
DNA Interaction with a Polyelectrolyte Monolayer at Solution—Air Interface

 , ,
, , 
Abstract
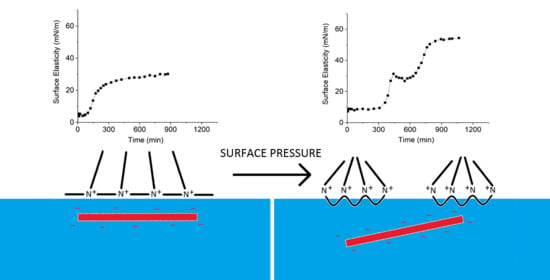
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sample Preparation
2.3. Methods
2.3.1. Dilatational Surface Elasticity and Surface Tension
2.3.2. Ellipsometry
2.3.3. IRRAS
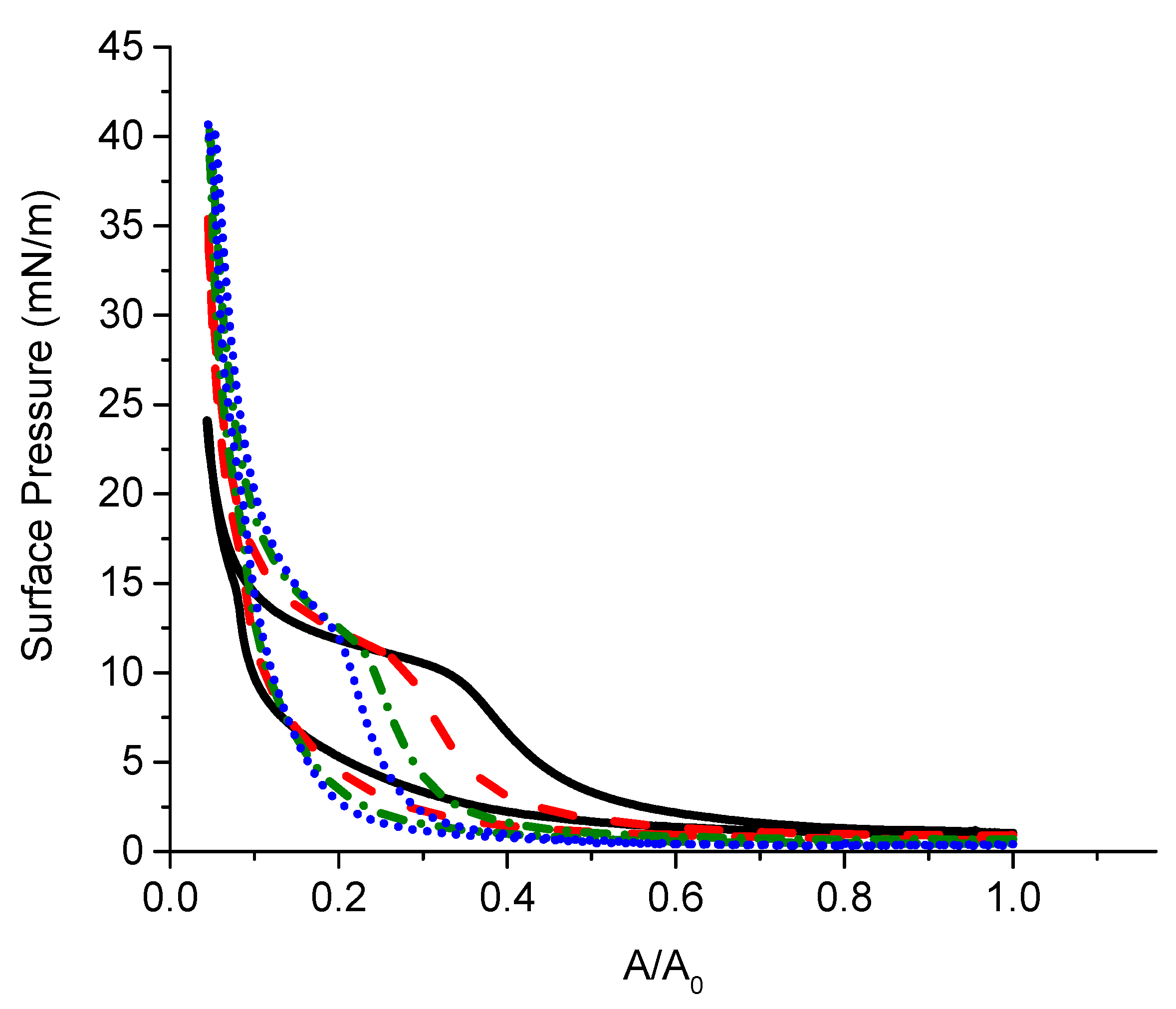
2.3.4. Surface Pressure-Area Isotherms
2.3.5. Atomic Force Microscopy
3. Results
3.1. PDAHMAC Layers on the Surface of Buffer Solutions
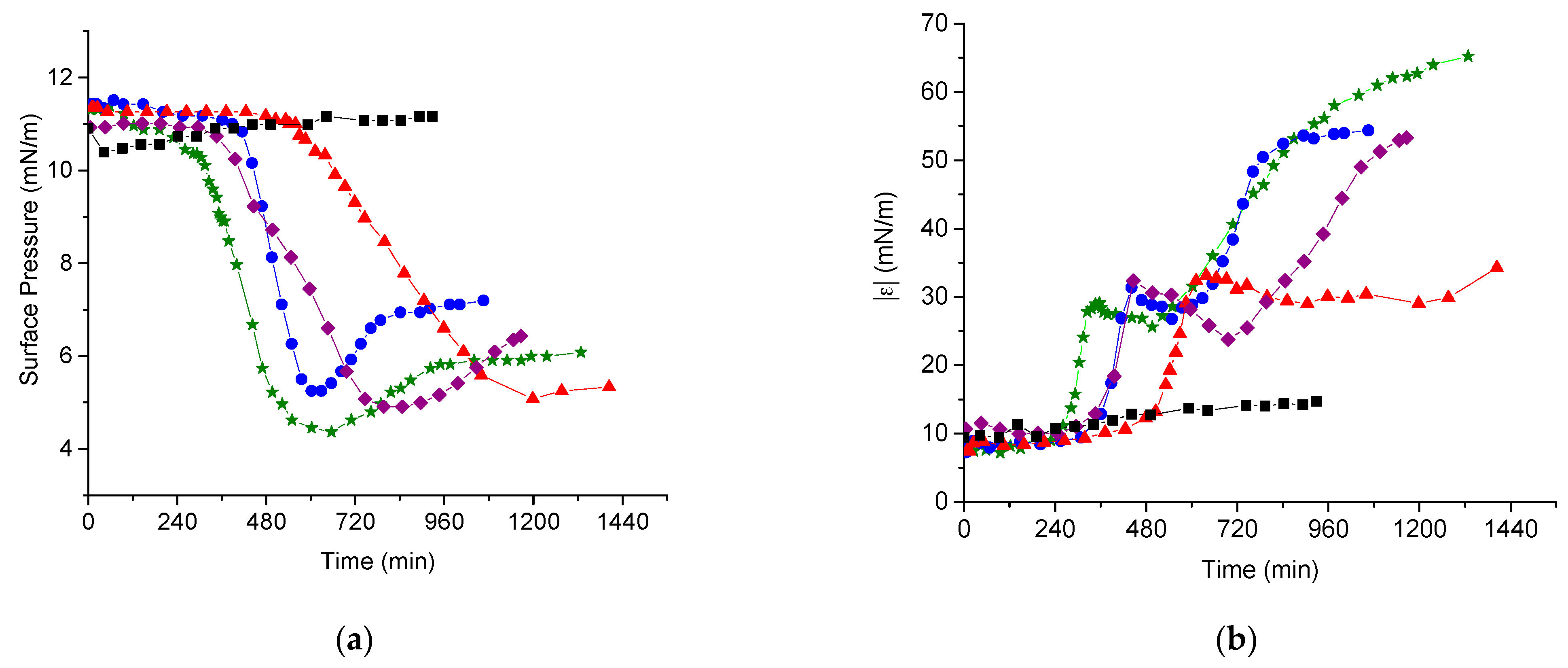
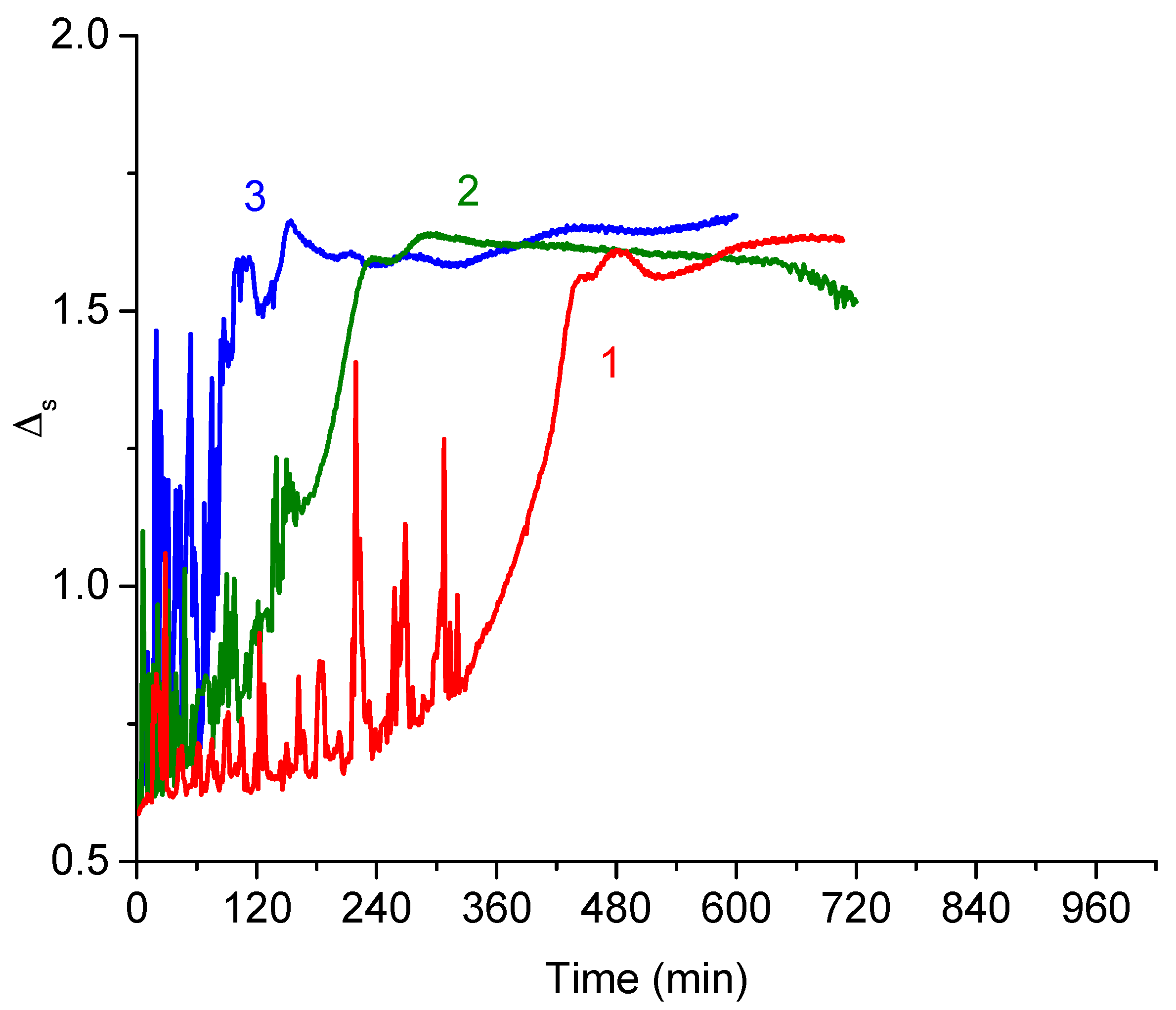
3.2. DNA Penetration into a PDAHMAC Layer, Plateau Region
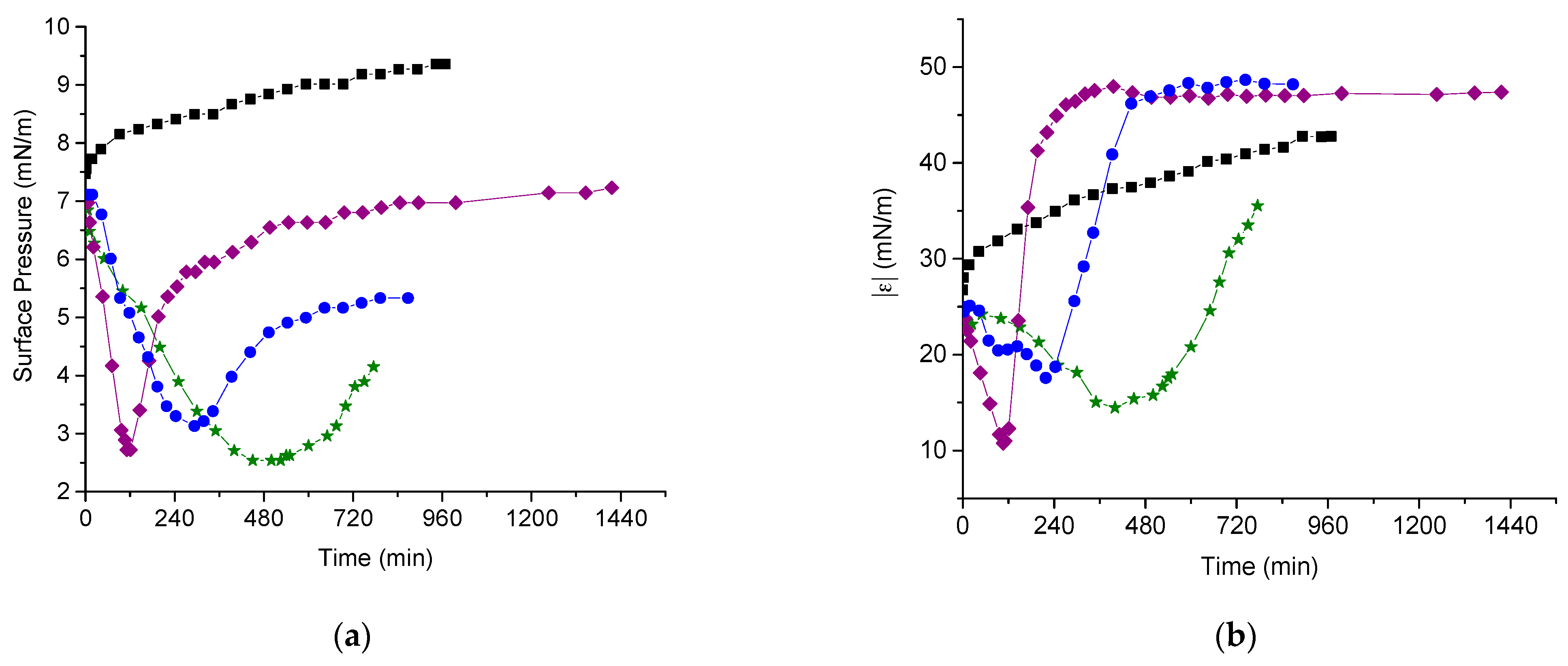
3.3. DNA Penetration into a PDAHMAC Layer, Low Surface Pressures
3.4. Microscopic Morphology of Mixed DNA/Layers
3.5. Ellipsometric Results
3.6. IRRAS
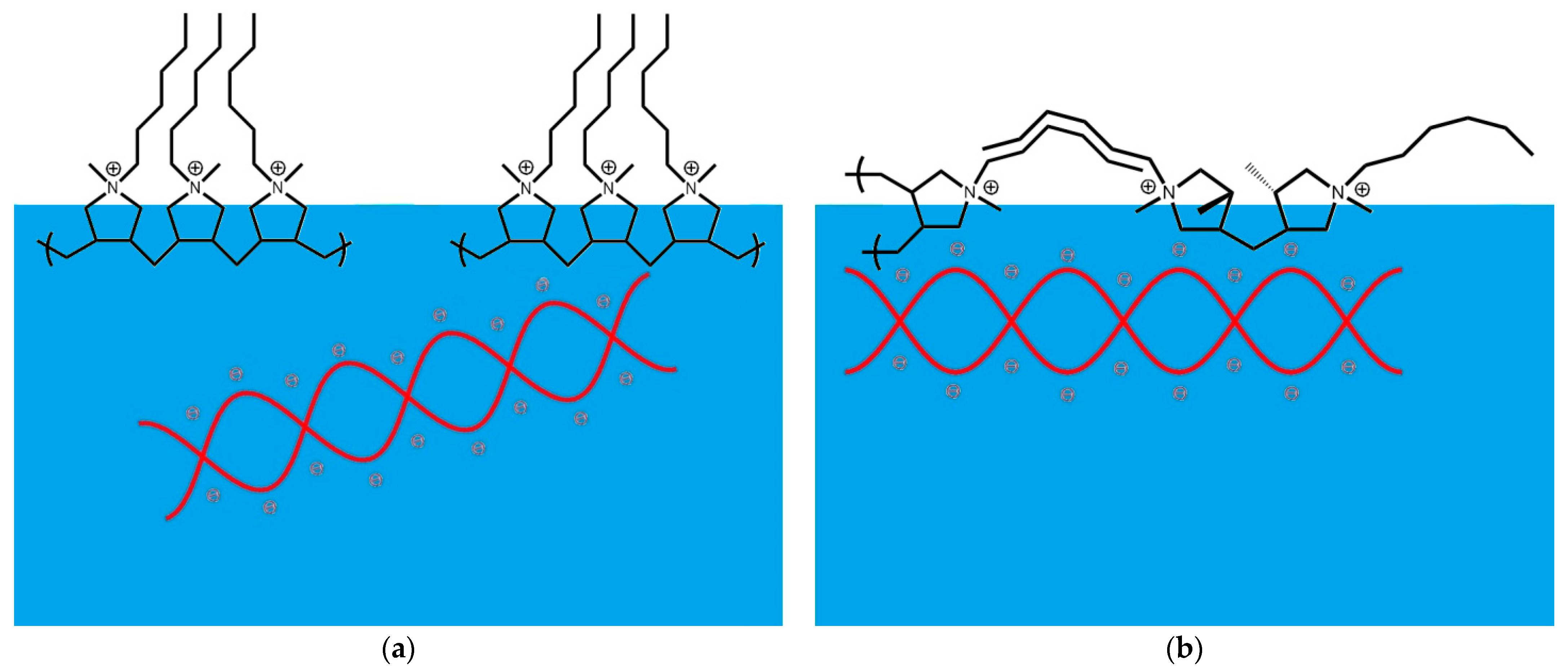
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dey, S.; Fan, C.; Gothelf, K.V.; Li, J.; Lin, C.; Liu, L.; Liu, N.; Nijenhuis, M.A.D.; Saccà, B.; Simmel, F.C.; et al. DNA Origami. Nat. Rev. Methods Prim. 2021, 1, 1–24. [Google Scholar]
- Kanno, T.; Tanaka, H.; Miyoshi, N.; Kawai, T. New Self-Fabrication of Large-Scale Deoxyribonucleic Acid Network on Mica Surfaces. Jpn. J. Appl. 2000, 39, L269. [Google Scholar] [CrossRef] [Green Version]
- Cristofolini, L.; Berzina, T.; Erokhin, V.; Tenti, M.; Fontana, M.P.; Erokhina, S.; Konovalov, O. The Structure of DNA-Containing Complexes Suggests the Idea for a New Adaptive Sensor. Colloids Surf. A Physicochem. Eng. Asp. 2008, 321, 158–162. [Google Scholar] [CrossRef]
- Bertin, A. Polyelectrolyte Complexes of DNA and Polycations as Gene Delivery Vectors. Adv. Polym. Sci. 2014, 256, 103–196. [Google Scholar]
