsiloxane) networks: Intrinsic contact angles via dynamic contact angle analysis. Macromolecules 2003, 36, 3689–3694. [Google Scholar] [CrossRef]
- Martinelli, E.; Pretti, C.; Oliva, M.; Glisenti, A.; Galli, G. Sol-gel polysiloxane films containing different surface-active trialkoxysilanes for the release of the marine foulant ficopomatus enigmaticus. Polymer 2018, 145, 426–433. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Copolymer | PEtEP/MPC/PEGMA | p d | Yield | Water Solubility | ||
|---|---|---|---|---|---|---|
| (mol%) a | (mol%) b | (wt%) c | (%) | (%) | ||
| p(SiMA) | 0 | 0 | 0 | 96 | 74 | no |
| p(PEtEPMA) | 100 | 100 | 100 | 76 | 32 | complete |
| SiMA-co-PEtEPMA20 | 25 | 20 | 44 | 94 | 66 | no |
| SiMA-co-PEtEPMA53 | 50 | 53 | 78 | 96 | 44 | up to 1 g/L |
| SiMA-co-PEtEPMA66 | 70 | 66 | 86 | 94 | 54 | up to 1 g/L |
| SiMA-co-MPC20 | 30 | 20 | 7 | 96 | 49 | no |
| SiMA-co-PEGMA28 | 25 | 28 | 10 | 85 | 62 | no |
| Copolymer | Tg1 b (°C) | ∆Cp1 b (J/g K) | Tg2 c (°C) | ∆Cp2 c (J/g K) |
|---|---|---|---|---|
| p(SiMA) a | −124 | 1.160 | - | - |
| p(MPC) | - | - | - | - |
| p(PEtEPMA) | - | - | −31 | 0.54 |
| PEtEPMA | - | - | −31 | 0.46 |
| SiMA-co-PEtEPMA20 | −121 | 0.26 | −29 | 0.27 |
| SiMA-co-PEtEPMA53 | - | - | −27 | 0.40 |
| SiMA-co-PEtEPMA66 | - | - | −26 | 0.40 |
| SiMA-co-MPC20 | −119 | 0.49 | - | - |
| SiMA-co-PEGMA28 | −119 | 0.47 | - | - |
| Polymer Films | θ (°) |
|---|---|
| p(MPC) | ~0 |
| p(PEtEPMA) | ~0 |
| SiMA-co-PEtEPMA20 | 81 ± 3 |
| SiMA-co-PEtEPMA53 | 87 ± 4 |
| SiMA-co-PEtEPMA66 | 85 ± 6 |
| SiMA-co-MPC20 | 95 ± 2 |
| SiMA-co-PEGMA28 | 89 ± 4 |
| PDMS-Based Films | 0 Days θ (°) | 7 Days θ (°) | 14 Days θ (°) | 21 Days θ (°) | 28 Days θ (°) |
|---|---|---|---|---|---|
| SiMA-co-PEGMA28_10 | 101 ± 1 | 98 ± 3 | 96 ± 2 | 88 ± 4 | 74 ± 3 |
| SiMA-co-PEGMA28_20 | 103 ± 1 | 97 ± 1 | 97 ± 2 | 93 ± 6 | 96 ± 5 |
| SiMA-co-MPC20_10 | 107 ± 3 | 96 ± 2 | 98 ± 2 | 88 ± 4 | 85 ± 5 |
| SiMA-co-MPC20_20 | 108 ± 4 | 95 ± 1 | 94 ± 3 | 86 ± 3 | 68 ± 3 |
| SiMA-co-PEtEPMA20_10 | 104 ± 3 | 95 ± 3 | 93 ± 4 | 72 ± 5 | 65 ± 2 |
| SiMA-co-PEtEPMA20_20 | 103 ± 2 | 96 ± 2 | 94 ± 4 | 75 ± 3 | 70 ± 4 |
| PDMS | 102 ± 1 | 100 ± 2 | 99 ± 2 | 97 ± 1 | 94 ± 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guazzelli, E.; Lusiani, N.; Monni, G.; Oliva, M.; Pelosi, C.; Wurm, F.R.; Pretti, C.; Martinelli, E. Amphiphilic Polyphosphonate Copolymers as New Additives for PDMS-Based Antifouling Coatings. Polymers 2021, 13, 3414. https://doi.org/10.3390/polym13193414
Guazzelli E, Lusiani N, Monni G, Oliva M, Pelosi C, Wurm FR, Pretti C, Martinelli E. Amphiphilic Polyphosphonate Copolymers as New Additives for PDMS-Based Antifouling Coatings. Polymers. 2021; 13(19):3414. https://doi.org/10.3390/polym13193414
Chicago/Turabian StyleGuazzelli, Elisa, Niccolò Lusiani, Gianfranca Monni, Matteo Oliva, Chiara Pelosi, Frederik R. Wurm, Carlo Pretti, and Elisa Martinelli. 2021. "Amphiphilic Polyphosphonate Copolymers as New Additives for PDMS-Based Antifouling Coatings" Polymers 13, no. 19: 3414. https://doi.org/10.3390/polym13193414
APA StyleGuazzelli, E., Lusiani, N., Monni, G., Oliva, M., Pelosi, C., Wurm, F. R., Pretti, C., & Martinelli, E. (2021). Amphiphilic Polyphosphonate Copolymers as New Additives for PDMS-Based Antifouling Coatings. Polymers, 13(19), 3414. https://doi.org/10.3390/polym13193414