- Seeman, N.C. Nucleic Acid Junctions and Lattices. J. Theor. Biol. 1982, 99, 237–247. [Google Scholar] [CrossRef]
- Rothemund, P.W.K. Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, S.; Maeda, Y.; Cai, L.T.; Tabata, H.; Kawai, T. Formation of Two Dimensional Network Structure of DNA Molecules on Highly Ordered Pyrolytic Graphite Surface. Jpn. J. Appl. Physics 2001, 40, 4217–4220. [Google Scholar] [CrossRef]
- Tanaka, S.I.; Cai, L.T.; Tabata, H.; Kawai, T. Formation of Two-Dimensional Network Structure of DNA Molecules on Si Substrate. Jpn. J. Appl. 2001, 40, 407–409. [Google Scholar] [CrossRef]
- Wu, A.; Li, Z.; Wang, E. DNA Network Structures on Various Solid Substrates Investigated by Atomic Force Microscopy. Anal. Sci. 2004, 20, 1083–1086. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Wei, C.; Li, Z.; Sun, Z.; Shen, R.; Zhang, Y. Self-Assembly of DNA Networks at the Air-Water Interface over Time. RSC Adv. 2013, 3, 16116–16121. [Google Scholar] [CrossRef]
- Okahata, Y.; Kobayashi, T.; Tanaka, K. Orientation of DNA Double Strands in a Langmuir-Blodgett Film. Langmuir 1996, 12, 1326–1330. [Google Scholar] [CrossRef]
- Kago, K.; Matsuoka, H.; Yoshitome, R.; Yamaoka, H.; Ijiro, K.; Shimomura, M. Direct in Situ Observation of a Lipid Monolayer—DNA Complex at the Air—Water Interface by X-Ray Reflectometry. Langmuir 1999, 15, 5193–5196. [Google Scholar] [CrossRef]
- Sastry, M.; Ramakrishnan, V.; Pattarkine, M.; Ganesh, K.N. Studies on the Formation of DNA-Cationic Lipid Composite Films and DNA Hybridization in the Composites. J. Phys. Chem. B 2001, 105, 4409–4414. [Google Scholar] [CrossRef]
- McLoughlin, D.; Langevin, D. Surface Complexation of DNA with a Cationic Surfactant. Colloids Surf. A Physicochem. Eng. Asp. 2004, 250, 79–87. [Google Scholar] [CrossRef]
- McLoughlin, D.; Dias, R.; Lindman, B.; Cardenas, M.; Nylander, T.; Dawson, K.; Miguel, M.; Langevin, D. Surface Complexation of DNA with Insoluble Monolayers. Influence of Divalent Counterions. Langmuir 2005, 21, 1900–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luque-Caballero, G.; Maldonado-Valderrama, J.; Quesada-Pérez, M.; Martín-Molina, A. Interaction of DNA with Likely-Charged Lipid Monolayers: An Experimental Study. Colloids Surf. B Biointerfaces 2019, 178, 170–176. [Google Scholar] [CrossRef]
- Luque-Caballero, G.; Maldonado-Valderrama, J.; Quesada-Pérez, M.; Martín-Molina, A. Atomic Force Microscopy as a Tool to Study the Adsorption of DNA onto Lipid Interfaces. Microsc. Res. Tech. 2017, 80, 11–17. [Google Scholar] [CrossRef]
- Dabkowska, A.P.; Barlow, D.J.; Campbell, R.A.; Hughes, A.V.; Quinn, P.J.; Lawrence, M.J. Effect of Helper Lipids on the Interaction of DNA with Cationic Lipid Monolayers Studied by Specular Neutron Reflection. Biomacromolecules 2012, 13, 2391–2401. [Google Scholar] [CrossRef]
- Erokhina, S.; Berzina, T.; Cristofolini, L.; Konovalov, O.; Erokhin, V.; Fontana, M.P. Interaction of DNA Oligomers with Cationic Lipidic Monolayers: Complexation and Splitting. Langmuir 2007, 23, 4414–4420. [Google Scholar] [CrossRef]
- Symietz, C.; Schneider, M.; Brezesinski, G.; Möhwald, H. DNA Alignment at Cationic Lipid Monolayers at the Air/Water Interface. Macromolecules 2004, 37, 3865–3873. [Google Scholar] [CrossRef]
- Antipina, M.N.; Schulze, I.; Heinze, M.; Dobner, B.; Langner, A.; Brezesinski, G. Physical-Chemical Properties and Transfection Activity of Cationic Lipid/DNA Complexes. ChemPhysChem 2009, 10, 2471–2479. [Google Scholar] [CrossRef] [Green Version]
- Gromelski, S.; Brezesinski, G. DNA Condensation and Interaction with Zwitterionic Phospholipids Mediated by Divalent Cations. Langmuir 2006, 22, 6293–6301. [Google Scholar] [CrossRef] [PubMed]
- Cristofolini, L.; Berzina, T.; Erokhina, S.; Konovalov, O.; Erokhin, V. Structural Study of the DNA Dipalmitoylphosphatidylcholine Complex at the Air-Water Interface. Biomacromolecules 2007, 8, 2270–2275. [Google Scholar] [CrossRef]
- Chen, Q.; Kang, X.; Li, R.; Du, X.; Shang, Y.; Liu, H.; Hu, Y. Structure of the Complex Monolayer of Gemini Surfactant and DNA at the Air/Water Interface. Langmuir 2012, 28, 3429–3438. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, J.; Shen, N.; Luo, Y.; Li, L.; Liu, M.; Thomas, R.K. Gemini Surfactant/DNA Complex Monolayers at the Air-Water Interface: Effect of Surfactant Structure on the Assembly, Stability, and Topography of Monolayers. Langmuir 2002, 18, 6222–6228. [Google Scholar] [CrossRef]
- Luque-Caballero, G.; Martín-Molina, A.; Sánchez-Treviño, A.Y.; Rodríguez-Valverde, M.A.; Cabrerizo-Vílchez, M.A.; Maldonado-Valderrama, J. Using AFM to Probe the Complexation of DNA with Anionic Lipids Mediated by Ca2+: The Role of Surface Pressure. Soft Matter 2014, 10, 2805–2815. [Google Scholar] [CrossRef] [Green Version]
- Lyadinskaya, V.V.; Lin, S.Y.; Michailov, A.V.; Povolotskiy, A.V.; Noskov, B.A. Phase Transitions in DNA/Surfactant Adsorption Layers. Langmuir 2016, 32, 13435–13445. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Zhang, X.; Du, Z.; Dang, H. Fabrication of Nanopatterned DNA Films by Langmuir-Blodgett Technique. Mater. Lett. 2005, 59, 423–429. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Wu, Y.J.; Gao, L.; Xing, T.L.; Ye, B.X. Electrochemical Behavior of 8-Azaguanine at DNA Langmuir—Blodgett Modified Glassy Carbon Electrode and Its Analytical Application. Electroanalysis 2009, 21, 1692–1698. [Google Scholar] [CrossRef]
- Wang, F.; Xu, Y.; Wang, L.; Lu, K.; Ye, B. Immobilization of DNA on a Glassy Carbon Electrode Based on Langmuir-Blodgett Technique: Application to the Detection of Epinephrine. J. Solid State Electrochem. 2012, 16, 2127–2133. [Google Scholar] [CrossRef]
- Hansda, C.; Hussain, S.A.; Bhattacharjee, D.; Paul, P.K. Adsorption Behavior of DNA onto a Cationic Surfactant Monolayer at the Air-Water Interface. Surf. Sci. 2013, 617, 124–130. [Google Scholar] [CrossRef]
- Lopes-Costa, T.; Gámez, F.; Lago, S.; Pedrosa, J.M. Adsorption of DNA to Octadecylamine Monolayers at the Air-Water Interface. J. Colloid Interface Sci. 2011, 354, 733–738. [Google Scholar] [CrossRef]
- Mora-Boza, A.; Lopes-Costa, T.; Gámez, F.; Pedrosa, J.M. Unveiling the Interaction of DNA-Octadecylamine at the Air-Water Interface by Ultraviolet-Visible Reflection Spectroscopy. RSC Adv. 2017, 7, 5872–5879. [Google Scholar] [CrossRef] [Green Version]
- Janich, C.; Hädicke, A.; Bakowsky, U.; Brezesinski, G.; Wölk, C. Interaction of DNA with Cationic Lipid Mixtures—Investigation at Langmuir Lipid Monolayers. Langmuir 2017, 33, 10172–10183. [Google Scholar] [CrossRef] [PubMed]
- Noskov, B.A.; Loglio, G.; Miller, R. Dilational Viscoelasticity of Polyelectolyte/Surfactant Adsorption Films at the Air/Water Interface: Dodecyltrimethylammonium Bromide and Sodium Poly (Styrenesulfonate). J. Phys. Chem. B 2004, 108, 18615–18622. [Google Scholar] [CrossRef]
- Noskov, B.A.; Loglio, G.; Miller, R. Dilational Surface Visco-Elasticity of Polyelectrolyte/Surfactant Solutions: Formation of Heterogeneous Adsorption Layers. Adv. Colloid Interface Sci. 2011, 168, 179–197. [Google Scholar] [CrossRef]
- Krycki, M.M.; Lin, S.Y.; Loglio, G.; Michailov, A.V.; Miller, R.; Noskov, B.A. Impact of Denaturing Agents on Surface Properties of Myoglobin Solutions. Colloids Surf. B Biointerfaces 2021, 202, 111657. [Google Scholar] [CrossRef]
- Chirkov, N.S.; Akentiev, A.V.; Campbell, R.A.; Lin, S.Y.; Timoshen, K.A.; Vlasov, P.S.; Noskov, B.A. Network Formation of DNA/Polyelectrolyte Fibrous Aggregates Adsorbed at the Water-Air Interface. Langmuir 2019, 35, 13967–13976. [Google Scholar] [CrossRef]
- Ainalem, M.L.; Campbell, R.A.; Nylander, T. Interactions between DNA and Poly(Amido Amine) Dendrimers on Silica Surfaces. Langmuir 2010, 26, 8625–8635. [Google Scholar] [CrossRef]
- Guo, X.; Zhang, H.; Wang, Y.; Pang, W.; Duan, X. Programmable Multi-DNA Release from Multilayered Polyelectrolytes Using Gigahertz Nano-Electromechanical Resonator. J. Nanobiotechnol. 2019, 17, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsvetkov, N.V.; Lezov, A.A.; Vlasov, P.S.; Lezova, A.A.; Samokhvalova, S.A.; Lebedeva, E.V.; Polushina, G.E. Copolymers of Diallyldimethylammonium Chloride and 2-(Diallyl(Methyl) Ammonio) Acetate: Effect of Composition and Ionic Strength on Conformational Properties. Eur. Polym. J. 2016, 84, 268–278. [Google Scholar] [CrossRef]
- Noskov, B.A.; Akentiev, A.V.; Bilibin, A.Y.; Zorin, I.M.; Miller, R. Dilational Surface Viscoelasticity of Polymer Solutions. Adv. Colloid Interface Sci. 2003, 104, 245–271. [Google Scholar] [CrossRef]
- Manning-Benson, S.; Bain, C.D.; Darton, R.C. Measurement of Dynamic Interfacial Properties in an Overflowing Cylinder by Ellipsometry. J. Colloid Interface Sci. 1997, 189, 109–116. [Google Scholar] [CrossRef]
- Tummino, A.; Toscano, J.; Sebastiani, F.; Noskov, B.A.; Varga, I.; Campbell, R.A. Effects of Aggregate Charge and Subphase Ionic Strength on the Properties of Spread Polyelectrolyte/Surfactant Films at the Air/Water Interface under Static and Dynamic Conditions. Langmuir 2018, 34, 2312–2323. [Google Scholar] [CrossRef] [PubMed]
- Assam, R.M.; Bashara, N.M. Ellipsometry and Polarised Light; North-Holland Publishing Company: Amsterdam, The Netherlands, 1977. [Google Scholar]
- Michailov, A.; Povolotskiy, A.; Kuzmin, V. Angular Invariance of the Contribution of an Anisotropic Thin Surface Layer to Reflectance and Reflectance-Absorbance. Opt. Express 2021, 29, 3090. [Google Scholar] [CrossRef]
- Langmuir, I.; Schaefer, V.J. Activities of Urease and Pepsin Monolayers. J. Am. Chem. Soc. 1938, 60, 1351–1360. [Google Scholar] [CrossRef]
- Campbell, R.A.; Tummino, A.; Varga, I.; Milyaeva, O.Y.; Krycki, M.M.; Lin, S.Y.; Laux, V.; Haertlein, M.; Forsyth, V.T.; Noskov, B.A. Adsorption of Denaturated Lysozyme at the Air-Water Interface: Structure and Morphology. Langmuir 2018, 34, 5020–5029. [Google Scholar] [CrossRef] [Green Version]
- Watson, J.D.; Crick, F.H.C. Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef]
- Erbe, A.; Bushby, R.J.; Evans, S.D.; Jeuken, L.J.C. Tethered Bilayer Lipid Membranes Studied by Simultaneous Attenuated Total Reflectance Infrared Spectroscopy and Electrochemical Impedance Spectroscopy. J. Phys. Chem. B 2007, 111, 3515–3524. [Google Scholar] [CrossRef] [Green Version]
- Tassler, S.; Pawlowska, D.; Janich, C.; Dobner, B.; Wölk, C.; Brezesinski, G. Lysine-Based Amino-Functionalized Lipids for Gene Transfection: The Influence of the Chain Composition on 2D Properties. Phys. Chem. Chem. Phys. 2018, 20, 6936–6944. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.W.; Lin, T.L.; Liu, I.T.; Hu, Y.; James, M. In Situ Neutron Reflectivity Studies of the Adsorption of DNA by Charged Diblock Copolymer Monolayers at the Air-Water Interface. Soft Matter 2012, 8, 7161–7168. [Google Scholar] [CrossRef]
- Ooi, Y.J.; Wen, Y.; Zhu, J.; Song, X.; Li, J. Surface Charge Switchable Polymer/DNA Nanoparticles Responsive to Tumor Extracellular PH for Tumor-Triggered Enhanced Gene Delivery. Biomacromolecules 2020, 21, 1136–1148. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Nagamani, S.; Liu, L.; Elghazaly, A.H.; Solin, N.; Inganäs, O. A DNA and Self-Doped Conjugated Polyelectrolyte Assembled for Organic Optoelectronics and Bioelectronics. Biomacromolecules 2020, 21, 1214–1221. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chirkov, N.S.; Campbell, R.A.; Michailov, A.V.; Vlasov, P.S.; Noskov, B.A. DNA Interaction with a Polyelectrolyte Monolayer at Solution—Air Interface. Polymers 2021, 13, 2820. https://doi.org/10.3390/polym13162820
Chirkov NS, Campbell RA, Michailov AV, Vlasov PS, Noskov BA. DNA Interaction with a Polyelectrolyte Monolayer at Solution—Air Interface. Polymers. 2021; 13(16):2820. https://doi.org/10.3390/polym13162820
Chicago/Turabian StyleChirkov, Nikolay S., Richard A. Campbell, Alexander V. Michailov, Petr S. Vlasov, and Boris A. Noskov. 2021. "DNA Interaction with a Polyelectrolyte Monolayer at Solution—Air Interface" Polymers 13, no. 16: 2820. https://doi.org/10.3390/polym13162820
APA StyleChirkov, N. S., Campbell, R. A., Michailov, A. V., Vlasov, P. S., & Noskov, B. A. (2021). DNA Interaction with a Polyelectrolyte Monolayer at Solution—Air Interface. Polymers, 13(16), 2820. https://doi.org/10.3390/polym13162820